
Abstract
The development of ischemic tolerance in the brain, whereby a brief period of sublethal ‘preconditioning’ ischemia attenuates injury from subsequent severe ischemia, may involve the activation of multiple intracellular signaling events that promote neuronal survival. In this study, the potential role of inducible DNA base-excision repair (BER), an endogenous adaptive response that prevents the detrimental effect of oxidative DNA damage, has been studied in the rat model of ischemic tolerance produced by three episodes of ischemic preconditioning (IP). This paradigm of IP, when applied 2 and 5 days before 2-h middle cerebral artery occlusion (MCAO), significantly decreased infarct volume in the frontal-parietal cortex 72 h later. Correlated with this protective effect, IP markedly attenuated the nuclear accumulations of several oxidative DNA lesions, including 8-oxodG, AP sites, and DNA strand breaks, after 2-h MCAO. Consequently, harmful DNA damage-responsive events, including NAD depletion and p53 activation, were reduced during postischemic reperfusion in preconditioned brains. The mechanism underlying the decreased DNA damage in preconditioned brain was then investigated by measuring BER activities in nuclear extracts. Betapolymerase-mediated BER activity was markedly increased after IP, and this activation occurred before (24 h) and during the course of ischemic tolerance (48 to 72 h). In similar patterns, the activities for AP site and 8-oxodG incisions were also upregulated after IP. The upregulation of BER activities after IP was likely because of increased expression of repair enzymes beta-polymerase, AP endonuclease, and OGG1. These results suggest that the activation of the BER pathway may contribute to IP-induced neuroprotection by enhancing the repair of endogenous oxidative DNA damage after ischemic injury.
Introduction
Oxidative damage to macromolecules may play a critical role in altering intracellular signaling pathways that determine the fate of injured cells after cerebral ischemia and reperfusion (Chan, 2001). Ischemia-induced oxidative damage to genomic DNA consists of lesions such as hydroxyl radical-modified bases, apurinic/apyrimidinic abasic site (AP site) lesions, and single-strand breaks (SSB) (Liu et al, 1996; Chen et al, 1997; Nagayama et al, 2000; Lan et al, 2003). These oxidative lesions, when unrepaired, may potently trigger various cell-killing signaling pathways (reviewed in Payne et al, 1995). Oxidative DNA damage occurs in neurons as early as minutes after transient cerebral ischemia (Chen et al, 1997; Cui et al, 2000; Lan et al, 2003; Liu et al, 1996), and, as an important distinctive feature, it is repairable in cells that eventually survive ischemia (Chen et al, 1997; Cui et al, 1999; Lan et al, 2003). In contrast, oxidative lesions are accumulated, mainly because of poor recovery, in cells of brain regions that ultimately develop DNA fragmentation and cell death (Cui et al, 2000; Fujimura et al, 1999; Kawase et al, 1999). Thus, accumulation of oxidative DNA damage is an important contributing factor in ischemic neuronal death (Cao et al, 2001; Eliasson et al, 1997; Endres et al, 1997).
The base-excision repair (BER) pathway is a critical mechanism for repair of oxidative DNA lesions in the brain. Base-excision repair is a highly coordinated process catalyzed by the sequential actions of a group of essential DNA repair enzymes (Frosina et al, 1996; Klungland and Lindahl, 1997; Srivastava et al, 1998), including specific DNA glycosylases, AP endonuclease, DNA polymerase-β, and DNA ligase I or DNA ligase III. In the brain, functional integrity of the BER pathway is essential for neurons to survive, as the neuronal genome constantly encounters oxidative damage (Ames et al, 1993; Nakamura and Swenberg, 1999). Since DNA damage is markedly increased in brain cells soon after ischemia/reperfusion, there are greatly increased demands for the BER pathway to repair the oxidative lesions in time to prevent their accumulation in injured cells. Recently, it has been shown that the rodent brain can quickly respond to oxidative DNA damage by markedly upregulating BER activities (Huang et al, 2000; Lan et al, 2003). Accordingly, it is speculated that BER, together with the endogenous antioxidant system, constitutes an important dual defense to protect against oxidative injury in the brain.
Ischemic tolerance (or ischemic preconditioning (IP)) is a phenomenon whereby a brief period of sublethal ‘preconditioning’ ischemia attenuates injury from subsequent severe ischemia. The precise mechanism of ischemic tolerance in the brain is unknown, but it likely involves a complex multiple-factor process requiring new protein synthesis and activation of intracellular prosurvival molecules or signaling pathways (reviewed in Chen and Simon, 1997; Dirnagl et al, 2003; Kirino, 2002; Perez-Pinzon, 2005). In this study, we have explored a new aspect of endogenous neuroprotective mechanism in the rat brain induced by IP. Using a well-characterized rat model of ischemic tolerance (Chen et al, 1996), we identified an inducible BER response in the brain after IP. Furthermore, we found that this inducible BER response may contribute to neuroprotection by markedly enhancing the repair of endogenous oxidative DNA lesions and, consequently, preventing the pro-death signaling pathways evoked by accumulated DNA damage.
Materials and methods
Animal Methods
All animal procedures were performed using protocols approved by the Animal Care Committee at the University of Pittsburgh and in accordance with the principles outlined in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Temporary focal ischemia was induced in male Sprague–Dawley rats weighing 275 to 310 g using the intraluminal vascular occlusion method as previously described (Chen et al, 1996, 1997). In brief, anesthesia was induced with 4% isoflurane in a mixture of 66% N2O and 30% O2. The animals were then intubated and ventilated with 1.5% isoflurane. Rectal and temporalis muscle temperatures were monitored and maintained between 36.5°C and 37.5°C using a heating pad and a temperature-regulated heating lamp throughout the experiment. The left femoral artery was cannulated for recording arterial blood pressure and for measurement of blood gases and blood glucose concentration. After isolation of the internal carotid artery (ICA), a 4 to 0 surgical monofilament nylon suture with a rounded tip was introduced into the ICA lumen through the external carotid artery stump and advanced 20 to 21 mm past the common carotid artery bifurcation. Preconditioning ischemia was induced, and the suture was gently withdrawn to permit reperfusion. Shamoperated rats underwent identical surgery except the suture was not inserted. At a later time, the suture was reinserted to induce prolonged middle cerebral artery occlusion (MCAO) for 2 h. After ischemia was induced, the suture was again gently withdrawn to permit reperfusion. The external carotid stump was ligated and the wound was closed.
To ensure the induction of ischemia by MCAO, regional cerebral blood flow (rCBF) was monitored in two groups of rats (n = 8 per group) using laser Doppler flowmetry (LDF, PeriFlux System 5,000, PERINMED). Two cortical regions were measured: the ipsilateral ischemic parietal cortex and the contralateral nonischemic parietal cortex. In brief, each animal was mounted in a stereotaxic frame. Under microscopic guidance, two burr holes (1 mm in diameter) were drilled in the following coordinates: 0.5 mm anterior and 5.0 mm lateral. Then, LDF probes (PROBE 403, Fiber separation = 0.25 mm) were placed on each cortical region with a micromanipulator. Blood flow was measured (2 Hz sampling rate) before, during, and up to 30 mins after MCAO.
The preconditioning paradigm used in this study was adopted from our previous study (Chen et al, 1996), which identified three 10-min episodes of transient focal ischemia with 45-min intervals as the optimal preconditioning to induce tolerance. The optimal interval between the preconditioning ischemia and prolonged ischemia was 2 to 3 days. Under this preconditioning paradigm, cortical infarct volume after 2 h of MCAO was reduced by half as compared with animals receiving sham operation (Chen et al, 1996).
Measurement of Infarct Volume
At 72 h after the 2-h MCAO, brains were removed and the forebrain was sliced into coronal sections of 2 mm thickness. Sections were stained with 2,3,5-triphenyl-tetrazolium (3%). Infarct volume was determined using the MCID image analysis system as described (Chen et al, 1996). Animals that showed a massive hematoma in the brain or no infarction in the brain were omitted from further histological analysis.
In a separate experiment, the effect of IP on infarct volume was confirmed using cresyl violet staining on freshly frozen sections. Twenty-micrometer-thick serial coronal sections were obtained every 0.4 mm between the levels of +5.0 and −5.0 mm (anterior–posterior) from the bregma. The sections were stained with cresyl violet for computerized image analysis of infarct volume as described previously (Chen et al, 1996).
Measurement of 8-OHdG/2-dG Ratio Using HPLC-EC Detection
The content of 8-hydroxyl-2′-deoxyguanosine (8-OHdG), a hallmark of oxidative DNA base damage, was measured in nuclear DNA extracts from the brain tissues using highperformance liquid chromatography with electrochemical (HPLC-EC) detection (Lan et al, 2000; Nagayama et al, 2000). Rats were killed at 0, 0.25, 0.5, 2, 8, 24, and 72 h after 2 h of focal ischemia, with or without preconditioning ischemia (n = 6 to 8 per time point). The brains were rapidly removed, and the parietal cortices and caudate putamina were separately dissected and stored at −80°C. The methods for nuclear DNA extraction and digestion were as previously described (Lan et al, 2000). This protocol avoided the use of phenol or any known oxidants that may induce background oxidative lesions, 8-OHdG or AP sites.
The isocratic analysis was performed on a CoulArray system (Model 5600) equipped with a dual piston (Model 580) and a PEEK pulse damper (ESA, Inc., Chelmsford, MA, USA). The analysis was performed using two coulometer array cell columns. The data were acquired and analyzed using the CoulArray software, and expressed as the number of 8-OHdG in 105 2-dG determined in the same sample.
Quantitative Measurement of AP Sites in Nuclear DNA
Nuclear DNA isolated from ischemic and sham brain tissues (n = 6 to 8 per time point) were subjected to quantitative measurement of AP sites using the calorimetric assay previously described (Nagayama et al, 2000). A biotin-labeled reagent specific for the aldehyde group in the ring-open form of AP site, designated as Aldehyde Reactive Probe (ARP), was used for the detection of AP sites (Dojindo Molecular Technologies, Gaithersburg, MD, USA). Aldehyde Reactive Probe specifically binds to AP sites in isolated genomic DNA, and the biotin molecule in ARP can then be detected calorimetrically using a streptavidin/biotin complex conjugated to horseradish peroxidase as the indicator enzyme (Kubo et al, 1992). All ARP assays were performed in triplicate, and the means were calculated. The data, expressed as the number of AP sites per 105 nucleotides, were calculated based on the linear calibration curve generated for each experiment using ARP-DNA standard solutions.
Detection of DNA Single-Strand Breaks by Nick-Translation
The DNA polymerase I-mediated biotin-dATP nicktranslation (PANT) assay was performed on fresh-frozen sections from brains subjected to 2 h of MCAO at 0.5, 4, 8, 24, and 72 h of reperfusion, with or without preconditioning ischemia (n = 6 per time point). The procedures for PANT have been described previously (Chen et al, 1997). In brief, sections were air-dried, fixed in ethanol/acetic acid (2:1 vol/vol) for 5 mins, and washed three times in PBS. The sections were permeabilized with 1% Triton X-100 for 20 mins, and were then incubated in a moist-air chamber at 37°C for 90 mins with the PANT reaction mixture containing 5 mmol/L MgCI2; 10 mmol/L 2-mercaptoethanol; 20 μg/mL bovine serum albumin; dGTP, dCTP, and dTTP at 30 μmol/L each; 29 μmol/L biotinylated dATP; 1 μmol/L dATP; and 40 U/mL Escherichia coli DNA polymerase I (Sigma) in phosphate-buffered saline (PBS) (pH 7.4). The biotin-dATP incorporated in DNA was detected using Texas Red Avidin D (cell sorting grade; Vector Laboratories, Burlingame, CA, USA). Polymerase I-mediated biotin-dATP nick-translation-positive cells were quantified using a computerized scanning program (MCID, St Catharines, Ontario, Canada) as described previously (Chen et al, 1997; Nagayama et al, 2000).
To colocalize DNA single- and double-strand breaks in ischemic cells, double-label of PANT and TUNEL was performed using sections obtained at 24 h after 2-h MCAO (n = 4 per group) as previously described (Nagayama et al, 2000). TUNEL staining was first performed using biotin-16-dUTP and fluorescein-avidin D (Vector Laboratories). This was followed by an avidin/biotin-blocking procedure according to the manufacturer's instructions (Vector Laboratories). Then, the sections were subjected to PANT staining as described above. Sections were examined by fluorescence microscopy using excitation/emission wavelengths of 550/565 nm (red) and 495/515 nm (green), respectively.
Detection of Cleavage Activities for 8-oxodG or AP Sites in Nuclear Extracts
The oligonucleotide incision assay was performed as described previously (Lan et al, 2003). This assay estimates the ability of nuclear proteins to recognize and remove two types of DNA lesions, 8-oxodG or AP sites. The repair substrate used in this assay was a 50-mer oligonucleotide with a precisely positioned 8-oxodG or synthetic AP site at position 26 (5′-TCG GTA CCC GGG GAT CCT CTA GAG TOG ACC TGC AGG CAT GCA AGC TTG GC-3′; 0 = 8-oxodG or AP site). The oligonucleotide was 5′-end-labeled using T4 polynucleotide kinase and [γ-32P]ATP, and then passed through a G-25 spin column to remove the free unlabeled [γ-32P]ATP. The labeled oligonucleotide was annealed to the complementary oligonucleotides in 100 mmol/L KCl, 10 mmol/L Tris (pH 7.8), and 1 mmol/L EDTA by heating the samples to 80°C (for 8-oxodG-containing oligonucleotides) or 55°C (for AP sitecontaining oligonucleotides), and then allowing them to cool down slowly to room temperature. The reaction mixture for the incision assay contained 40 mmol/L HEPES–KOH (pH 7.6), 75 mmol/L KCl, 2 mmol/L DTT, 1 mmol/L EDTA, 0.1 mg/mL BSA, 2 mmol/L CaCl, 20 μmol/L zinc acetate, 10% glycerol, 400 fmol of 32P-labeled DNA duplex, and nuclear protein extracts in the indicated amounts. The reaction was incubated at 32°C for 60 mins, and then terminated. The DNA was ethanolprecipitated and then resuspended in formamide dye containing 90% formamide, 0.002% bromphenol blue, and 0.002% xylene cyanol. The samples were subjected to electrophoresis on a denaturing 20% Polyacrylamide gel containing 7 mol/L urea. The incision products were analyzed by autoradiography and densitometry analysis.
Detection of DNA Polymerase-β Activity in Nuclear Extracts
The principle of this repair assay has been described previously (Lan et al, 2003). To perform the assay, the 50-mer oligonucleotide (400 fmol) containing a uracil at position 26 (sequence: 5′-TCG GTA CCC GGG GAT CCT CTA GAG TUG ACC TGC AGG CAT GCA AGC TTG GC-3′) was annealed to the complementary oligonucleotide. The DNA duplex was subjected to lesion-digestion using purified UDG (5U) and endonuclease IV (10 U) at 37°C for 15 mins, and then the mixture was heated to 55°C for 10 mins to inactivate UDG and endonuclease IV. This reaction produced a single-nucleotide nick at position 26 in the DNA duplex, which subsequently served as the repair substrate for DNA polymerase-β. Nuclear extracts at the indicated protein concentrations were then incubated with this repair substrate in the same buffer as above with the additions of 2 μCi of [α-32P]CTP, 40 mmol/L phosphocreatine, 2.5 μg creatine Phosphokinase, 3% glycerol, 2 mmol/L NAD+, and 1 mmol/L β-mercaptoethanol. The reaction was performed at 32°C for 60 mins before it was terminated by adding an equal volume of loading buffer and heating to 80°C for 2 mins. The reaction products were separated in a 15% Polyacrylamide gel containing 7 mol/L urea, and detected using autoradiography.
Detection of DNA Ligase Activity in Nuclear Extracts
DNA ligase activity was examined using the oligonucleotide ligation assay as previously described (Chen et al, 2000, 2002). This was performed by incubation of nuclear extracts with the DNA substrates prepared by annealing 10 pmol of 5′-32P-oligo(dT)18-l μg of poly(rA)100 in reaction mixtures containing DNA ligase buffer and 1 mmol/L ATP, 40 mmol/L phosphocreatine, and 2.5 μg creatine Phosphokinase. The ligase buffer consisted of 20 mmol/L Tris (pH 8.0), 40 mmol/L NaCl, 5 mmol/L MgCl2, 5 mmol/L DTT, 8% glycerol, and 0.02% Triton X-100. The reaction was continued for 30 mins at 32°C, and the ligation products were analyzed by electrophoresis on a 10% Polyacrylamide gel containing 8 mol/L urea and detected by autoradiography.
Western Blot Analysis
The levels of selective BER enzymes were examined in nuclear protein extracts from ischemic brains using Western blot analysis. The tested essential BER enzymes included DNA polymerase-β, AP endonuclease (APE), OGG1, DNA ligase I, and DNA ligase III. Rats were killed at 2, 8, 24, 48, 72, or 168 h after preconditioning ischemia or sham operation (n = 4 per experimental condition). Cortices and caudate putamina were dissected separately, and subjected to nuclear protein extraction and Western blot analysis using the standard method. The working dilutions for primary antibodies were as follows: DNA polymerase-β (1:500) and OGG1 (1:500) monoclonal antibodies (NeoMarkers); APE (1:1,000), DNA ligase (1:1,000), and DNA ligase III (1:500) monoclonal antibodies (Novus Biologicals, Littleton, CO, USA).
Northern Blot Analysis
The cDNA encoding the open reading frame of the rat DNA polymerase-β or AP endonuclease was obtained using PCR from a rat brain cDNA library previously compiled in this laboratory (Chen et al, 1998). The sequences of the obtained cDNAs were confirmed by sequencing reaction on both strands (University of Pittsburgh Sequencing Service Facility).
To perform Northern blot analysis, total cellular RNA was extracted from brains subjected to IP followed by 24- or 48-h reperfusion, or extracted 24 h after sham operation (3 animals per experimental condition). Northern blot analysis was performed using 32P-labeled cDNA probes as described previously (Chen et al, 1998).
In Situ Hybridization
In situ hybridization was performed using brains subjected to IP followed by 24- or 48-h reperfusion or on sham-operated brains 24 h after sham operation (4 animals per experimental condition). The procedures were the same as described previously (Chen et al, 1998). Briefly, the 35S-labeled single-stranded RNA probe was prepared from plasmids containing the rat DNA polymerase-β or AP endonuclease cDNA inserts. The brain sections were hybridized with the labeled RNA probe (1 × 107 c.p.m./mL) in a hybridization cocktail for 18 h at 55°C. All slides were exposed to the same sheet of Kodak SB-5 film for 3 weeks, and then developed.
Determination of NAD Content in Rat Brain
Brain NAD content was quantitatively measured in the cortex and caudate putamina, respectively, at 2 and 8 h after 2 h of MCAO with or without IP (n = 5 per group). The in situ freezing technique described by Ponten et al (1973) was adopted in this study to minimize the changes of labile cerebral metabolites during decapitation and brain dissecting procedures. In brief, the rats were anesthetized, intubated, and ventilated with 1.5% isoflurane in a mixture of 68.5% N2O and 30% O2. A skin incision was made in the midline to expose the skull, and a plastic funnel with a bottom diameter of approximately 15 mm was fitted into the skin incision and secured by pulling the skin up around the funnel with sutures. Brain freezing was achieved by liquid N2 through the funnel, which continued for 3 mins; thereafter the respiratory tubing was disconnected and the whole head was immersed in liquid N2. Dissection of brains was performed at −22°C, and approximately 50 mg of brain tissues from the cortex and caudate putamina within the MCA territory was used for NAD measurement. The tissue was homogenized in 400 μL of 0.5 mol/L HClO4 at 4°C and centrifuged (3,000g for 15 mins, −2°C). The pellet was used for protein determination and the supernatant was neutralized with 800 μL of 2 mol/L KOH/0.2 mol/L K2HPO4–KH2PO4 NAD content in the supernatant was measured using an enzymatic cycling method in which NAD is converted to NADH by alcohol dehydrogenase (Zhang et al, 1994). The absorbance was measured at 556 nm using a spectrophotometer (Beckman DU 7400). The data, expressed as pmol/mg protein, were calculated based on the linear calibration curve generated for each experiment using NAD standard solutions.
Immunohistochemistry
To detect the distribution of poly(ADP-ribosyl)ation after ischemia, immunohistochemistry was performed using fresh frozen brain sections obtained at 2 and 8 h after 2 h of MCAO, with or without IP. Standard protocols were used for immunohistochemistry. The primary antibody was the rabbit anti-poly(ADP-ribose) polyclonal antibody (BIOMOL Research Laboratories, Plymouth Meeting, PA, USA), used at a dilution of 1:100, followed by the biotinylated goat anti-rabbit IgG secondary antibody (1:1,000) and the avidin-biotin–peroxidase method. Alternate sections were immunoreacted without the primary antibody as a control.
To detect the cellular changes in β-pol protein expression, immunohistochemistry was performed using fresh frozen brain sections obtained at 48 h after sham operation or preconditioning (n = 4 per group). The primary antibody (monoclonal anti-β-pol) was used at a dilution of 1:250, followed by the standard procedures. As a control, alternative sections were incubated with the primary antibody that had been reabsorbed with purified β-pol at the antigen/antibody ratio of 10:1.
Statistical Analysis
The statistical significance between groups was determined with analysis of variance (ANOVA). Post hoc testing used the Bonferroni t-test, and P < 0.05 was accepted as statistically significant. Differences in physiologic parameters and rCBF were analyzed using repeated measures ANOVA. All values are expressed as mean ± s.d.
Results
Preconditioning Ischemia Affects Infarct Volume
The effect of preconditioning ischemia on the infarct volume is summarized in Figure 1A. Control animals received only the 2 h of MCAO; their infarct volume was the same as animals receiving 2 h of ischemia but previously subjected to a sham operation 3 days before (data not shown). However, infarct volumes of animals with 3-day intervals between the preconditioning ischemia and 2 h of focal ischemia were significantly reduced. A significant reduction in infarct volume was also shown when the interval was 2 or 5 days; but the 1-day or 7-day interval after the preconditioning ischemia showed no protection. There were no statistically significant differences in the physiologic variables (temporalis temperature, blood pressure, blood glucose, and blood gases) among any of the experimental groups (Table 1).
Physiologic variables in ischemic preconditioning experiments
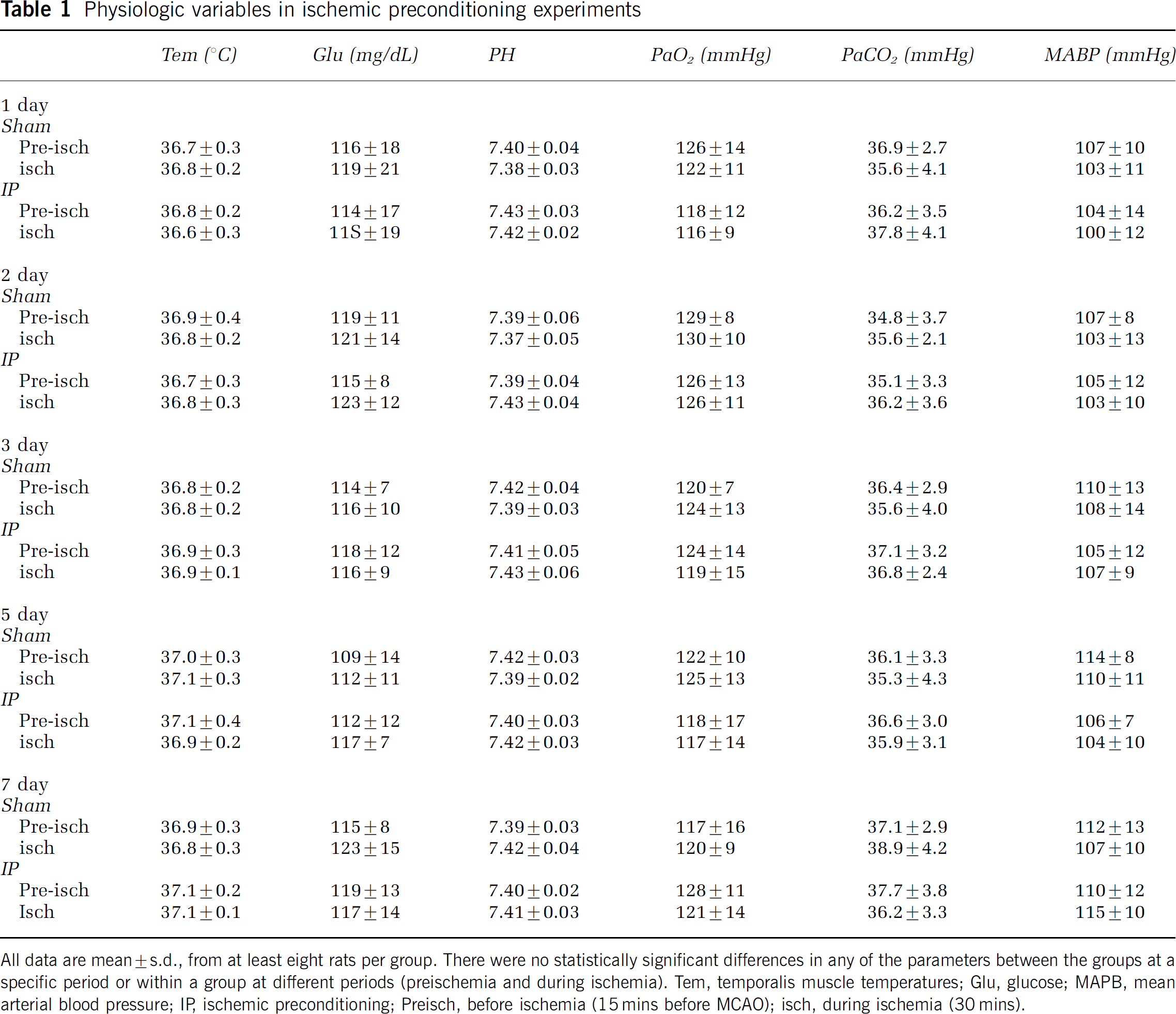
All data are mean ± s.d., from at least eight rats per group. There were no statistically significant differences in any of the parameters between the groups at a specific period or within a group at different periods (preischemia and during ischemia). Tern, temporalis muscle temperatures; Glu, glucose; MAPB, mean arterial blood pressure; IR ischemic preconditioning; Preisch, before ischemia (15 mins before MCA0); isch, during ischemia (30 mins).
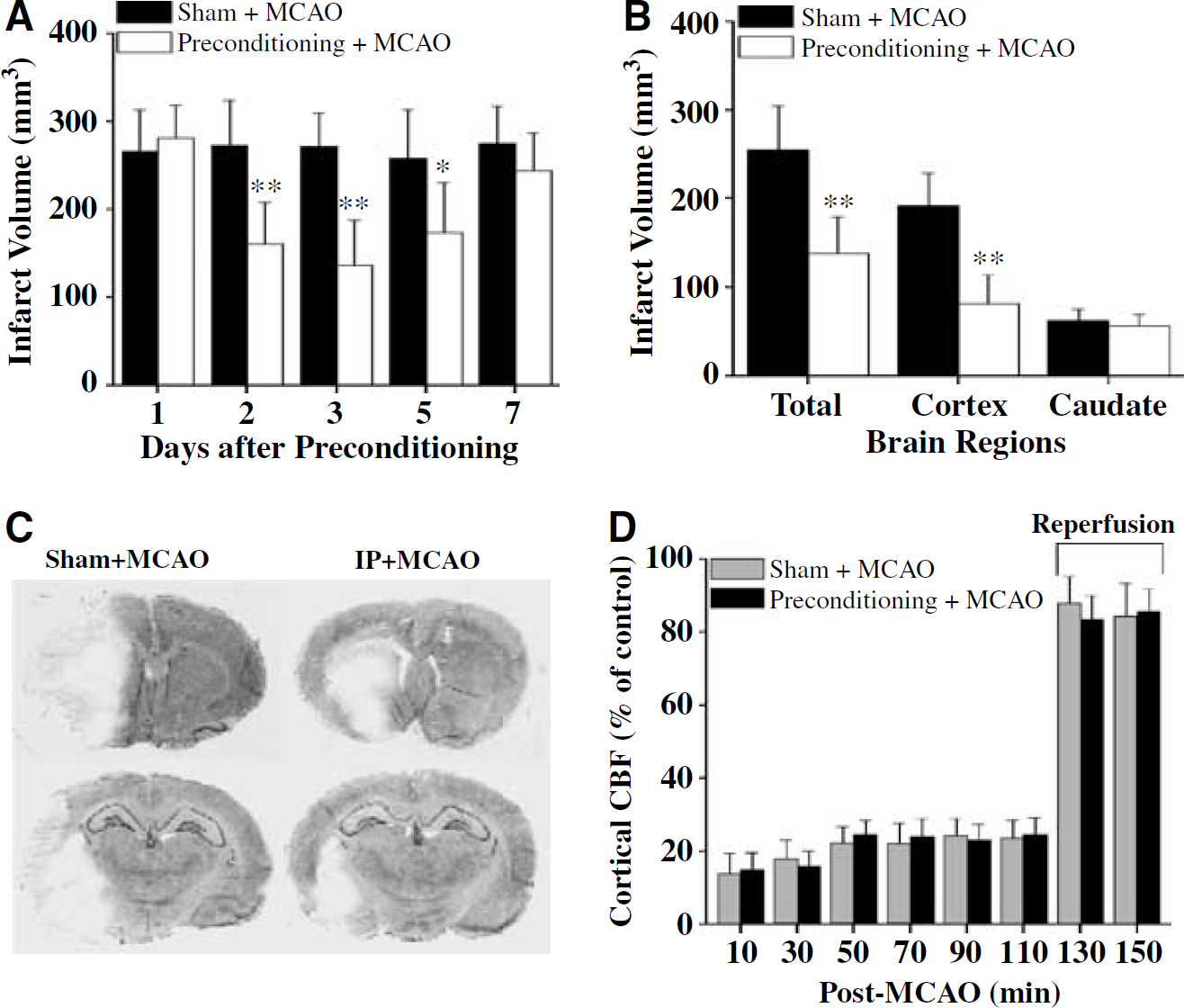
Verification of the rat ischemic tolerance model. (
To confirm the TTC-staining results, infarct volume was also measured in cresyl violet-stained sections from rats killed 3 days after 2 h of focal ischemia with or without preconditioning ischemia. Figures 1B and 1C illustrates the effect of preconditioning ischemia with the 3-day interval on infarct volume. Significant reduction in infarct volume was detected in the cortex, but not in the caudate putamina, of brains receiving preconditioning ischemia. Regional cortical blood flow measured using LDF during and after 2 h of focal ischemia showed no significant differences between the preconditioning and sham groups (Figure 1D).
Ischemic Preconditioning Enhances the Repair of Endogenous Oxidative DNA Damage
To investigate the effect of IP on the induction profiles of oxidative DNA damage after severe focal ischemia and reperfusion, the frontal/parietal cortices and caudate-putamina were dissected separately at various time points after 2 h of focal ischemia with prior preconditioning or sham operation (n = 8 per group), and then subjected to quantitative measurement for nuclear contents of 8-oxodG and AP site lesions. Figures 2 and 3 illustrate the profiles of 8-oxodG and AP site induction, respectively, in the cortex and caudate during postischemic reperfusion. Both types of lesions were significantly increased in the cortex and caudate within 15 mins of reperfusion; the levels were not significantly different at that time point between groups receiving either preconditioning ischemia or sham operation 3 days before. Thereafter, in sham-treated animals, the levels of 8-oxodG and AP sites were persistently increased in both the cortex and caudate throughout the time course of reperfusion (0.5 to 72 h). In contrast, in preconditioned animals the levels of 8-oxodG and AP site lesions in the cortex were significantly decreased compared with sham controls. However, IP had insignificant effects on the levels of 8-oxodG and AP sites in the caudate after 2 h of focal ischemia. These results show that IP diminished the nuclear accumulation of 8-oxodG and AP site lesions in the cortex but not in the caudate after subsequent severe focal ischemia. Thus, the effect of preconditioning on postischemic DNA base damage in the cortex is correlated with its protective effect in this region.
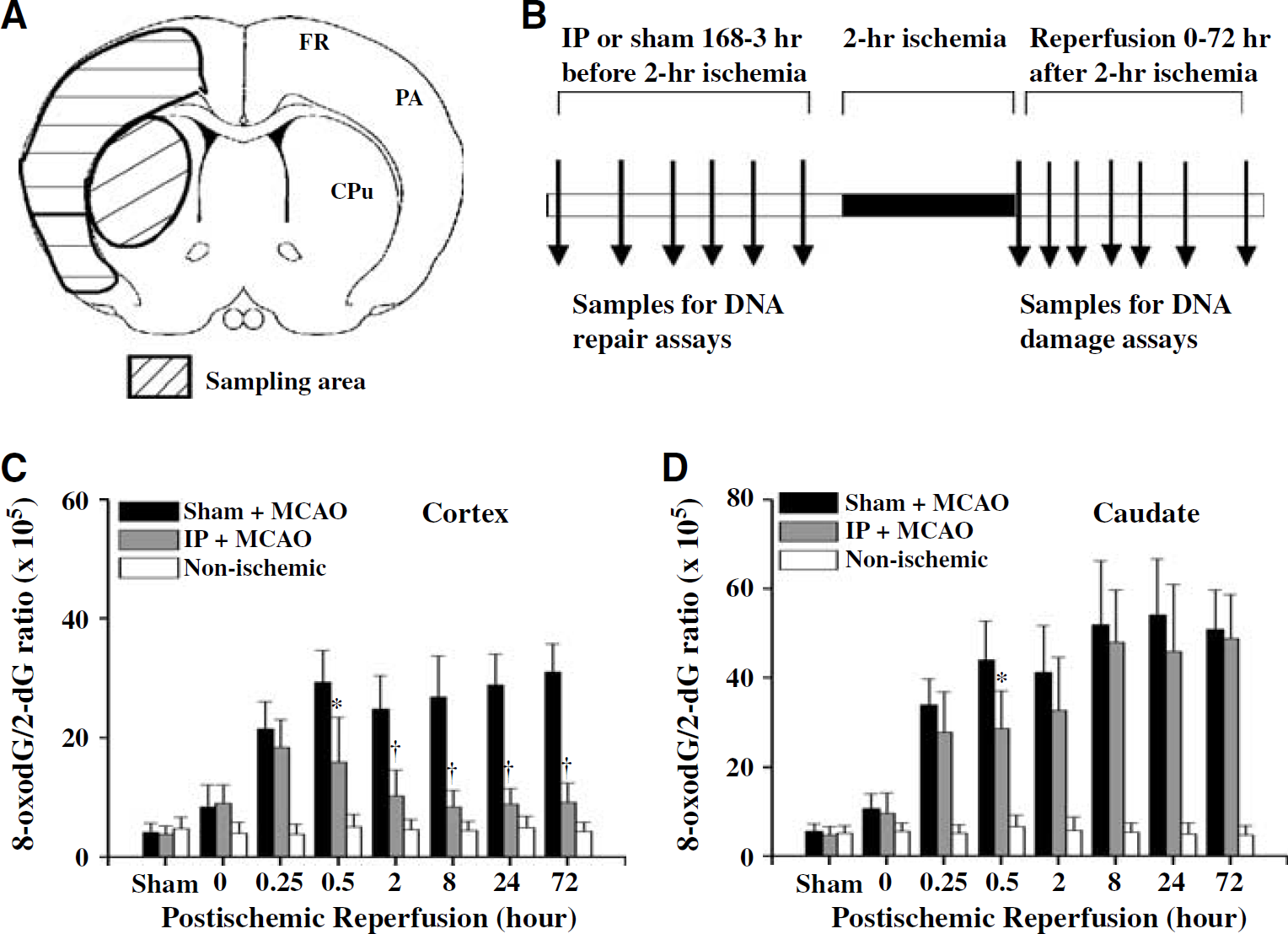
Ischemic preconditioning (IP) attenuates the induction of DNA 8-oxodG in the cerebral cortex after focal ischemia and reperfusion. (
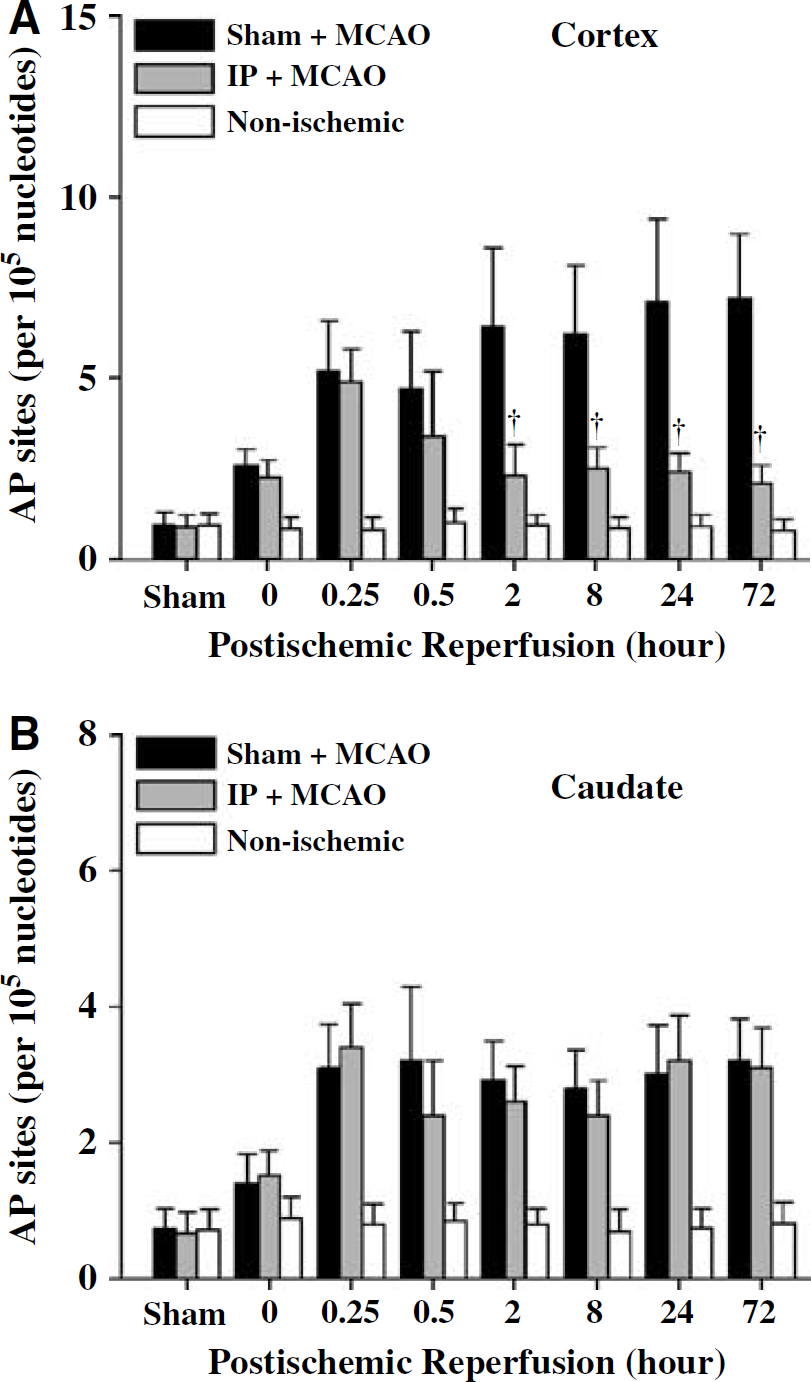
Ischemic preconditioning (IP) attenuates the induction of DNA AP sites in the cerebral cortex after focal ischemia and reperfusion. Induction of AP sites in the cortex (
Single-strand breaks are another form of oxidative DNA damage that occurs during early reperfusion periods after severe ischemia and are thought to directly contribute to ischemic cell death (Chen et al, 1997; Nagayama et al, 2000). In this study, the effect of IP on the induction of SSB after subsequent 2-h focal ischemia was also investigated. The PANT assay was performed to detect SSB using freshly prepared coronal brain sections at various time points after ischemia, and the results are summarized in Figures 4A and 4B. Ischemic preconditioning significantly attenuated the induction of SSB in the cortex during postischemic reperfusion. This protective effect by preconditioning was incomplete, though, as significant amounts of SSB-containing cells (approximately 20% to 40% of the amounts in the sham controls) were detectable at each time point after ischemia. Moreover, consistent with the 8-oxodG and AP site data, preconditioning had an insignificant effect on SSB induction in the caudate.
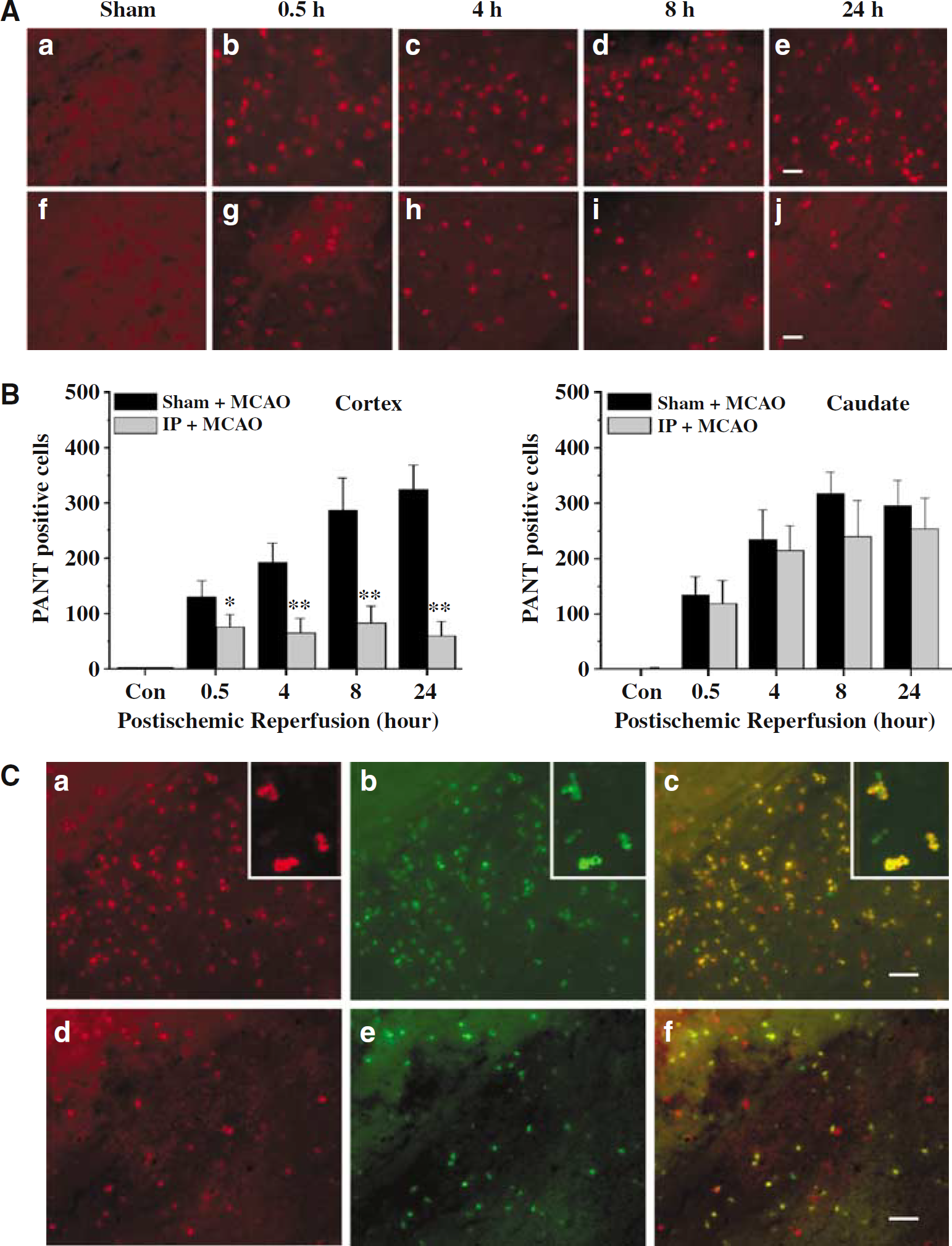
Ischemic preconditioning (IP) attenuates the induction of DNA single-strand breaks (SSB) in cerebral cortex after focal ischemia and reperfusion. (
Double-label staining of PANT and TUNEL was performed to colocalize SSB and DNA fragmentation or double-strand breaks, a marker of cell death, in brains 24 h after 2-h focal ischemia. At this time point, at least 80% of PANT-positive cells in the ischemic brain were also TUNEL-positive (Figure 4C). In agreement with the effect of IP on cortical induction of SSB, the amounts of TUNEL-positive cells were decreased in preconditioned cortices to less than 30% of those in sham-pretreated brains.
Ischemic Preconditioning Attenuates Cellular Signaling Induced by DNA Damage
PARP-1 activation and consequent NAD depletion are a well-established signaling pathway that mediates neuronal cell death resulting from genomic DNA damage (Eliasson et al, 1997; Ying et al, 2001; Yu et al, 2002; Zhang et al, 1994). Based on the observations that IP is associated with decreased accumulation of oxidative DNA lesions in the brain, we hypothesized that DNA damage-induced cellular signaling would be decreased in preconditioned brains after ischemia. Therefore, two signaling events, poly(ADP-ribose)polymer formation and NAD depletion, which are directly related to PARP-1 activation, were examined in brains at 2 and 8 h after 2-h focal ischemia with or without preconditioning. As shown (Figure 5A), markedly increased immunoreactivity for poly(ADP-ribose)-polymer was observed in ischemic cerebral cortex 2 and 8 h after 2-h ischemia, and morphological examination indicated that poly(ADP-ribose)polymer-positive cells were mostly neurons. The formation of poly(ADP-ribose)polymer was decreased in cerebral cortex that had received IP 3 days before. Consistent with the inhibitory effect of preconditioning on PARP activation, NAD depletion (41% and 28% decreases at 2 and 8 h, respectively, after 2 h ischemia without prior preconditioning) was significantly attenuated in the cerebral cortex receiving prior IP (Figure 5B).
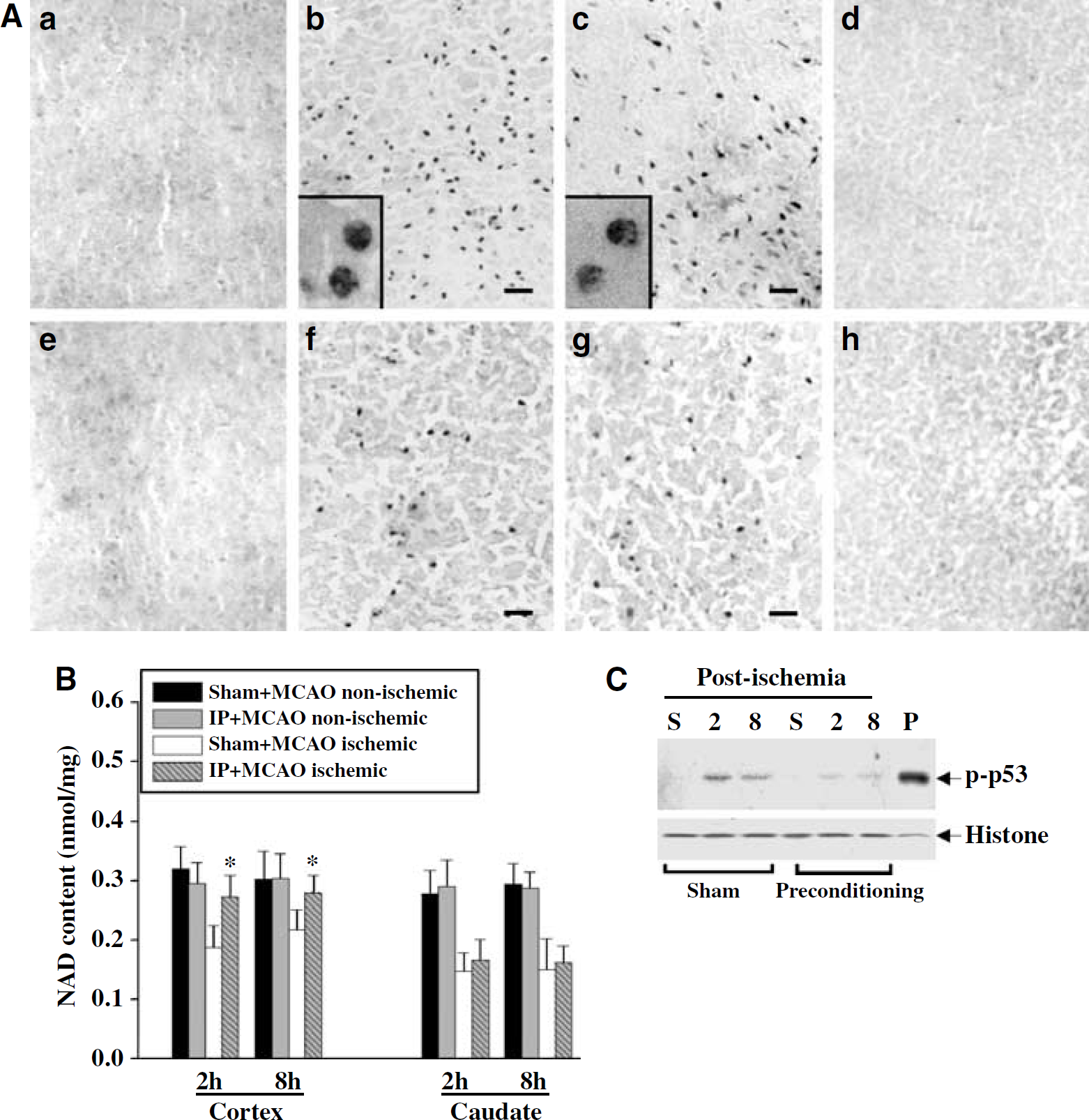
Ischemic preconditioning (IP) attenuates DNA damage-signaling responses after focal ischemia and reperfusion. (
Activation of the nuclear protein p53 is another important DNA damage signaling event that contributes to cell death after ischemia. Accordingly, the phosphorylation of p53 (p-p53, Ser46), an indicator of activated p53, was examined in cortical extracts at 2 and 8 h after 2-h ischemia with or without preconditioning. The ischemia-induced increases in p-p53 were reduced in brains that had received IP (Figure 5C).
Ischemic Preconditioning Activates Multiple Components of the Base-Excision Repair Pathway
The results presented above suggest that IP enhances the recovery of endogenous oxidative DNA damage by increasing the cellular DNA repair capacity instead of by reducing the generation of DNA lesions, as the amounts of DNA lesions were not significantly different during early periods of postischemic reperfusion between groups receiving preconditioning and sham operation. To test this hypothesis, we performed DNA repair assays to determine the repair activity in the cerebral cortex at each major step along the BER pathway. Cortical nuclear extracts were obtained at 3, 6, 12, 24, 48, 72, and 168 h after IP or sham operation. First, the capacity for DNA repair synthesis, the main rate-limiting step in BER, was examined. DNA polymerase-β is the predominant DNA polymerase responsible for the ‘gap-filling’ and ‘dRP-lysis’ steps in the BER pathway in the brain. In this study, we examined its activity in nuclear extracts using a repair assay employing synthesized DNA duplex that contains a precisely positioned single-nucleotide nick (Lan et al, 2003). This assay generates both 25-mer and 50-mer repair products, and the latter is the mature product (Lan et al, 2003). Titration assays showed that brain nuclear extracts resulted in protein concentration-dependent radiolabeling of the repair substrate (Figure 6A). Using this assay, we detected markedly increased DNA polymerase-β activity (increased formation of the 50-mer mature products) in the preconditioned cortex, beginning at 24 h after preconditioning, peaking at 48 h, and lasting for at least 72 h (Figures 6B and 6C). DNA polymerase-β activity was not changed at any time points after sham operation. The DNA repair synthesis rate calculated from the 50-mer product yields in the linear range of reaction (Figure 6D) was 0.45 ± 0.068 and 0.16 ± 0.033 fmol/μg min at 72 h after preconditioning and after sham operation, respectively (mean ± s.d., n = 5; P < 0.01). Thus, the upregulation of DNA polymerase-β activity occurred before (24 h) and during the course of ischemic tolerance (48 to 72 h).
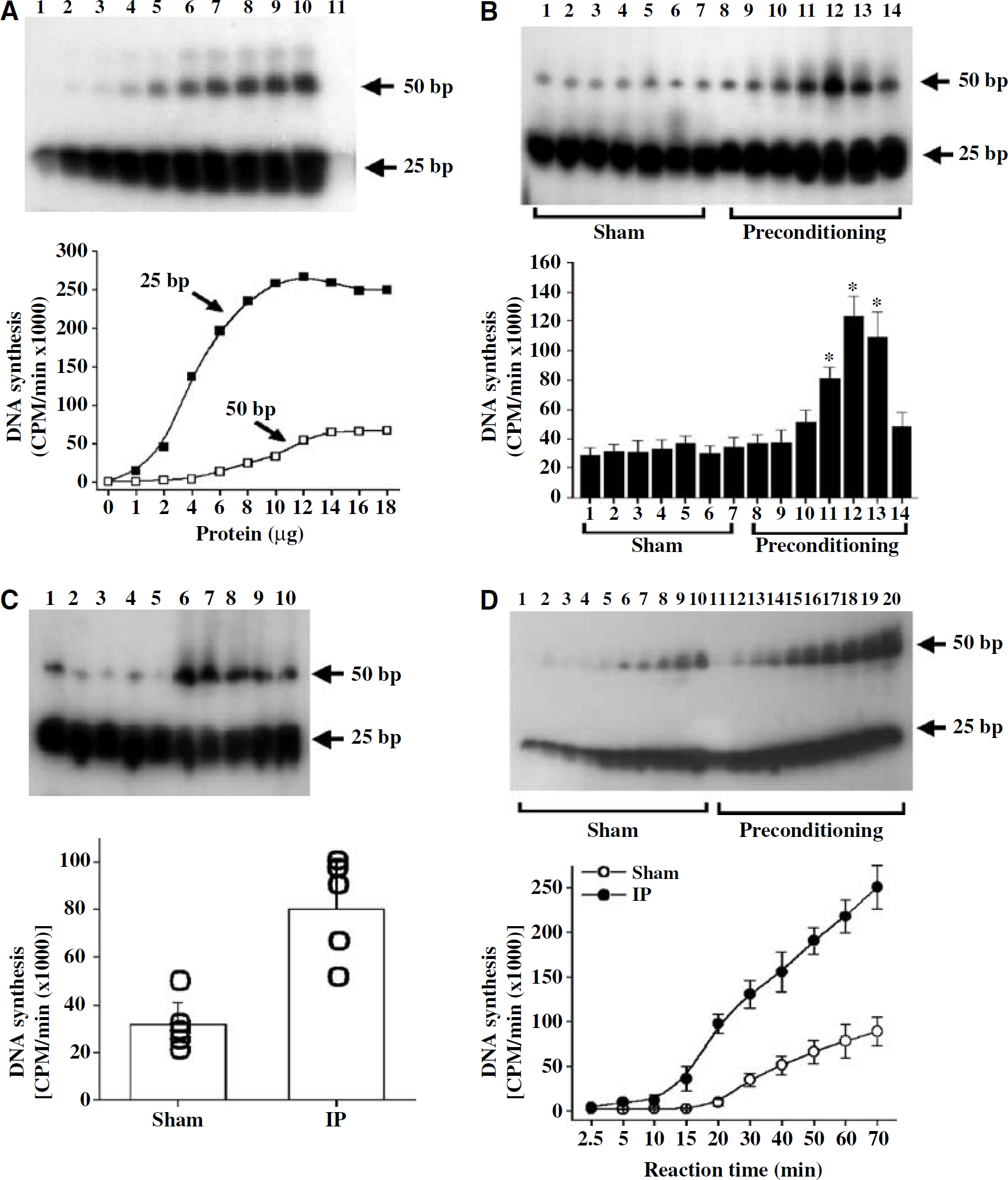
Ischemic preconditioning (IP) stimulates β-pol-dependent base-excision repair (BER) in the cerebral cortex. (
In subsequent experiments, oligonucleotide incision assays were performed to estimate the 8-oxodG-and AP site-incision activity in cortical nuclear extracts. These assays employed specific substrates containing precisely positioned 8-oxodG or AP sites (Chen et al, 2000; Lan et al, 2003). The results are summarized in Figure 7. Both 8-oxodG-incision and AP site-incision activities were upregulated in brains receiving IP but not sham operation. Increases in the 8-oxodG-incision and AP site-incision activities began at 24 h, peaked 48 to 72 h, and subsided at 168 h after preconditioning. Thus, the patterns for the induced 8-oxodG-incision and AP site-incision activities after preconditioning coincided with that of DNA polymerase-β activity.
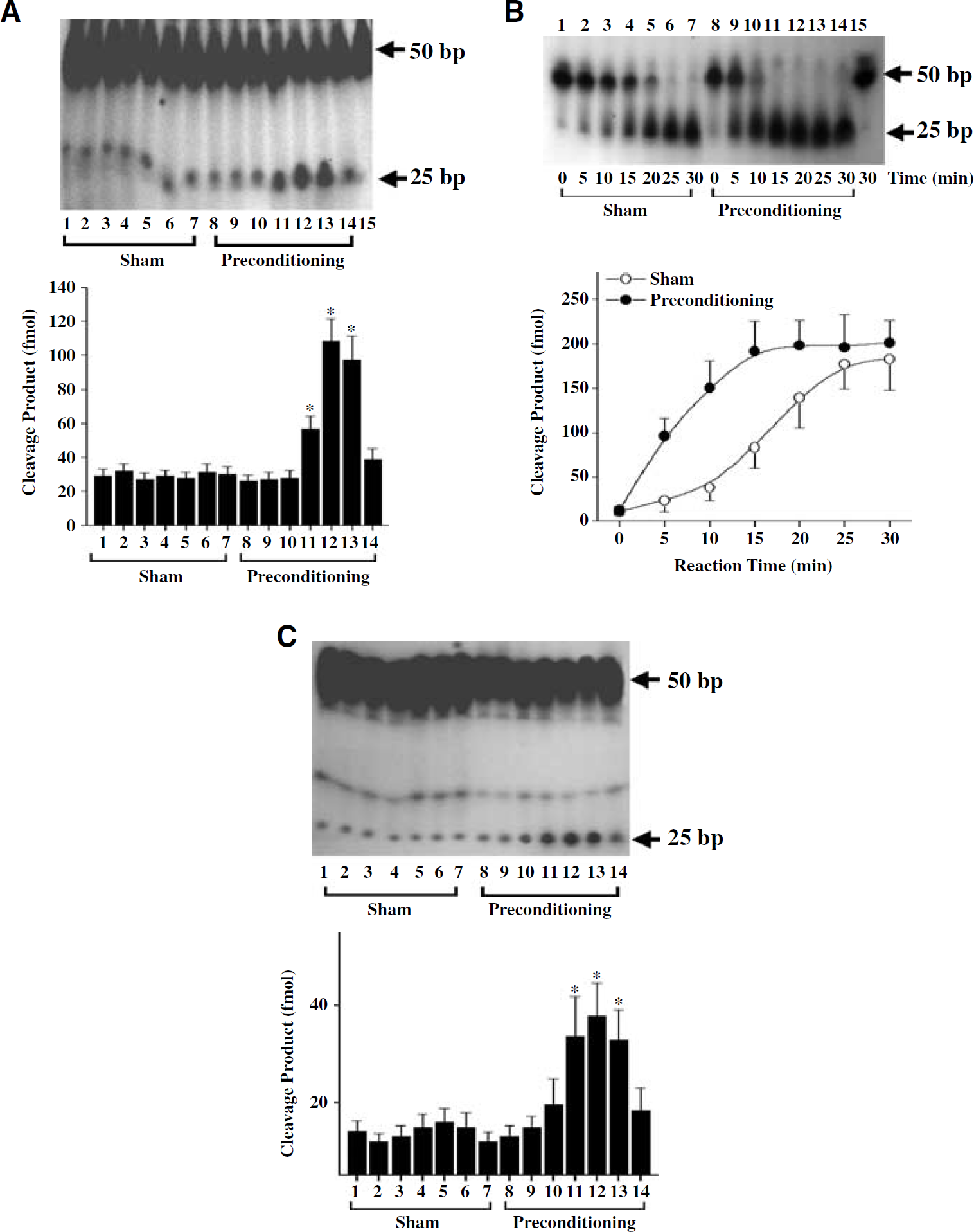
Ischemic preconditioning (IP) stimulates DNA base-incision activities in the cerebral cortex. (
Finally, to determine if the increased DNA polymerase-β activity after IP is associated with a comparable increase in DNA ligase activity, we performed the standard oligonucleotide ligation assay using cortical nuclear extracts. No significant alteration in DNA ligase activity was detected at any time point after preconditioning (data not shown).
Ischemic Preconditioning Enhances Base-Excision Repair Enzyme Expression in the Brain
Western blot was performed using nuclear protein extracts to determine whether the enhanced BER activities in the preconditioned brain are associated with increased expression of BER enzymes (Figure 8A). Consistent with the results of the repair assays, the levels of APE, DNA polymerase-β, and OGG1 (which is responsible for the incision of 8-oxodG) were significantly increased at 24 to 72 h after preconditioning, whereas the levels of DNA ligase I and DNA ligase III were not changed at any time point tested (Figure 8B). Northern blot analysis (Figure 8C) and in situ hybridization (Figure 8D) confirmed that the upregulation of APE and DNA polymerase-β proteins after IP was accompanied by increased expression of their mRNAs, suggesting that the regulation of these BER enzymes after preconditioning is initiated at the transcriptional level. The increased APE and DNA polymerase-β mRNAs were distributed mainly in the frontal-parietal cortex, consistent with the increased BER activity in this region (Figure 8D). Finally, immunohistochemistry was performed to confirm the cellular localization of DNA polymerase-β 48 h after IP. Increased DNA polymerase-β immunoreactivity, showing mainly a nuclear localization, was found in widespread cortical neurons after preconditioning (Figure 8E).
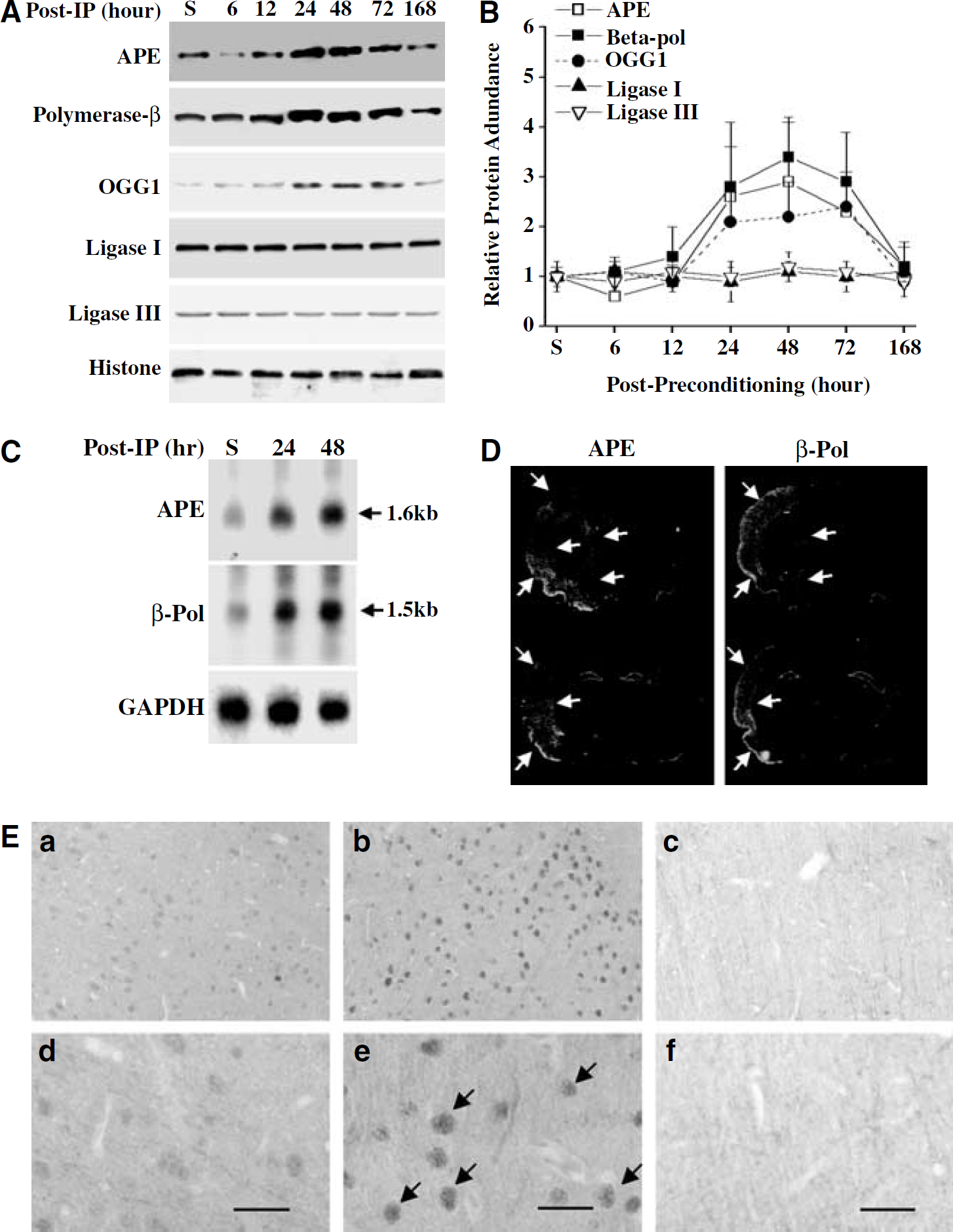
Ischemic preconditioning (IP) stimulates the expression of base-excision repair (BER) enzymes in the cerebral cortex. (
Discussion
Investigations of IP have helped gain insights into the mechanisms of endogenous inducible brain neuroprotection. Recent reviews on this topic suggest that IP is a complex, multiple-factor adaptive process involving various transcriptional, translational, and posttranslational changes that enable brain cells to be markedly more resistant to subsequent severe ischemia (Chen and Simon (1997); Dawson and Dawson, 2000; Dirnagl et al, 2003; Kirino, 2002; Schaller and Graf, 2002; Perez-Pinzon, 2005). In this study, using an established rat model of focal IP, we identified a novel cellular response that could contribute to the mechanism of endogenous neuroprotection against ischemic brain injury. The major observations resulting from this study are that (1) IP elicits a marked activation of the BER pathway, the predominant repair mechanism for oxidative DNA damage in brain, by upregulating the expression and activity of essential BER enzymes; and that (2) IP enhances endogenous DNA repair and consequently prevents the accumulation of cell-killing oxidative DNA lesions in brain cells after severe focal ischemia. These results suggest that the inducible BER activity may be part of the adaptive responses that contribute to the induction of ischemic tolerance.
Emerging evidence suggests that induction of oxidative DNA damage plays an important role in mediating neuronal death after cerebral ischemia (Chen et al, 1997; Cui et al, 1999; Huang et al, 2000; Lin et al, 2000; Liu et al, 1996; Nagayama et al, 2000). Oxidative DNA damage results from direct attacks by reactive oxygen species, such as hydroxyl radicals and nitric oxide derivatives, which are overproduced robustly during postischemic reperfusion (Chan, 1996, 2001). Several types of oxidative DNA damage that have been detected in ischemic brain may have specific detrimental effects on cell survival. Formation of 8-oxodG is associated with gene mutagenesis (Dizdaroglu, 1991; Grollman and Moriya, 1993), and the accumulation of 8-oxodG may result in partial or complete loss of the functions of the damaged genes (Cui et al, 1999; Mazzarello et al, 1992; Schneider et al, 1990). AP sites, when accumulated, prevent the process of DNA synthesis or gene transcription through the lesions, and thus are directly lethal to cell survival (Janssen et al, 1993; Moran and Wallace, 1985; Schaaper and Loeb, 1981). DNA strand damage, especially SSB, is also a potent blocker of DNA synthesis or gene transcription. In addition, the accumulation of DNA strand breaks may directly trigger cell death by activating PARP-, p53-, or/and CDK-dependent pathways (Eliasson et al, 1997; Ghahremani et al, 2002; Payne et al, 1995; Ying et al, 2001; Yu et al, 2002; Zhang et al, 1994). The results from a number of studies have suggested that blockage of DNA damage-triggered prodeath signaling pathways can offer remarkable neuroprotection against ischemic brain injury (Culmsee et al, 2001; Cao et al, 2001; Eliasson et al, 1997; Endres et al, 1997; Iwashita et al, 2004; Lo et al, 1998; O'Hare et al, 2002; Wang et al, 2002).
The results presented here show that the postischemic accumulation of oxidative DNA lesions is markedly attenuated in brains receiving IP. Interestingly, this effect was seen in the cerebral cortex, where tolerance was induced, but not in the caudate, a region that was not spared preconditioning in this model. Consistent with this inhibitory effect by preconditioning, the data also show that the two DNA damage-responsive signaling events, PARP-1 and p53 activation, were diminished in preconditioned cortices after 2 h of focal ischemia. PARP-1 is potently activated by SSB, and it mediates cell death by depleting intracellular NAD+ and ATP (Zhang et al, 1994), which leads to necrotic cell death, or by activating the Bax-dependent apoptosis pathway (Cao et al, 2001; Cregan et al, 2002; Yu et al, 2002). Both AP sites and SSB can activate the p53 pathway (Fritz et al, 2003; Martin and Liu, 2002; Nur et al, 2003; Robertson et al, 2001), which then induces apoptosis through the actions of several pro- apoptotic molecules, including Bax, PUMA and Noxa (Villunger et al, 2003). Therefore, the decreases in oxidative DNA damage by IP may contribute to the attenuation of both necrosis and apoptosis after focal ischemia and reperfusion.
In the present study, the inhibitory effect of preconditioning on the accumulation of oxidative lesions manifests at 0.5 h and longer duration of reperfusion, and, likely, this effect is achieved by accelerating the DNA repair process. All three forms of lesions examined in this study, including 8-oxodG, AP site, and SSB, were induced in the brain during early reperfusion (0–15 mins), and the magnitudes of induction in the groups receiving IP were similar to those subject to sham operation, indicating that preconditioning does not prevent the initial induction of oxidative DNA damage. These results suggest that the cerebral tissues are vulnerable to oxidative DNA damage after severe ischemia and reperfusion regardless of preconditioning. Several previous studies examined the levels of antioxidants such as superoxide dismutase-1 and −2 in the brain after preconditioning, but showed conflicting results (Kis et al, 2004; Mitchell et al, 2001; Toyoda et al, 1997), suggesting that an upregulation of antioxidant enzymes may not be the universal mechanism underlying the induction of ischemic tolerance. Nevertheless, the antioxidant system in the brain, which constitutes the first line of defense against oxidative neuronal injury, is insufficient to prevent all attacks by ROS on genomic DNA after a severe ischemic insult.
Based on the observation that oxidative DNA damage was efficiently repaired in the ischemic cortex after receiving preconditioning but not after sham operation, we speculated that IP might achieve this effect by enhancing the DNA repair capacity. To directly test this hypothesis, DNA BER activities were measured in nuclear protein extracts using specific DNA repair substrates. We found that 8-oxodG glycosylase, AP endonuclease, and DNA polymerase-β activities along the BER pathway were significantly increased in cerebral cortex after preconditioning, ranging from 2.5- to 5-fold over the levels after sham operation; however, DNA ligase I and DNA ligase III activities were not changed. The induced BER activities after preconditioning were the direct result of the upregulation of the expression of BER enzymes because the time course of changes in activities and enzyme expression coincided (24 to 72 h after preconditioning). Strikingly, the increases in BER activities occurred before (24 h) and during the course of ischemic tolerance (48 to 72 h). Although a cause–effect relationship between the upregulation of the BER pathway and ischemic tolerance has yet to be established, the results suggest that markedly enhanced BER activity resulting from preconditioning may be responsible for the improved recovery of endogenous oxidative DNA damage after severe ischemia and, possibly, together with other mechanisms, contributes to the overall neuroprotective effect of IP.
The increased enzymatic activities of AP endonuclease and DNA polymerase-β are instrumental in preconditioning-induced enhancement of the endogenous DNA repair process. AP endonuclease and DNA polymerase-β form the backbone of the BER pathway (Frosina et al, 1996; Klungland and Lindahl, 1997; Srivastava et al, 1998). The sequential actions of these two enzymes occur downstream of lesion-specific glycosylases but upstream of the final DNA ligation step, and thus are essential for successful repair of various DNA base damage lesions and the highly cytotoxic lesions AP site and SSB (Janssen et al, 1993; Payne et al, 1995). The direct role of inducible expression of AP endonuclease and DNA polymerase-β in promoting cell survival has been partially studied, mainly in non-neuronal systems. A number of studies have shown that cells deficient in AP endonuclease or DNA polymerase-β develop remarkably increased vulnerability to oxidative stress or hypoxic injury (Clairmont and Sweasy, 1996; Horton et al, 2002; Kaina et al, 1998; Ochs et al, 1999; Sobol et al, 1996; Walker and Sikorska, 1994). Several studies have used gene transfection approaches to enhance BER capacity in mammalian cells and have found a direct link between the expression levels of AP endonuclease or DNA polymerase-β and cell protection (Grosch et al, 1998; Horton et al, 2002; Sobol et al, 2000; Tomicic et al, 1997, 2001). For example, transfection of the human APE cDNA driven by an inducible promoter into CHO cells markedly reduced cytotoxicity of oxygen-derived free radicals (Grosch et al, 1998), whereas the expression of DNA polymerase-β in mouse fibroblasts protected against the cytotoxicity of hydrogen peroxide (Horton et al, 2002). Taken together, these results suggest that an alteration in cellular BER activity can substantially influence the fate of genome-damaged cells. Therefore, based on the observations presented here, we further suggest that IP-induced upregulation of BER activity may play a role in enhancing cell survival in the ischemic cortex by limiting the genomic effect of oxidative stress. However, to directly prove the role of BER activity, especially AP endonuclease and DNA polymerase-β, in ischemic tolerance, one needs to specifically alter the expression and activity of BER enzymes in the brain, which will be an important objective for future studies.
The mechanism underlying the induced expression of BER enzymes after IP is unclear. In this study, results of Northern blot analysis show that the mRNA expression of AP endonuclease and DNA polymerase-β was increased 24 and 48 h after preconditioning, suggesting that transcriptional regulation was involved. In situ hybridization using AP endonuclease- and DNA polymerase-β-specific probes confirmed their upregulation in the brain. Several factors involved in ischemic injury, such as hypoxia and oxidative stress, could activate the transcriptional process of DNA polymerase-β by enhancing the DNA binding of ATF-1/CREB. ATF-1/CREB is the major promoter activator of the DNA polymerase-β gene (Narayan et al, 1996), and several recent studies have confirmed its upregulation in neurons that survived ischemia or oxidative stress (Mabuchi et al, 2001; Tanaka et al, 2000; Zaman et al, 1999; Meller et al, 2005). Similarly, CREB may also activate the transcription of AP endonuclease (Grosch and Kaina, 1999). In addition, a CCAAT box and two inverted CCAAT motifs have been found in the promoters of AP endonuclease and OGG1 gene, respectively (Lee et al, 2004; Seki et al, 2002). NF-YA is the transcriptional factor that binds to the inverted CCAAT motifs and is essential for DNA damage-induced expression of human OGG1 (Lee et al, 2004). Whether the above mechanisms are responsible for preconditioning-induced upregulation of BER enzymes in the brain needs further investigation. Nevertheless, the increased gene transcription of BER enzymes coupled with continued protein translation in the cerebral cortex is essential for the induced BER activity and enhanced DNA repair in the region after preconditioning. However, the caudate could not tolerate the insults imposed by IP itself and showed a decreased BER enzyme expression at the protein levels (data not shown). Failure to express BER enzymes in the caudate may contribute directly to the extremely poor recovery of oxidative lesions in this region after ischemia. These results are consistent with the previous reports that the expression of AP endonuclease was markedly decreased in the brain after severe focal ischemia and that this decrease might contribute to DNA fragmentation and cell death (Fujimura et al, 1999; Kawase et al, 1999).
In summary, the present study has identified a novel cellular response associated with IP. Preconditioning elicits a marked activation of the BER pathway for DNA repair in the cerebral cortex protected against infarction after subsequent severe focal ischemia and reperfusion. This cellular response may contribute to neuroprotection by enhancing the repair of oxidative DNA damage and preventing the prodeath signal transduction triggered by DNA damage. These results suggest that preconditioning activation of the BER pathway is a potentially important endogenous mechanism for neuroprotection.
Footnotes
Acknowledgements
The authors thank Dr Thaddeus Nowak, University of Tennessee, for kindly offering expertise on brain tissue-sampling methodology for NAD measurement. The authors also thank Shuping Wang for technical support, Carol Culver for editorial assistance, and Pat Strickler for secretarial support.