
Abstract
The relation between cerebral ischemia and local release of angiogenic factors was investigated after subarachnoid hemorrhage (SAH) in humans. Time-dependent concentration-changes of vascular endothelial growth factor (VEGF), sFlt-1 and sTie-2 extracted from plasma, serum, and cerebrospinal fluid (ventricular, cisternal, and lumbar) were analyzed in 15 patients surgically treated for ruptured aneurysms of the anterior circulation (Hunt and Hess grades I-V). Data were related to brain Po2 (Pbro2) and cerebral energy metabolites (extracellular lactate, pyruvate, glutamate, and glycerin concentrations) as well as clinical and radiologic reference data. Delayed impairment of cerebral perfusion secondary to progressive microcirculatory alterations was associated with reduced local Pbro2 and energy metabolism (increased lactate-pyruvate ratio, glutamate and glycerine levels). Elevated serum/plasma and CSF concentrations of VEGF, sFlt-1, and sTie-2 matched the scale of ischemic tissue hypoxia. Excessive VEGF/sFlt-1 and sTie-2 levels were related to Pbro2 values consistently less than 5 mm Hg, glutamate concentrations greater than 300 μmol/L, lactate-pyruvate ratio greater than 300, cerebral infarction, and reduced outcome (P < 0.01). Delayed microcirculatory impairment was mirrored by distinct elevation of cisternal and arterial VEGF and sFlt-1 concentrations, suggesting local induction of angiogenesis. Arterial levels of VEGF, sFlt-1, and sTie-2 reflect both extent and time course of compensatory, yet clinically inefficient, angiogenesis in the absence of general hypoxia.
Keywords
Delayed tissue ischemia caused by complex cerebrovascular alterations involving smooth muscle contraction and inflammatory responses to a number of extravascular blood components is an important cause of morbidity and mortality after subarachnoid hemorrhage (SAH) (Dietrich and Dacey, 2000; Kassell et al., 1985; van Gijn and Rinkel, 2001; Yin et al., 2001). Numerous research efforts have been directed to elucidate various pathogenetic pathways (Dreier et al., 2000; Dietrich and Dacey, 2000; van Gijn and Rinkel, 2001). Microcirculatory impairment caused by SAH appears to follow a biphasic pattern with an initial acute phase of 1 to 3 days after aneurysm rupture and a subsequent delayed phase, beginning at day 3 and reaching a maximum at day 6 to 8 after SAH. Exact definition of the causative factors for each phase is still lacking. Although acute arterial vessel narrowing is observed during diagnostic angiography in humans and in various animal models of SAH (Megyesi et al., 2000), its pathophysiologic role and significance in humans remains unclear. Microcirculatory impairment of delayed onset, often (but not always) matched by significant increases of flow velocity in major cerebral arteries and their branches with corresponding decreases in tissue oxygenation and clinical deterioration (Kassell et al., 1985; Seiler et al., 1986) is deemed to be responsible for significant secondary neurologic morbidity and mortality (Kassell et al., 1985). It is frequently refractory to current rheologic, pharmacologic, and physiologic treatment modalities. The delayed onset of the second phase of cerebral microcirculatory impairment (as primary target of contemporary therapeutic strategies (Yang et al., Abbreviations used: CT, computed tomography; DIND, delayed ischemic neurologic deficits; ELISA, enzyme-linked immunosorbent assay; SAH, subarachnoid hemorrhage; VEGF, vascular endothelial growth factor. 2001), however, potentially opens a time window for early, preemptive intervention aiming at improved collateral circulation and timely adaptation to reduced microvascular cerebral blood flow.
The molecular mechanisms of angiogenesis under physiologic as well as hypoxic conditions is currently subject to systematic experimental research (Dor et al., 2001; Ferrara 2001; Jin et al., 2000; Lenmyr et al., 1998; Lin et al., 2001; Matsuzaki et al., 2001; Shibuya 2001; Yokota et al., 2001). Vasculogenesis and remodeling is regulated by paracrine signals from transmembrane tyrosine kinase receptors of endothelial cells. The physiologic ligand of the tyrosine kinase receptors Flk-1 and Flt-1 is vascular endothelial growth factor (VEGF). VEGF and its receptors are required for blood vessel development during embryogenesis (Fong et al., 1995) as well as postnatal vascular remodeling and neovascularization (Asahara et al., 1998; Isner et al., 1996; Li et al., 1996). Tie-1 and Tie-2 constitute another family of endothelial cell-specific tyrosine kinase receptors. Angiopoietins (Ang-1 and Ang-2) have been identified as physiologic ligands of Tie-2 (Sato et al., 1995; Wong et al., 1997), whereas the endogenous ligand of Tie-1 remains unknown. Tie-1, however, is involved in maintaining vascular endothelial cell integrity (Sato et al., 1995). VEGF modulates the Tie-2-Tie-1 receptor complex by inducing a switch between two different functional states (Tsiamis et al., 2002). Binding of Ang1 to Tie-2 induces stabilization and maturation of newly established vessels by recruiting and sustaining periendothelial support cells (pericytes, smooth muscle cells) (Suri et al., 1996), whereas binding of Ang-2 to Tie-2 has been shown to promote vascular destabilization and sprouting during VEGF-induced neovascularization (Asahara et al., 1998). Thus, Ang-2 is presumed to collaborate with VEGF at the leading edge of newly sprouting vasculature by blocking constitutive stabilization and maturation physiologically mediated by Ang-1 (Maisonpierre et al., 1997). Consequently, the relative ratio of Ang-1 versus Ang-2 is critical in regulating the overall angiopoietin-VEGF system effect on angiogenesis.
Evidence of a close coupling between hypoxia-induced gene expression and sequential activation of proangiogenic cellular signaling pathways is accumulating (Dor et al., 2001; Slevin et al., 2000b; Zhang et al., 2000, 2001). Ischemic diseases of the central nervous system comprise a group of conditions potentially amenable to proangiogenic therapy (Alafaci et al., 2000; Hayashi et al., 1998; Jin et al., 2000b, c; Wang et al., 2000). Although insight into the molecular response to tissue hypoxia is growing (Hayashi et al., 2001; Jin et al. 2000a, b; Wu et al., 2000), sparse clinical data exist concerning the temporal expression profile of angiogenic factors within different physiologic compartments (peripheral blood, extracellular fluid, CSF) during cerebral ischemia in humans (Belgore et al., 2000, 2001; Gunsilius et al., 2001; Slevin et al., 2000a, b). Recently, Slevin et al. were able to demonstrate a correlation between VEGF, but not transforming growth factor-β1, and infarct volume after ischemic stroke in 29 patients (Slevin et al., 2000a). They suggested that the cerebral angiogenic response to an ischemic insult was reflected by increased systemic VEGF levels. Interpretation of these findings, however, remains controversial (Gunsilius et al., 2001).
To clarify whether local cerebral ischemia coincides with a measurable increase of angiogenic factors in systemic circulation, we investigated VEGF and soluble tyrosine-kinase receptors sFlt-1 (Barleon et al., 1997; Kendall et al., 1996) and sTie-2 (Reusch et al., 2001) concentration profiles in CSF and peripheral blood during ischemic and nonischemic periods after aneurysmal SAH in 15 patients. VEGF, sFlt-1, and sTie-2 concentration profiles are compared with several biochemical and physiologic parameters of cerebral perfusion and energy metabolism.
PATIENTS AND METHODS
During a period of 10 months (February 2000 to November 2000), 15 patients referred to our institution within 12 hours and surgically treated within 24 hours of aneurysmal SAH entered our prospective study. The study protocol was approved by the local institutional review board. Selection criteria for inclusion in our study were as follows: (1) age between 20 and 75 years; (2) sufficient anamnestic information to exactly define the onset of hemorrhage; (3) admission within 12 hours of bleeding; (4) surgical management (clip-ligation) within 24 hours of initial symptoms; (5) no significant comorbidity (malignant disease, severe hepatic, renal, cardiopulmonary disorders, cerebrovascular diseases); (6) definitive angiographic demonstration of an aneurysm as bleeding cause; and (7) no signs of extensive cerebral ischemia on admission computed tomography scan (CT). On demonstration of a ruptured intracranial aneurysm by four-vessel cerebral angiography and obtaining an interdisciplinary consensus decision in favor of surgical treatment, informed consent was obtained from eligible patients before inclusion in the study protocol. All patients were graded according to the Glasgow Coma Scale (Teasdale and Jennet, 1974) and Hunt and Hess clinical score systems (Hunt and Hess, 1968). Semiquantitative radiographic evaluation of SAH according to the Fisher grading system (Fisher et al., 1980) was conducted using the admission CT scan (Table 1). Depending on clinical and radiologic signs of impaired CSF circulation, ventricular (Codman and Shurtleff Inc., Raynham, MA, U.S.A.) or lumbar CSF drains (Portex Ltd., Hythe Kent, U.K.) were inserted on admission. Ventricular drains were preferred in those cases with manifest ventricular dilatation evidenced by the initial CT scan, whereas lumbar drains were placed in the remaining patients. Patients presenting with significant comorbidity, i.e., malignancies, systemic hypoxia caused by pulmonary dysfunction, or peripheral ischemic vascular diseases or impaired systemic circulation (e.g., cardiac failure) were excluded from the study protocol. Patient characteristics, including outcome scores, are shown in Table 1. The Glasgow Coma Scale score on admission ranged from 15 to 3 points, corresponding to Hunt and Hess grades between I and V. Standard institutional treatment guidelines were followed during perioperative care including prophylactic intravenous administration of Ca2+ antagonists (nimodipine 1 to 2 mg/h), frequent neurologic monitoring to detect clinical signs of microcirculatory impairment and a modified triple H (hypervolemic hemodilution and induced hypertension) therapy protocol. Hematocrit was adjusted to 33%. Depending on neurologic status and respiratory function, grade III-V patients were managed by analgosedation and ventilation using an O2/air mixture to maintain arterial oxygen tensions (Pao2) between 100 and 140 mm Hg. Ventilation was adjusted to maintain arterial CO2 levels (Paco2) between 35 and 40 mm Hg. In nonventilated patients, oxygen was administered via face masks according to continuous analysis of Spo2 and assessment of pH, Paco2 and Pao2 at 2- to 4-hour intervals (ABL; Radiometer, Copenhagen, Denmark) to maintain constant conditions with respect to gas exchange and acid-base homeostasis. Central venous catheters and arterial lines were routinely placed, and mean arterial pressure was recorded continuously. Central venous pressures were obtained at least every 6 hours to adjust fluid management. Continuous registration of vital parameters was performed in all patients, using a dedicated monitor setup allowing synchronous data transfer from patient monitor (CS/3; Datex-Ohmeda, Duisburg, Germany), Licox-CMP monitor and CMA 600 microdialysis system to a multi-modal PC-based data logger (ICU Pilot; CMA Microdialysis, Solna, Sweden).
Clinical summary of 15 patients
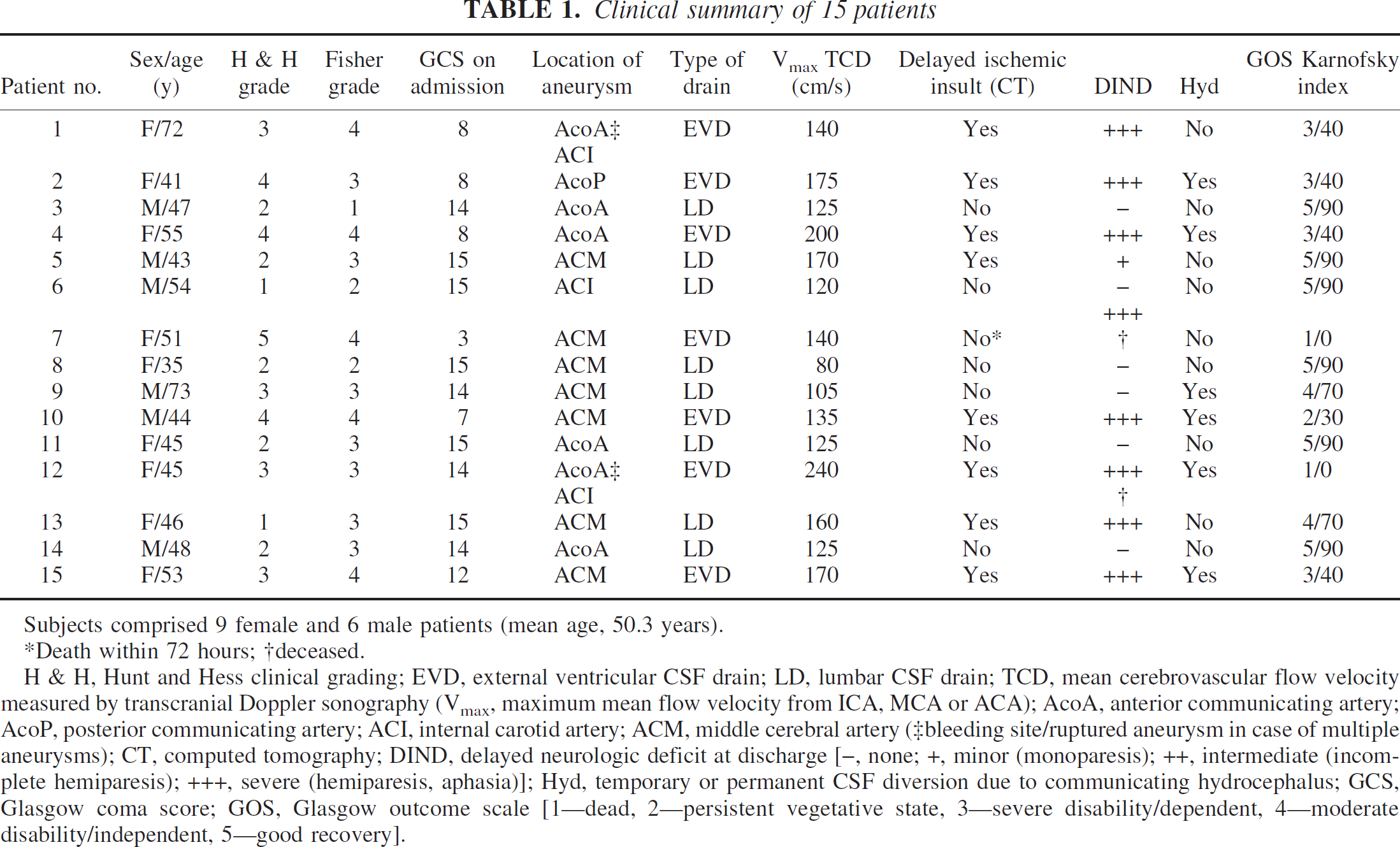
Subjects comprised 9 female and 6 male patients (mean age, 50.3 years).
Death within 72 hours; †deceased.
H & H, Hunt and Hess clinical grading; EVD, external ventricular CSF drain; LD, lumbar CSF drain; TCD, mean cerebrovascular flow velocity measured by transcranial Doppler sonography (Vmax, maximum mean flow velocity from ICA, MCA or ACA); AcoA, anterior communicating artery; AcoP, posterior communicating artery; ACI, internal carotid artery; ACM, middle cerebral artery (‡bleeding site/ruptured aneurysm in case of multiple aneurysms); CT, computed tomography; DIND, delayed neurologic deficit at discharge [–, none; +, minor (monoparesis); ++, intermediate (incomplete hemiparesis); +++, severe (hemiparesis, aphasia)]; Hyd, temporary or permanent CSF diversion due to communicating hydrocephalus; GCS, Glasgow coma score; GOS, Glasgow outcome scale [1—dead, 2—persistent vegetative state, 3—severe disability/dependent, 4—moderate disability/independent, 5—good recovery].
Surgical treatment and perioperative monitoring
Surgical treatment of ruptured aneurysms was conducted via a standard pterional craniotomy in all patients. The sylvian fissure was opened, attaining control of all major arteries of the anterior circle of Willis. Stepwise dissection of the aneurysm was carried out using standard microsurgical techniques with minimal use of brain retractors to preserve local microcirculatory conditions after SAH within the dissected sylvian fissure. Meticulous care was taken to avoid coagulation of cortical microvessels, including veins during access to the aneurysm through the lateral fissure. Before and after application of a suitable titanium clip to exclude the aneurysm from cerebral circulation, arterial flow proximal and distal to the aneurysm was assessed using a 16-MHz EME microvascular Doppler probe (Eden Medizinische Elektronik, Ueberlingen, Germany) to prevent significant perfusion deficiencies resulting from stenosis or occlusion of the parent artery. After generous irrigation of the operative field, a Clark-type Po2-microelectrode (Revoxode; GMS, Kiel-Mielkendorf, Germany) and a micro-dialysis catheter (CMA 70; Axel-Semrau GmbH, Sprockhövel, Germany) were implanted tangentially within the frontal cortex of the ipsilateral sylvian fissure. Catheter placement within the vascular territory of ipsilateral middle cerebral artery perforating branches was chosen based on identification of the ipsilateral peri/intrasylvian cortex as one of the preferred locations of delayed ischemic insults caused by cerebral vasospasm within our recent 5-year institutional series (n > 300) of surgically treated aneurysms of the anterior cerebral circulation. The middle cerebral artery and its branches were involved in the development of delayed ischemia caused by microcirculatory impairment after SAH in more than 90% of patients of our 5-year series and in all patients exhibiting delayed ischemia in our study cohort. No systematic differences between anterior-communicating and middle cerebral artery aneurysms were observed regarding location and extent of delayed ischemic insults during our study. Microdialysis catheters were preflushed with Ringer's solution (147 mval Na+, 4 mval K+, 155.5 mval Cl−, 2.25 mval Ca++) using a CMA 106 microdialysis pump (flow rate: 0.3 μL/min), yielding recovery rates of approximately 70% (Hutchinson et al., 2000). In addition, a silicon ventricular drain (Codman and Shurtleff Inc.) was placed in the carotid cistern, after copious irrigation of the cistern to clear visible blood remnants. Both catheters were passed subcutaneously for several centimeters to exit through individual small skin incisions. The CSF drains were connected to closed port systems to provide sterile access to cisternal CSF. The outflow pressure of the sylvian cisternal drainage was set to 10 cm H2O. Three to four milliliters of CSF was collected daily during the entire observation period. Lumbar and ventricular CSF catheters were connected to standard drainage systems for continuous CSF diversion (Codman and Shurtleff Inc.). Correct location of all catheters (including Revoxode and CMA 70 catheters) could be verified by thin-slice (3 mm) postoperative CT scans along with assessment of postoperative perisylvian edema formation in all but two patients (3, 7; Table 1), in whom the CMA 70 catheter was not visualized. Follow-up CT scans were conducted at regular intervals (2 to 4 days) to screen for manifestation of secondary tissue ischemia (infarction). Clear delineation of localized and diffuse edema as well as intracerebral hemorrhage was feasible. In addition, ventricular size and configuration, signs of transependymal CSF sequestration, and time-dependent resolution of the subarachnoid blood clot were documented. Cerebrospinal fluid dynamics were assessed by daily documentation of drainage volume, cell count, and determination of CSF protein content. Proper function of all Po2 electrodes was confirmed by observation of an adequate brain oxygen tension (Pbro2) response after raising FIo2 to 100%. In addition, each probe was tested for zero drift (<1 mm Hg) using an absolute zero solution (Na2S2O4) and for sensitivity drift (<5%) at a mean room air Po2 of 156.9 ± 1.7 mm Hg. Pbro2 was registered continuously (Licox system; GMS), while microdialysis samples were collected at 120-minute (differing from the usual 30 to 60 minutes) intervals and frozen at −70°C. These intermediate sampling intervals appeared justified because of the 24-hour sampling interval for the angiogenesis parameters, whereas continuous assessment of Pbro2 within the same tissue volume was used to detect any short transient ischemic periods. Off-line analysis of lactate, pyruvate, glycerol, and glutamate using double-step enzymatic assays and subsequent photometric quantification of the enzymatic end-product quinone-diimine at 546 nm (CMA 600; Axel-Semrau GmbH) was performed within 24 hours. To exclude systematic errors during cerebral microdialysis, arterial glucose and lactate concentrations were determined every 120 minutes serving as systemic reference (ABL; Radiometer). Arterial red blood cell, leukocyte, and thrombocyte counts were conducted at regular intervals (24 to 48 hours). Markers of systemic or myocardial ischemia (arterial pH and lactate concentration, troponin-t, creatine-kinase isoform MB, lactate-dehydrogenase) were repetitively screened in addition to continuous ECG recordings to exclude potential confounding effects related to noncerebral ischemia during the course of SAH. Cerebrovascular flow velocities (mean flow velocity; centimeters per second) in the internal carotid and vertebro-basilar system were bilaterally assessed twice daily using transcranial Doppler sonography (Multidop P; DWL Elektronische Systeme GmbH, Sipplingen, Germany) until discharge from the hospital.
Analysis of VEGF, soluble VEGF-receptor 1 (sFlt-1), and soluble angiopoietin receptor (sTie-2)
Cisternal, ventricular, or lumbar CSF as well as arterial blood samples were collected daily (standardized time interval between 10 and 12 a.m.) for the entire treatment period at our institution (range: 3 to 28 days, mean: 16.8 days). Plasma and serum samples and CSF were gently agitated and ice-cooled for 25 minutes before centrifugation at 1,140g. Aliquots of each sample were frozen and stored at −70°C. The sTie-2 enzyme-linked immunosorbent assay (ELISA) (Reusch et al., 2001) is designed to measure sTie-2 levels in serum and uses a quantitative sandwich enzyme immunoassay technique. The monoclonal antibody aTEK16 specific for sTie-2 was used as a capture antibody. Standards and samples were incubated on microplates (Nunc MaxiSorp; Nunc GmbH, Wiesbaden, Germany) to allow adsorption of sTie-2 molecules to the immobilized antibody. Plates were washed after incubation and blocking period. A mixture of two biotinylated monoclonal antibodies (aTEK2 and aTEK9) was added to all wells, including Streptavidin-AP (ExtrAvidin; Sigma-Aldrich, Munich, Germany). After a final incubation period, plates were developed using p-nitrophenylphosphate detection buffer. Photometric quantification was performed by detection of absorption changes at 405 and 620 nm. A standard curve was established for each plate, allowing exact quantitative detection within a concentration interval between 0.8 and 50 ng/mL. A similar sandwich enzyme immunoassay technique was used for quantitative detection of sFlt-1 (Kendall et al., 1996) in serum (2-day ELISA). The monoclonal antibody 11G2 specific for sFlt-1 was coated onto microplates (Nunc MaxiSorp). After the incubation and blocking periods, all microplates were washed followed by distribution of standards and samples into the wells to permit adsorption of Flt-1 to the immobilized antibody. After washing (clearance of any unbound molecules), the polyclonal antibody rabbit-anti-Flt-1 (detection antiserum) specific for Flt-1 was added to the wells. After another washing period (clearance of any unbound antibody-enzyme reagent), the microplates were incubated with biotinylated conjugate goat-antirabbit IgG (BAR), followed by washing. Thereafter, streptavidin-AP (ExtrAvidin) was added. After a final incubation period, the plate was developed with the substrate p-nitrophenylphosphate detection buffer. Photometric quantification was performed by detection of absorption changes at 405 and 620 nm. A standard curve was established for each plate, allowing quantitative detection within a concentration interval between 0.2 ng/mL and 12.8 ng/mL. A quantitative sandwich immunoassay kit (Human VEGF Duo-Set DY293; R&D Systems, Wiesbaden, Germany) was used for detection of VEGF. This assay is a 4.5-hour solid-phase ELISA designed to measure VEGF165 levels in cell culture, supernates, serum, and plasma. The plates were precoated with a monoclonal antibody specific for VEGF. Microplates were loaded with standards and samples to allow adsorption of VEGF to the immobilized antibody.
After the washing cycle, an enzyme-linked polyclonal antibody specific for VEGF was added, followed by another washing period. Subsequently, the plates were incubated with conjugate (horseradish peroxidase) and finally washed again. Then, substrate solution (hydrogen peroxide and stabilized chromogen) was added to all wells. After a standard incubation time, color development was stopped by adding the stop solution (2 N sulfuric acid) provided with the kit. Photometric quantification was performed by detection of absorption changes at 450 and 540 nm. A standard curve was established for each plate, allowing quantitative detection within a concentration interval between 31.3 ng/mL and 2 μg/mL.
Samples were stored at −80°C. Before analysis, they were stored at −20°C for several days, to be thawed on the morning of application. Serum samples were diluted 1:6 for the sTie-2 analysis and 1:3 for the sFlt-1 analysis.
STATISTICAL ANALYSIS
Data are expressed as the mean ± SEM or as median and range in those instances with substantial deviation from normal distribution of data. Two-sample significance was tested using the Mann-Whitney U test. Pearson's correlation was used to describe the relation between continuous variables. Nonparametric analysis of variance (Kruskal-Wallis test) was used to describe differences in distribution among the various groups. Statistical significance was inferred at P < 0.05. All statistical analyses were performed using the Statistica 5.0 software package (StatSoft, Hamburg, Germany).
RESULTS
No complications from local bleeding or infection because of multiple catheter placements were observed during our study. Temporary postoperative tissue ischemia (local edema related to dissection of the lateral fissure) in the vicinity of the probes was documented by thin-slice (3 mm) serial postoperative CT scans. No Pbro2 or microdialysis catheter malfunction occurred in our study population. Revoxode drifts did not exceed 1.2 mm Hg. The mean duration of invasive cerebral monitoring was 8.7 ± 4.1 days. In one patient, a temporary CSF leakage around a cisternal CSF catheter was noted, prompting removal of the drain after 4 days. Significant and sustained (duration >48 hours) cerebrovascular flow accelerations beyond 160 cm/s were observed in 6 patients (40%). Seven patients (47%), however, developed delayed ischemic neurologic deficits (DIND), whereas patient 7 died within 72 hours from progressive cerebral ischemia, thus formally precluding development of delayed microcirculatory impairment. DIND, including moderate to severe hemiparesis, aphasia, and neuropsychologic impairments, remained permanent except for patient 5, who recovered completely from transient global aphasia within 1 week after onset of his neurologic deficit.
Brain oxygen tension and cerebral energy metabolite concentrations
Two parameters of glioneuronal energy metabolism and cellular integrity (lactate-pyruvate ratio and extracellular glutamate concentration) were closely related to Pbro2 (Fig. 1). Each plot in Fig. 1 illustrates pooled data from all 15 patients. Data from patients 2, 4, 7, 12 and 15 (Table 1) contributed to Pbro2 values less than 10 mm Hg and corresponding peak values for lactate-pyruvate ratio, VEGF, sFlt-1, and sTie-2. Pbro2 values were statistically unrelated to corresponding Pao2, mean arterial pressure, and cerebral glycerol concentrations (r < 0.25).
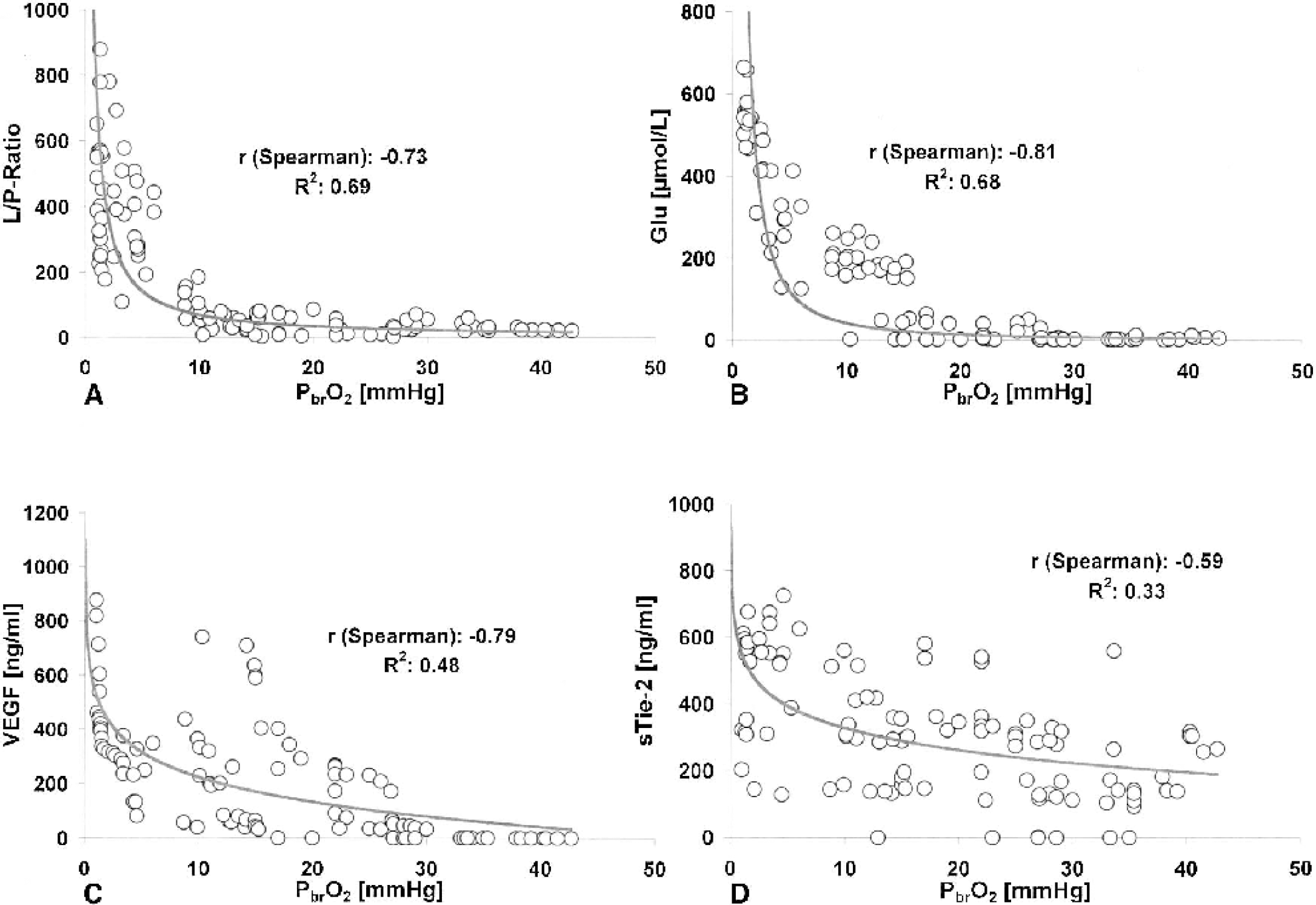
Corresponding variations of lactate-pyruvate ratio (L/P ratio;
Serum, plasma, and cerebrospinal fluid levels of VEGF, sFlt-1, and sTie-2
Serum (CSF) sTie-2 levels between 0 and 1,000 (0 and >50) ng/mL were observed, whereas sFlt-1 levels in serum (CSF) ranged from 0 to 6 (1 and >12) ng/mL. VEGF concentrations in plasma (CSF) varied between 0 and 900 (0 and >2,000) ng/mL. An inverse relation was observed between corresponding Pbro2 and plasma VEGF values (r: −0.8) as well as between Pbro2 and sTie-2 concentrations (r: −0.6) in serum (Fig. 1). In addition, sTie-2 and sFlt-1 concentrations in serum appeared to be related (r: 0.6) to plasma VEGF levels. Neither VEGF nor sFlt-1 and sTie-2 levels observed in serum/plasma or CSF samples were correlated to corresponding leukocyte or thrombocyte concentrations as well as C-reactive protein levels in serum (Fig. 2).
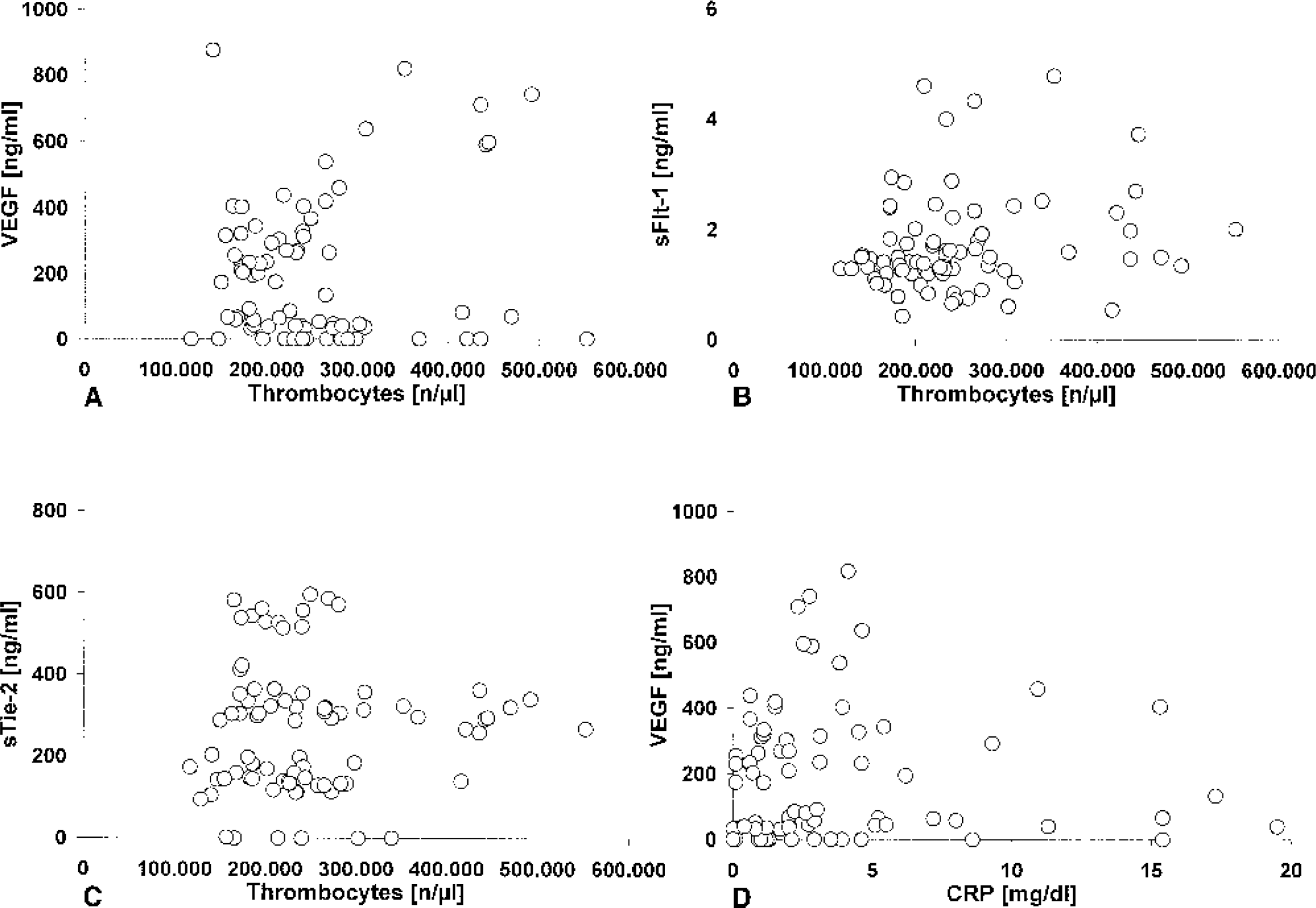
Relation between systemic thrombocyte concentration and plasma vascular endothelial growth factor (VEGF;
Relation between clinical condition, metabolic and angiogenic parameters
Corresponding data obtained from simultaneous Pbro2 measurements, microdialysis, arterial blood, and CSF sampling were allotted to groups according to clinical grade assigned during admission (Hunt and Hess score). Furthermore, pooled data from all patients were allocated to five groups according to the following (arbitrary) categories: (1) admission (day 1, pre- and/or intraoperative measurement); (2) postinterventional (day 1, postoperative period); (3) uneventful follow-up (absence of pathologic findings during postinterventional clinical and radiologic assessment); (4) transient ischemic events (indicated by cortical Pbro2 values less than 15 mm Hg and/or transitory hypodense territories observed on follow-up CT studies); and (5) tissue infarction, defined as cortical Pbro2 values persistently (>12 hours) less than 5 mm Hg and/or permanent hypodensities (manifest tissue infarction) on follow-up CT scans.
The prognostic implication of Hunt and Hess clinical admission scores IV and V was reflected by significantly lower perioperative Pbro2 values (9.4 ± 3.7 mm Hg), elevated glutamate concentrations (105.8 ± 38.3 μmol/L) and L/P ratios (131.7 ± 51.5), as compared to Pbro2 (22.1 ± 9.8 mm Hg), glutamate values (22.7 ± 16.8 μmol/L) and L/P ratio (28.6 ± 18.7) from patients graded Hunt and Hess I-III. Mean Pbro2, glutamate levels and L/P ratios recorded during admission and in the early postoperative period differed significantly from nonischemic follow-up values (P < 0.05), whereas no significant variation was observed between admission, postoperative, and transient-ischemic follow-up levels (Fig. 3) The latter were distinctly lower than those values observed during tissue infarction (P < 0.01). Accordingly, significant variations in ventricular, cisternal, and arterial VEGF concentrations (P < 0.05) were observed between admission, postoperative, and nonischemic follow-up periods (Fig. 3). Arterial, ventricular, and cisternal VEGF concentrations during infarction were markedly elevated (P < 0.05) compared with uneventful follow-up and transient ischemia. Similar sFlt-1 levels were observed in all compartments during admission, postoperative, and non-ischemic follow-up periods (Fig. 4). In cisternal sFlt-1, however, concentrations increased significantly during transient cerebral ischemia and tissue infarction (P < 0.05). Similarly, serum sTie-2 concentrations in all compartments remained essentially unchanged from admission to uneventful follow-up (except for a steady decline in cisternal levels). For periods with transient and permanent severe cerebral ischemia, only arterial concentrations were available in sufficient number for statistical analysis, showing significant differences (P < 0.05) between these two categories (Fig. 4). Table 2 illustrates the associations observed between corresponding Pbro2 and arterial VEGF, sFlt-1, and sTie-2 levels (pooled data from patients of all clinical categories).
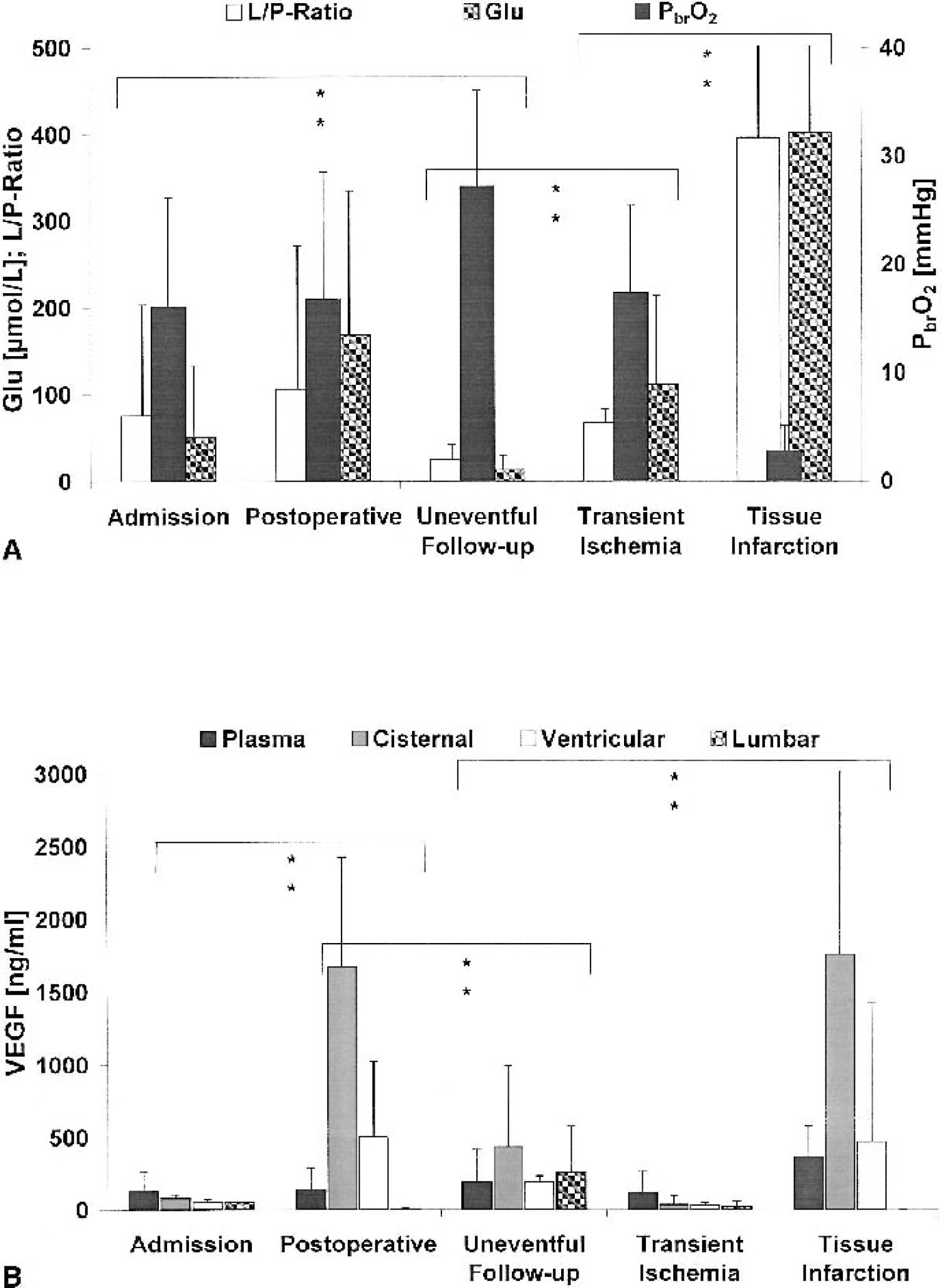
Variation of
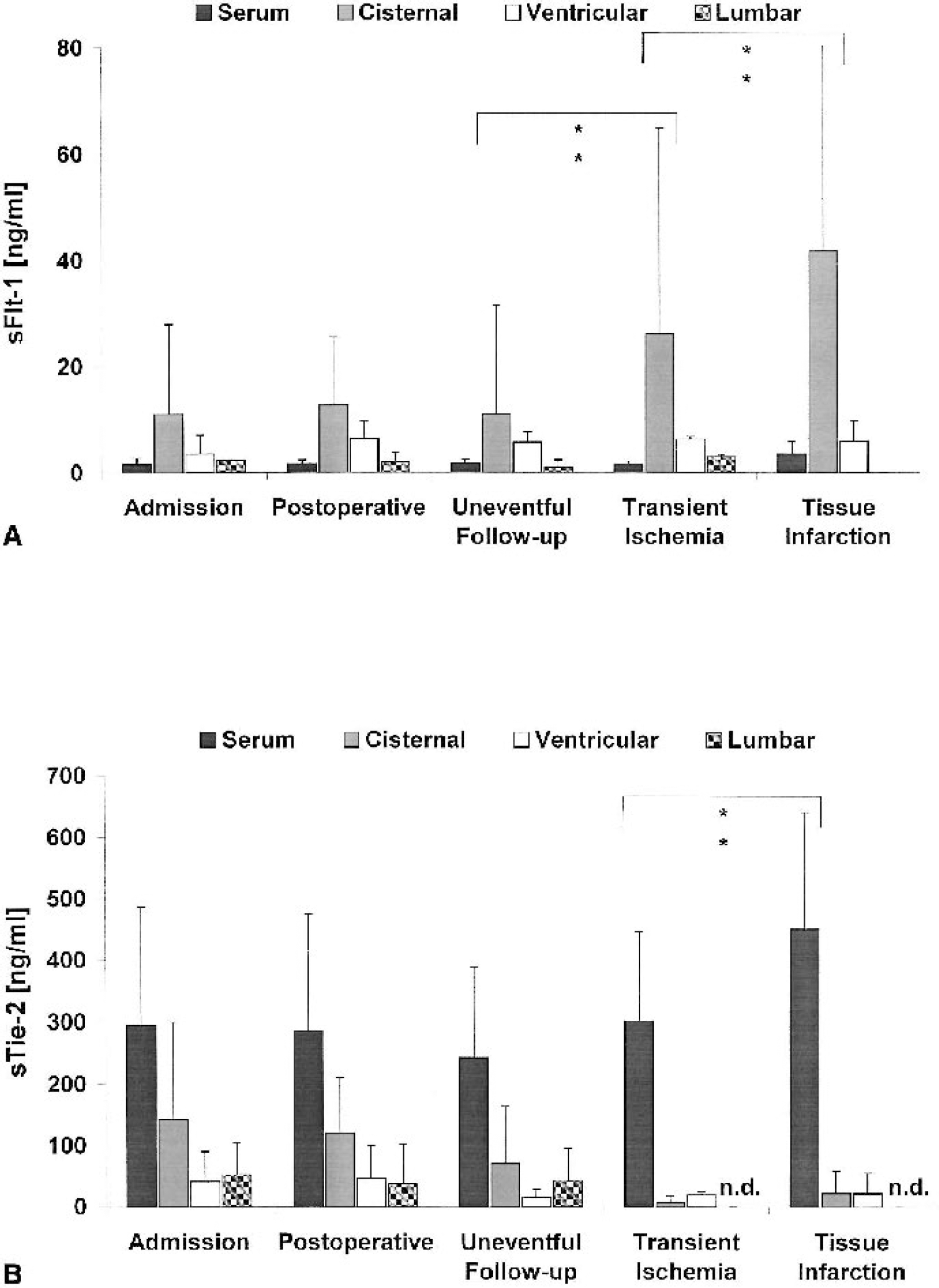
Variation of serum sFlt-1
Relation between brain oxygen tension and arterial vascular endothelial growth factor, sFlt-1, and sTie-2 concentrations
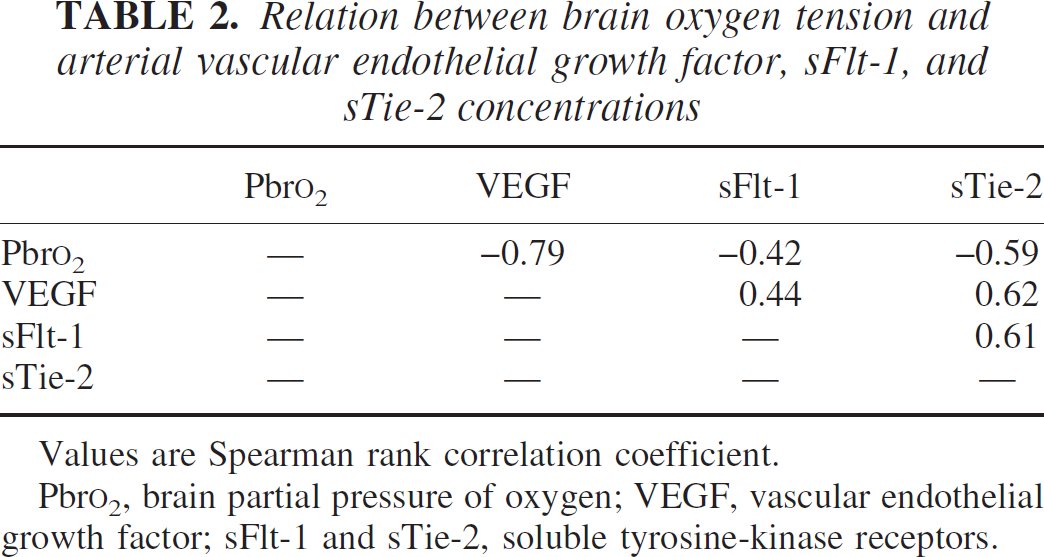
Values are Spearman rank correlation coefficient.
Pbro2, brain partial pressure of oxygen; VEGF, vascular endothelial growth factor; sFlt-1 and sTie-2, soluble tyrosine-kinase receptors.
DISCUSSION
After a previous report of association between cerebral ischemia and raised VEGF levels in systemic blood (Slevin et al., 2000), doubts have been raised regarding the source and specificity of VEGF elevation (Gunsilius et al., 2001). Platelets have been shown to constitute a major source of VEGF in circulating blood (Salgado et al., 2001). Sequestration and destruction of platelets within areas of ischemic infarction were not convincingly ruled out as a possible cause of raised VEGF levels, considering data from previous investigations demonstrating a reduction of circulating platelets in patients with large ischemic strokes (Kim et al., 1996) as well as disintegration of the microvascular basal lamina and blood—brain barrier leakage, associated with dynamic platelet aggregation in cerebral microvessels distal to the primary occlusion site (Zhang et al., 2001). However, upregulation of VEGF, Flt-1, Flk-1, angiopoietins, and Tie receptors in response to stroke has been observed locally in endothelial cells, neurons, and astrocytes within brain tissue surrounding the ischemic core (Beck et al., 2000; Lin et al., 2001; Jin et al., 2000b; Lenmyr et al., 1998; Slevin et al., 2000) as well as during global cerebral ischemia (Jin et al., 2000b).
Samples of CSF, if taken directly from an intracranial compartment close to the region of interest (i.e., the sylvian cistern, which is closely surrounded by brain tissue), should (theoretically) correspond to measurements from directly adjacent brain tissue (i.e., the intrasylvian cortex). In contrast, samples taken from the ventricular or lumbar CSF are expected to be less predictive of any ischemic cerebral event, even with expedient convection of metabolites within the CSF. Principally, even more caution is warranted during interpretation of systemic variables regarding their value in predicting or monitoring cerebral ischemia. The aim of this study was therefore to provide further evidence for the specificity of elevated systemic as well as cisternal and ventricular VEGF, sFlt-1, and sTie-2 levels with respect to detection of cerebral ischemic events. Although the number of patients admitted to the study was limited due to the stringent inclusion criteria, we attempted to present data obtained under well-defined and controlled conditions, avoiding confounding systemic influences. By allowing assessment of both preischemic, baseline conditions and episodes of manifest cerebral ischemia in individual patients, development of delayed microcirculatory impairment after SAH may provide more suitable study conditions than acute ischemic stroke.
Complex morphologic and biochemical changes occur in cerebral microvessels after SAH, including increased expression of interleukins, tumor necrosis factor-α, basic fibro-blast growth factor, and intercellular and vascular adhesion molecules (Aihara et al., 2001). Impaired microvascular flow will lead to local thrombosis and subsequent ischemic infarction of dependent brain areas, with a preponderance for the perisylvian region ipsilateral to the primary bleeding site and operative approach as well as basal ganglia, which are supplied by long perforating branches (Kistler et al., 1983).
Extent and time course of delayed cerebral ischemia caused by secondary microcirculatory disturbances occurring in a significant proportion of our study cohort were quantified by complementary reference parameters of cerebral oxygenation and energy metabolism (Pbro2, tissue glycerol, pyruvate, lactate, and glutamate concentrations) assessed within the frontal cerebral cortex. Morphologic assessment using serial CT scans was useful only to volumetrically quantify manifest cerebral infarction, whereas more sensitive imaging methods such as xenon-CT and positron emission tomography (which were logistically not accessible for serial examinations during the study) would have allowed quantitative analysis of focal hypoperfusion. In distinction to previous studies (Charbel et al., 2000; Hutchinson et al., 1999, 2000; van den Brink et al., 2000), we provide data from intracortical registration of Pbro2 and energy metabolites. Assessment of Pbro2, lactate, and pyruvate within the frontal sylvian cortex appears to yield values slightly higher, yet comparable to those observed during standard measurements within the frontal white matter of the brain (Charbel et al., 2000; Hutchinson et al., 1999, 2000; Reinstrup et al., 2000; Schulz et al., 2000; van den Brink et al., 2000). Significant and sustained elevation of cerebrovascular flow velocities (>160 cm/s) in arterial vessels of the circle of Willis was highly predictive of DIND (r: 0.99; P < 0.01) in the observed group of patients. Similarly, the amplitude of cerebrovascular flow velocity elevation was related to the amount of blood present in the basal cisterns observed on the admission CT scan (r: 0.52; P < 0.01). As an important prerequisite for meaningful interpretation of VEGF, sFlt-1, and sTie-2 concentrations, we minimized potential confounding effects by monitoring and immediately correcting changes in cerebral oxygen delivery resulting from fluctuations of mean arterial pressure and Pao2.
Pbro2 values less than 15 mm Hg (observed in >50% of our patients) were clearly related to both increasing extracellular glutamate concentrations and L/P ratio and to significant amplification of systemic arterial VEGF and sTie-2 levels. A similar, yet less stringent inverse association was observed between Pbro2 and arterial sFlt-1 concentrations. Metabolites of the glycolytic pathway have been shown to produce an angiogenic response with endothelial cell proliferation and migration in vivo and in vitro, possibly independent from induction of hypoxia-inducible genes (Murray and Wilson, 2001). Interestingly, arterial sTie-2 and VEGF levels were more closely coupled than corresponding VEGF and sFlt-1 concentrations. Cisternal and plasma VEGF levels obtained intra-operatively as well as during ischemic and nonischemic periods in the postoperative follow-up period display corresponding peaks and troughs. The postoperative cisternal and ventricular VEGF peak was paralleled by transient impairment of local tissue oxygen supply (low Pbro2 values, elevated glutamate levels and L/P ratio), most likely because of surgical manipulation during dissection of the sylvian fissure. The scale and compartmentalization of these concentration changes, however, favor local VEGF release, possibly from thrombocytes accumulating in ventricular and cisternal CSF during the initial hemorrhage. This would not only exculpate the surgeon, but also explain both the significant consecutive decrease toward baseline levels associated with uneventful follow-up, during which ventricular and cisternal CSF are successively cleared from blood remnants, and the absence of a corresponding plasma peak. The conspicuously low VEGF concentrations in all compartments observed during periods of transient cerebral ischemia as well as the high lumbar CSF concentrations during nonischemic follow-up may possibly be explained by the limited number of samples. In contrast, significantly elevated cisternal sFlt-1 concentrations were observed during both transient ischemic periods and manifest cerebral infarction, whereas serum sFlt-1 was significantly increased after cerebral infarction only, and ventricular concentrations remained stable under all investigated conditions. Similarly, serum sTie-2 levels were significantly increased only after manifest brain infarction, whereas initially elevated posthemorrhagic cisternal sTie-2 concentrations steadily decreased, remaining unaffected by changes in tissue oxygenation throughout the complete observation period.
The temporal dynamics of cisternal and ventricular sTie-2 concentrations are potentially explained by initial release from local clot and/or brain tissue during primary hemorrhage, followed by progressive resolution of ventricular and basal subarachnoid blood clots. In contrast, the stable sFlt-1 concentrations in ventricular and lumbar CSF were suitable to be used as controls for time-dependent changes of cisternal and systemic arterial concentrations. The exact reasons, however, for the significant differences between serum/plasma and cisternal VEGF and sFlt-1 concentrations based on our findings remain speculative. We suspect that VEGF in plasma originates from intravascular release by both aggregated thrombocytes and endothelial cells located in the ischemic brain region. In turn, sFlt-1 is released into systemic circulation by endothelial cells. Usually, only 10% of sFlt-1 remains detectable as free, uncomplexed receptor (Hornig et al., 2000), whereas the major fraction is occupied by its ligand. Additional dilution of both VEGF and sFlt-1 in systemic circulation may render their concentrations considerably lower than corresponding cisternal levels. Demonstrating the presence of sFlt-1 in serum and plasma of a mixed-sex control group, Barleon et al. (2001) concluded that it might play an important role as a physiologic VEGF antagonist, regulating and restricting biologic VEGF availability to a target volume. To this respect, it is notable that corresponding VEGF and sFlt-1 concentration changes in cisternal CSF (r: 0.59) and arterial blood (r: 0.44) were correlated, whereas no associations were observed between ventricular (r: −0.07) and lumbar CSF concentrations (0.16). It is neither known nor apparent from our data whether VEGF, sFlt-1, and sTie-2 are released into cisternal CSF (extracellular fluid) primarily by endothelial cells, the glioneuronal pool, or both.
Potential implications for future therapeutic strategies
Assessment of VEGF, sFlt-1, and sTie-2 in plasma/serum does not appear to be suitable for clinical monitoring of cerebral ischemia. However, the extent of cerebral ischemia and consecutive activation of compensatory tissue revascularization is reflected by time-dependent concentration changes of these angiogenesis parameters. This may have important implications for potential future therapeutic strategies incorporating early prophylactic and specifically targeted augmentation of arteriogenesis (collateralization) in response to impending ischemic insults caused by SAH, vascular diseases such as moyamoya or progressive atherosclerotic occlusion of cerebral vessels. Therapeutic use of proangiogenic substances is currently been evaluated in multiple tissues, including virus-mediated gene transfer of VEGF-mRNA in coronary artery disease (Su et al., 2000), direct topical application in cultured cortical and hippocampal neurons as well as rat brain in vivo (Hayashi et al., 1998; Lin et al., 2001; Matsuzaki et al., 2001). Direct neuroprotective effects mediating improved cell survival during hypoxia and glucose deprivation (Jin et al., 2000a) as well as glutamate-induced neurotoxicity (Matsuzaki et al., 2001) were observed in the neuronal in vitro experiments. The latter effects were mediated exclusively by binding to Flk-1 (KDR) and activation of the PI3-K/Akt and MEK/ERK pathways, whereas the Flt-1 receptor did not appear to be involved in VEGF-induced cellular protection (Jin et al., 2000a, c; Matsuzaki et al., 2001). Neurotrophic properties of VEGF in retinal development, however, seem to be mediated by both Flt-1 and Flk-1 (Robinson et al., 2001), and multiple pathways are involved in the biologic action of VEGF. Additional evidence regarding the potential value of therapeutic angiogenesis was provided by revealing that ischemic brain damage may be attenuated by ischemic preconditioning, likely through activation of collateral vessel maturation (Masada et al., 2001). Colocalization of neuropilin-1 and VEGF has been observed in cerebral blood vessels and activated astrocytes of ischemic rat brain, suggesting that both VEGF and its receptors may contribute to neovascular formation (Jin et al., 2000b, c). Moreover, undesirable effects from VEGF, like enhancement of vascular permeability, may be blocked physically (Fischer et al., 1999) pharmacologically (Fischer et al., 2001), or by selective inhibition of specific subsystems, such as Src-kinase (Paul et al., 2001; van Bruggen et al., 1999; Zhang et al., 2000). Both angiogenic and direct neurotrophic properties may be enhanced by combined administration of additional growth factors (Sugimori et al., 2001). The delayed yet predictable onset of ischemia secondary to progressive microvascular impairment after SAH theoretically provides ideal conditions for implementation of these rapidly advancing techniques in experimental therapy trials (Alafaci et al., 2000; Ooboshi et al., 2001; Wang et al., 2000).
CONCLUSIONS
Analysis of local cisternal and arterial VEGF and Flt-1 as well as arterial sTie-2 concentrations yield information on extent and time course of delayed focal cerebral ischemia after SAH. Elevated plasma/serum concentrations of VEGF and sFlt-1 are probably caused by release from local thrombocyte aggregations within microvessels of ischemic brain regions, whereas the origin of systemic sTie-2 remains elusive. Cisternal VEGF and sFlt-1 may originate from both endothelial cells and the glioneuronal pool, whereas cisternal sTie-2 concentrations decrease with progressive resolution of the subarachnoid blood clot, indicating that sTie-2 is released from clot components rather than brain tissue. Whereas local and systemic concentration changes of these parameters of angiogenesis reflect development of collaterals, and potentially, new capillaries, these natural compensatory mechanisms appear to be insufficient to prevent progressive cerebral ischemia caused by microcirculatory impairment. Further time-resolved structural investigation of cerebral microvessel remodeling triggered by ischemia will aid in development of future strategies implementing the therapeutic potential of angiogenesis.