
Abstract
Oxygen and glucose deprivation (OGD) in cell cultures is generally studied in a medium, such as artificial cerebrospinal fluid (CSF), with an ion composition similar to that of the extracellular fluid of the normal brain (2 to 4 mmol/L K+, 2 to 3 mmol/L Ca2+; pH 7.4). Because the distribution of ions across cell membranes dramatically shifts during ischemia, the authors exposed mouse organotypic hippocampal tissue cultures to OGD in a medium, an ischemic cerebrospinal fluid, with an ion composition similar to the extracellular fluid of the brain during ischemia in vivo (70 mmol/L K+, 0.3 mmol/L Ca2+; pH 6.8). In ischemic CSF, OGD induced a selective and delayed cell death in the CA1 region, as assessed by propidium iodide uptake. Cell death was glutamate receptor dependent since blockade of the N-methyl-D-aspartate and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors mitigated cell damage. Hyperglycemia aggravates ischemic brain damage in vivo, whereas in vitro glucose in artificial CSF prevents oxygen deprivation-induced damage. The authors demonstrate that glucose in ischemic CSF significantly exacerbates cell damage after oxygen deprivation. This new model of in vitro “ischemia” can be useful in future studies of the mechanisms and treatment of ischemic cell death, including studies using genetically modified mice.
The integrated mechanisms of ischemic brain damage and the effect of drug interventions are readily studied in rodent in vivo models, which for these purposes are more suitable than in vitro models (Ginsberg and Busto, 1989). The intact brain preserves the blood-brain barrier, the complex neuronal networks, and interactions among neurons and nonneuronal cells. On the other hand, this complexity does not permit detailed studies of particular molecular mechanisms and isolated cellular events. These limitations are overcome in the in vitro models of brain ischemia where the contribution of blood components are eliminated, and where tissue temperature and the extracellular environment, including ion and nutrient availability, can be standardized (Lipton 1999). In most in vitro models of ischemia a combination of oxygen and glucose deprivation (OGD) is used to imitate in vivo ischemic conditions (Sick and Somjen, 1998). In these models the ionic composition of the incubation medium, such as the artificial cerebrospinal fluid (aCSF), is similar to that found in normal brains.
It is evident, however, that the in vitro models do not fully reproduce the pathophysiologic events that occur in the brain after in vivo ischemia. The hippocampus has been extensively studied after global ischemia in the rat and gerbil. The damage is characterized by selective neuronal death in the CA1 region appearing at 48 to 72 hours of recovery after 10 to 15 minutes of ischemia (Ito et al., 1975; Kirino, 1982; Pulsinelli et al., 1982). Although dissociated neuronal cultures or organotypic hippocampal tissue cultures are readily damaged by OGD, the temporal and special pattern of cell death is not similar to that seen in vivo.
One of the hallmarks of cerebral ischemia is the loss of ion homeostasis across cell membranes due to inhibition of ATP synthesis. This has been extensively studied in animal models of global and focal ischemia (Siesjö, 1992). The membrane depolarization results in an increase in extracellular potassium, a decrease in extracellular calcium, and a decrease in pH (Hansen, 1985). We reasoned that in tissue cultures the medium could be regarded as an extension of the extracellular space. The volume of the extracellular space in vitro (i.e., the medium) is at least 100 times larger than that of the volume of the individual neurons. Since the ion concentrations are equilibrated across the plasma membrane during OGD owing to membrane depolarization (Siesjö, 1985), the intracellular ion concentrations in neurons can be assumed to approach those of the medium. Therefore, in order to mimic in vivo conditions, we used a medium with an ion composition similar to that determined in the extracellular space in the brain during ischemia.
The aim of this study was to develop a model of OGD-induced cell death in mouse hippocampal tissue cultures that, to a larger degree, mimic the pathophysiologic sequence of events occurring in vivo. We used organotypic cultures of brain tissue that offer an integrated system with preserved synaptic connections and a diversity of cells including neurons, astrocytes, and microglia (Gähwiler, 1981; Stoppini et al., 1991). These cultures have been extensively used in studies of OGD in rats (Laake et al., 1999; Newell et al., 1995; Perez Velazquez et al., 1997; Pringle et al., 1997) but not in mice, except for one investigation using chemical hypoxia (Bernaudin et al., 1998). Using this approach we were able to mimic the pathophysiologic events in the mouse hippocampus after ischemia in vivo, including selective and delayed neuronal death in the CA1 region and increased damage by hyperglycemia.
MATERIALS AND METHODS
Preparation and maintenance of organotypic hippocampal tissue cultures
The Malmoe/Lund ethical committee on animal experiments approved all animal experiments (Approval nr M106–98). Organotypic hippocampal cultures were prepared according to the methods described by Stoppini et al. (1991), with some modifications. Brains of 6-day-old Balb/c mice were removed and gently immersed in ice-cold dissection medium containing HBSS (Hank balanced salt solution) with 20 mmol/L HEPES, 100-U penicillin-streptomycin/mL, and 6 mg/mL D-glucose. Hippocampi were dissected out on ice and cut into 250-μm-thick transverse sections using a McIlwain Tissue Chopper. Cultures with even margins and clear, uniform, and well-defined pyramidal cell layers were selected and plated onto Millicell culture inserts (0.4 μm Millicell-CM, 12 mm in diameter; Millipore Corp., Bedford, MA, U.S.A.), one culture per insert. Sections from the caudal and most frontal parts of the hippocampus were discarded. Cultures were maintained at 35°C, 90% to 95% humidity in a CO2 incubator (Thermo Forma Scientific, Marietta, MA, U.S.A.) for 10 or 11 days before the experiments were performed. The culture medium consisted of 50% MEM (Eagles with Earl balanced salt solution), 25% heat-inactivated horse serum, 18% HBSS, 2% B27 supplemented with 4 mmol/L L-glutamine, 50-U penicillin-streptomycin/mL, and 6-mg/mL D-glucose. The pH was adjusted to 7.4 with sodium bicarbonate. Culture medium was changed on the second day in vitro (div 2) and then every other day. All substances used for preparation and maintenance of cultures were obtained from GibcoBRL, Life Technologies (both now Invitrogen, Carlsbad, CA, U.S.A.), with the exception of D-glucose, which was from Sigma-Aldrich (St. Louis, MO, U.S.A.).
Histologic evaluation of cultures
For examination of histology the cultures were washed in phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde in PBS for 1 hour at room temperature, and then stored in PBS at 4°C. Cultures on their membranes were dehydrated and embedded in Agar100 (LinkNordiska, Lidingö, Sweden). Cultures were cut out of the insert and sectioned into 2.5-μm-thick sections on an ultratome. Sections were then stained with 0.5% methylene blue/0.5% azure blue dissolved in 1% borax.
Electrophysiologic experiments
For electrophysiologic recordings, the slice on its membrane was placed in a recording chamber and submerged in aCSF consisting of (in millimolar concentrations): 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 26.2 NaHCO3, 1 NaH2PO4, and 11 glucose, which was gassed with 95% O2 to 5% CO2 The temperature of the recording chamber was kept between 21°C and 23°C, which is the standard in our laboratory. Extracellular field EPSPs were recorded via a glass pipette containing 3 mol/L NaCl (0.5–1 MΩ). Field potentials were amplified and filtered at 1 kHz and sampled at 10 kHz with an EPC-9 patch-clamp amplifier (HEKA Electronics, Lambrecht, Germany) and stored on a Power Macintosh computer for off-line analysis. The amplitude of the field EPSP at its peak was measured over a period of 1 or 2 milliseconds. Both recording and stimulating electrodes were placed under visual guidance in the stratum radiatum (stimulating and extracellular recording electrodes) or in the cell layer (whole-cell recording pipettes) of the CA1 region. Whole-cell recording pipettes (4–6 MΩ) were filled with the following (in millimolar concentrations): 122.5 K gluconate, 17.5 KCl, 10 HEPES, 0.2 EGTA, 8 NaCl, 2 MgATP, and 0.3 GTP (pH 7.2; osmolarity, 295 mOsm). Both current-clamp and voltage-clamp recordings were used. Membrane currents were amplified and filtered at 2.9 kHz and sampled at 10 kHz with an EPC-9 patch-clamp amplifier. The holding potential was −80 mV for recording AMPA receptor-mediated EPSCs and 30 mV for recording N-methyl-D-aspartate (NMDA) receptor-mediated EPSCs. Junctional potentials were not corrected.
Induction of oxygen-glucose deprivation
All cultures used in one experiment were prepared from mice pups from one to three females with litters born on the same day. Cultures were grown for 10 or 11 days and grouped before experiment so that each experimental group contained cultures from six different pups. Cultures showing distinct PI uptake in the pyramidal cell band were excluded. A scarce PI uptake was sometimes observed in the dorsal blade of the dentate gyrus. Before OGD, cultures were washed once in glucose-free medium and transferred to the anaerobic incubator. The anaerobic incubator (Elektrotek Ltd., Keighley, U.K.), equipped with a palladium catalyst to remove traces of oxygen, was custom made for the hypoxia experiments and had an aperture for rapid entry of 24-well plates. It had an atmosphere of 10% H2, 5% CO2, and 85% N2 and the temperature was maintained at 35.0°C ± 0.3°C. Inside the incubator cultures were transferred to wells containing OGD medium, which had been placed in the anaerobic atmosphere overnight or bubbled with the anoxic gas mixture for at least 1 hour before use. Oxygen and glucose deprivation was terminated when cultures were transferred to new wells with oxygenated culture medium and then transferred to the CO2 incubator. Culture controls were maintained in the CO2 incubator throughout the experiment. Two OGD mediums were used: (1) aCSF with the following composition (in millimolar concentrations): 2 CaCl2, 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 MgSO4, and 10 sucrose (pH 7.4); and (2) iCSF with the following composition: 0.3 CaCl2, 70 NaCl, 5.25 NaHCO3, 70 KCl, 1.25 NaH2PO4, 2 MgSO4, and 10 sucrose (pH 6.8). For studies of glutamate receptors, 20 μmol/L noncompetitive NMDA-receptor antagonist dizocilpine maleate (MK-801) was added to the culture medium 1 hour before OGD. The competitive NMDA receptor antagonist D-2 amino-5-phosphonopentanoic-acid (D-AP5, 150 μmol/L) and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-receptor blocker 2,3-Dihydro-6-nitro-7-sulphamoyl-benzo(F)quinoxaline (NBQX, 100 μmol/L) were added to the medium 1 hour before OGD, and to the OGD and culture media during the 24-hour recovery period. MK-801 was purchased from Sigma, and D-AP5 and NBQX-disodium salt were obtained from Tocris Cookson (St. Louis, MO, U.S.A.).
Quantification of cell damage
Propidium iodide (PI; 1 μg/mL) was added to the culture medium on the day before the insult and was included throughout the duration of the experiment (Fig. 1). Cultures were examined with an inverted fluorescence microscope. Excitation was at 510 to 550 nm and emission was at 590 nm and above. Images were captured using a 12-bit monochrome-cooled fluorescence camera (Apogee Instruments, Logan, UT, U.S.A.) and processed with Image-Pro Plus 4.0 software (Media Cybernetics, Maryland, U.S.A.).
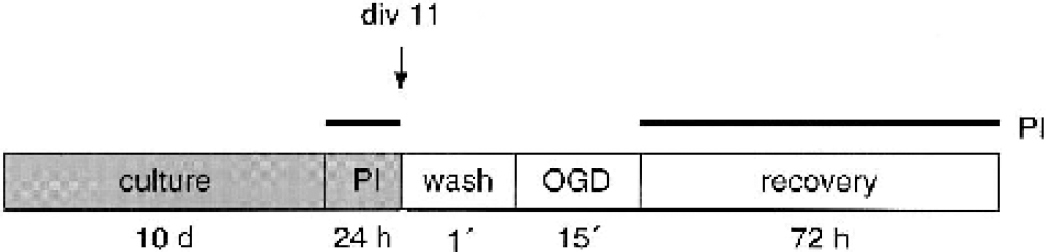
Protocol for oxygen-glucose deprivation (OGD) experiments in mouse hippocampal organotypic cultures. Organotypic cultures were grown for 11 days before the experiments were performed. Propidium iodide (PI; 1 μg/mL) was added to the culture medium 24 hours before the start of OGD and maintained throughout the experiment. Cultures were washed once before the induction of OGD. Images were captured at variable time points during the 72 hours of recovery.
Measurements of cell damage were made on PI-fluorescence images. Images were obtained before and 4, 8, 12, 24, 48, and 72 hours after the insult. Fluorescence intensity was measured in a standardized area in CA1 (Fig. 2). At all time points a faint, diffuse staining was observed throughout the pyramidal cell layer. Background staining was measured in a small hexagon placed in the CA2/CA3 region where no damage was observed. As this background staining increased with time it was subtracted from the value measured in the standardized CA1 area. Values are expressed as mean fluorescence intensity (MFI).
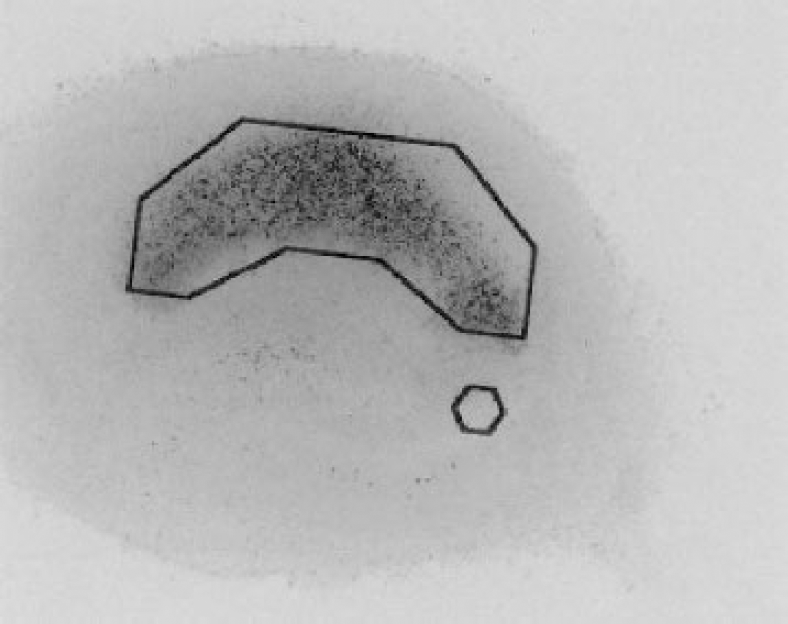
Method for quantification of cell damage. Inverted fluorescence image of a propidium iodide (Pl)-stained cell culture 48 hours after oxygen-glucose deprivation; PI intensity is measured as mean fluorescence intensity (MFI) in standardized area in the CA1 region as indicated. Background MFI is measured in a standardized hexagon area in the CA2-CA3 region and subtracted from the MFI measured in the CA1 area.
Statistics
For statistical analyses the commercial software Statview 4.0 (Abacus Concepts Inc, Berkley, CA, U.S.A.) was used. Data are expressed as mean ± SD. All statistical groups consist of data obtained from at least three separate experiments. All compared groups were run in parallel inside the anaerobic incubator. Two-way analysis of variance (ANOVA) with Scheffé post hoc test was used to evaluate differences between groups (with the exception of Fig. 6B, where a repeated-measures ANOVA with Scheffé post hoc test was used). Variability between experimental dates was compensated for by including date as a factor.
RESULTS
Electrophysiologic characteristics of the mouse hippocampal organotypic tissue culture
In order to assess whether our culture conditions affected the properties of the mouse hippocampal organotypic tissue culture, we investigated the electrophysiologic properties of CA1 neurons. Using whole-cell recordings in current-clamp mode, it was found that all cells tested were able to fire action potentials in response to depolarizing current injections (Fig. 3A). In voltage-clamp mode, in which the cell was clamped at −80 mV and inhibitory synaptic activity was blocked with the GABAA antagonist picrotoxin, spontaneous excitatory synaptic currents were recorded (Fig. 3B). When evoking excitatory synaptic signals by activating the Schaffer collaterals, both AMPA- and NMDA-receptor mediated synaptic signals could be obtained (Fig. 3C). Spontaneous inhibitory synaptic currents were recorded when the AMPA and NMDA receptors were blocked with NBQX and D-AP5, respectively (data not shown). These two findings indicate that CA1 pyramidal cells have both functional excitatory and inhibitory synaptic inputs. The afferent excitatory synapses to the CA1 pyramidal cells were able to express long-term synaptic plasticity, which was tested using extracellular recordings. As seen in Fig. 3D two brief, high-frequency stimulations (two 100-impulse trains, 100 Hz, 20 seconds apart) of the afferents induced a long-lasting increase in synaptic efficacy. Taken together these findings suggest that the CA1 pyramidal cells from mouse organotypic slices (div 11) display normal histologic and electrophysiologic properties and maintain both functional excitatory and inhibitory synaptic inputs, which are able to express synaptic plasticity.
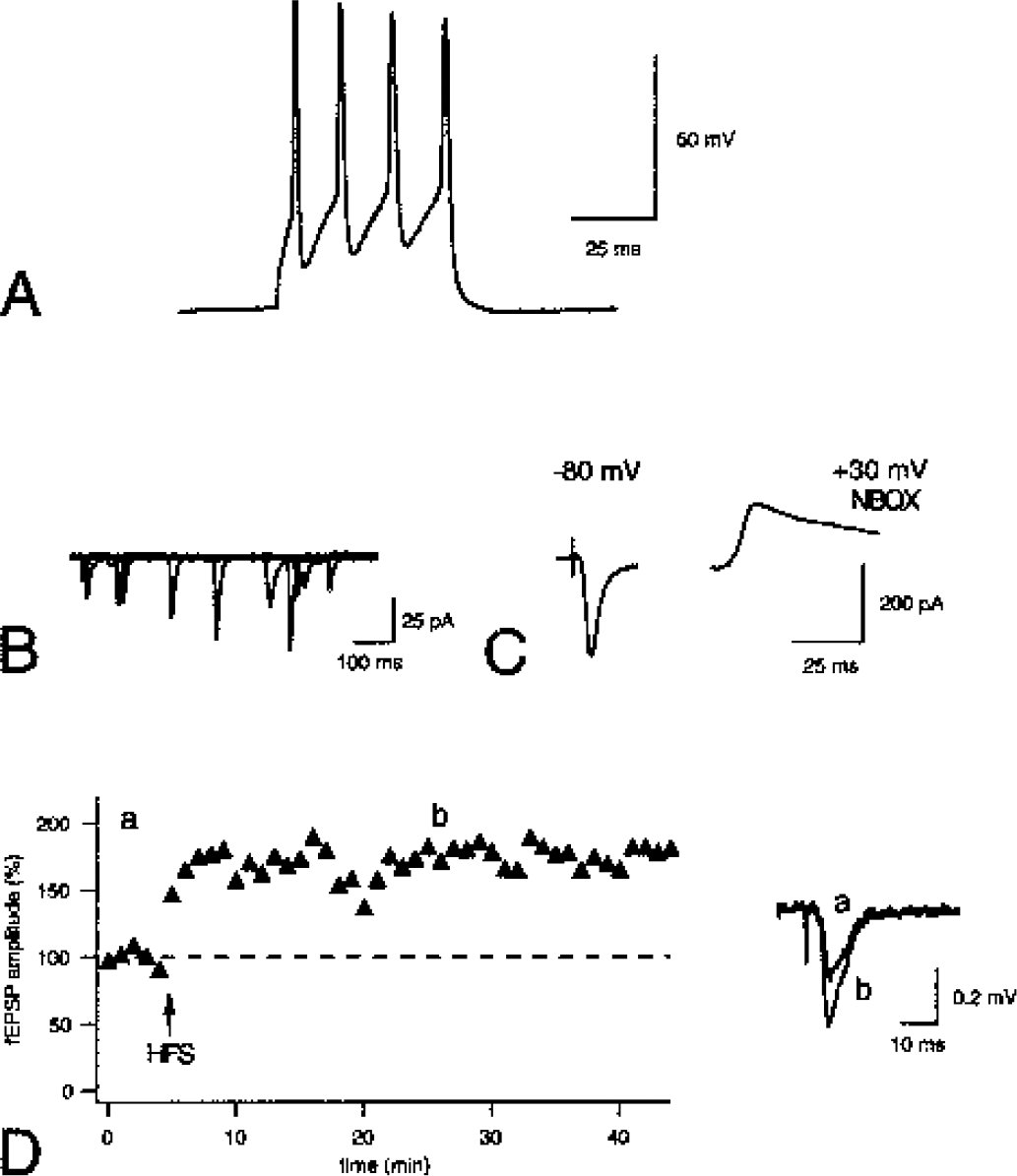
Electrophysiologic characteristics of CA1 pyramidal cells in mouse hippocampal organotypic tissue cultures.
Morphologic response to oxygen-glucose deprivation
Figure 4 shows sections of two hippocampal organotypic tissue cultures stained with PI (Figs. 4A and 4B) and with methylene blue/azure blue (Figs. 4C–4F). Photomicrographs of a control culture (Figs. 4A, 4C, and 4E) on div 11 and a culture exposed to 15-minute OGD in iCSF with 48-hour recovery (div 13; Figs. 4B, 4D, and 4F). The cultures have preserved their organotypic appearance and the pyramidal and granule cell layers are clearly visible (Figs. 4C and 4D). In control cultures the pyramidal cell bodies have a typical owl's-eye look and the neuropil is compact with no evident swollen structures (Fig. 4E). After OGD, damage to CA1 neurons is seen as a clearing of the cell body layer, whereas cells in CA3 are intact. In the CA1 region, pyknotic dead neurons with dark condensed nuclei are seen among surviving neurons (Fig. 4E). Approximately 25% cell death is seen in the CA1 region after 15-minute OGD in iCSF.
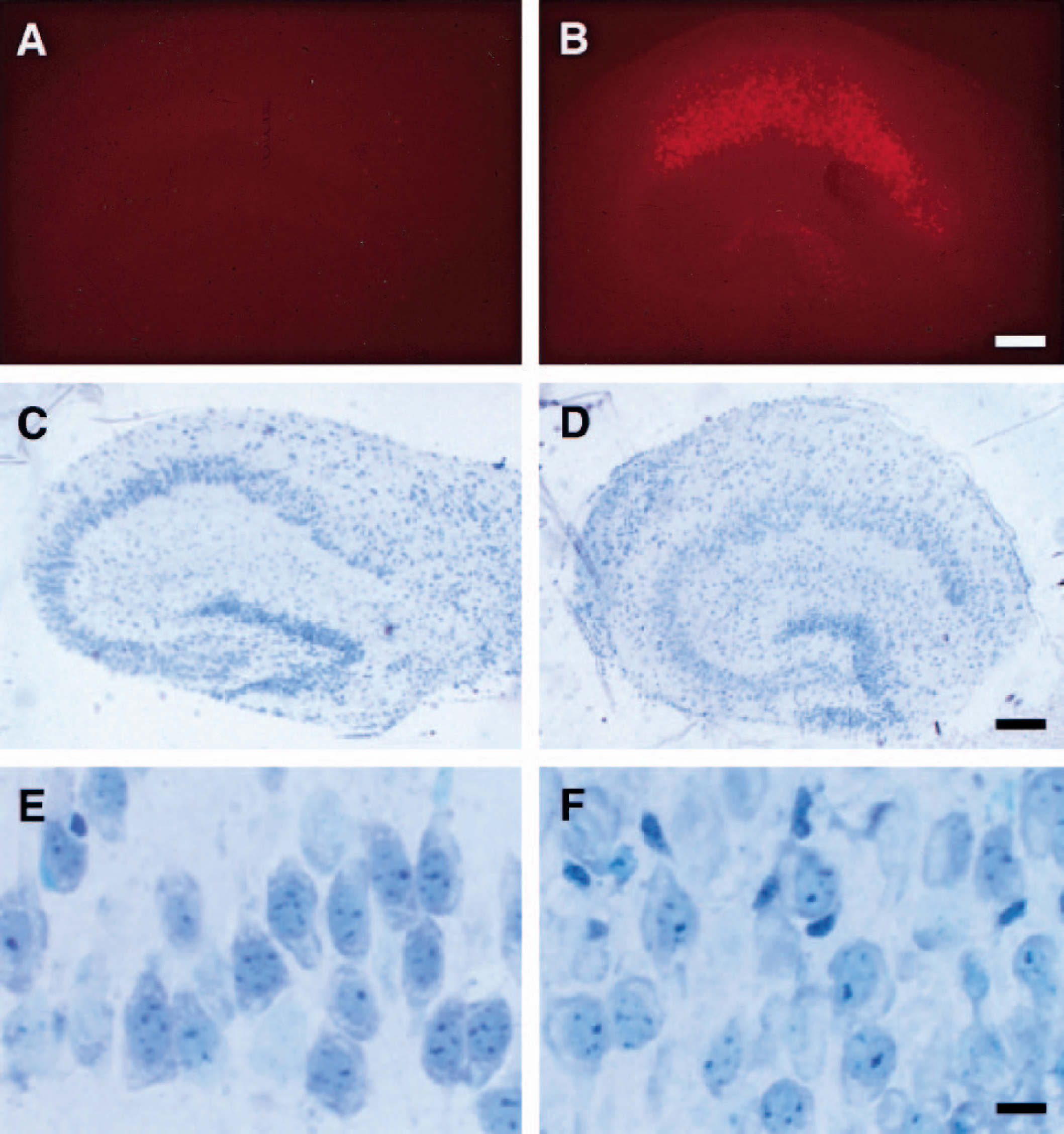
Histologic evaluation of cell damage. Control tissue culture
Influence of potassium, calcium, and hydrogen ions on the development of cell damage after oxygen-glucose deprivation
Complete deprivation of oxygen and glucose for 15 minutes in glucose-free aCSF led to a homogenous uptake of PI in the CA1 region (Fig. 5A). This PI uptake was not restricted to the CA1 area, as the CA3 area also displayed significant PI uptake. The increase in PI uptake in the CA1 area was rapid, clearly visible after 4 hours of recovery and increasing for 24 hours (Fig. 5B). The PI fluorescence in the CA1 area was very dense and homogenous. As our aim was to establish an in vitro model resembling the in vivo ischemia conditions, we decided to change the concentration of potassium, calcium, and hydrogen ions in the OGD medium to those reported in vivo. We first examined the contribution of the individual ions by modifying aCSF. The temporal development of damage and the density of PI uptake in the CA1 region were similar when the potassium concentration was increased from 2.5 to 70 mmol/L; however, PI uptake in the CA3 region also increased. A decrease in calcium to 0.3 mmol/L significantly reduced cell damage. Lowering pH to 6.8 led to significantly less PI uptake at all time points from 4 to 72 hours. In cultures subjected to OGD in pH-6.8 aCSF, the area in CA1 taking up PI was smaller compared with the aCSF-treated group and was selective to the CA1 region. At 0.3 mmol/L calcium and pH 6.8, no damage to CA3 was seen.
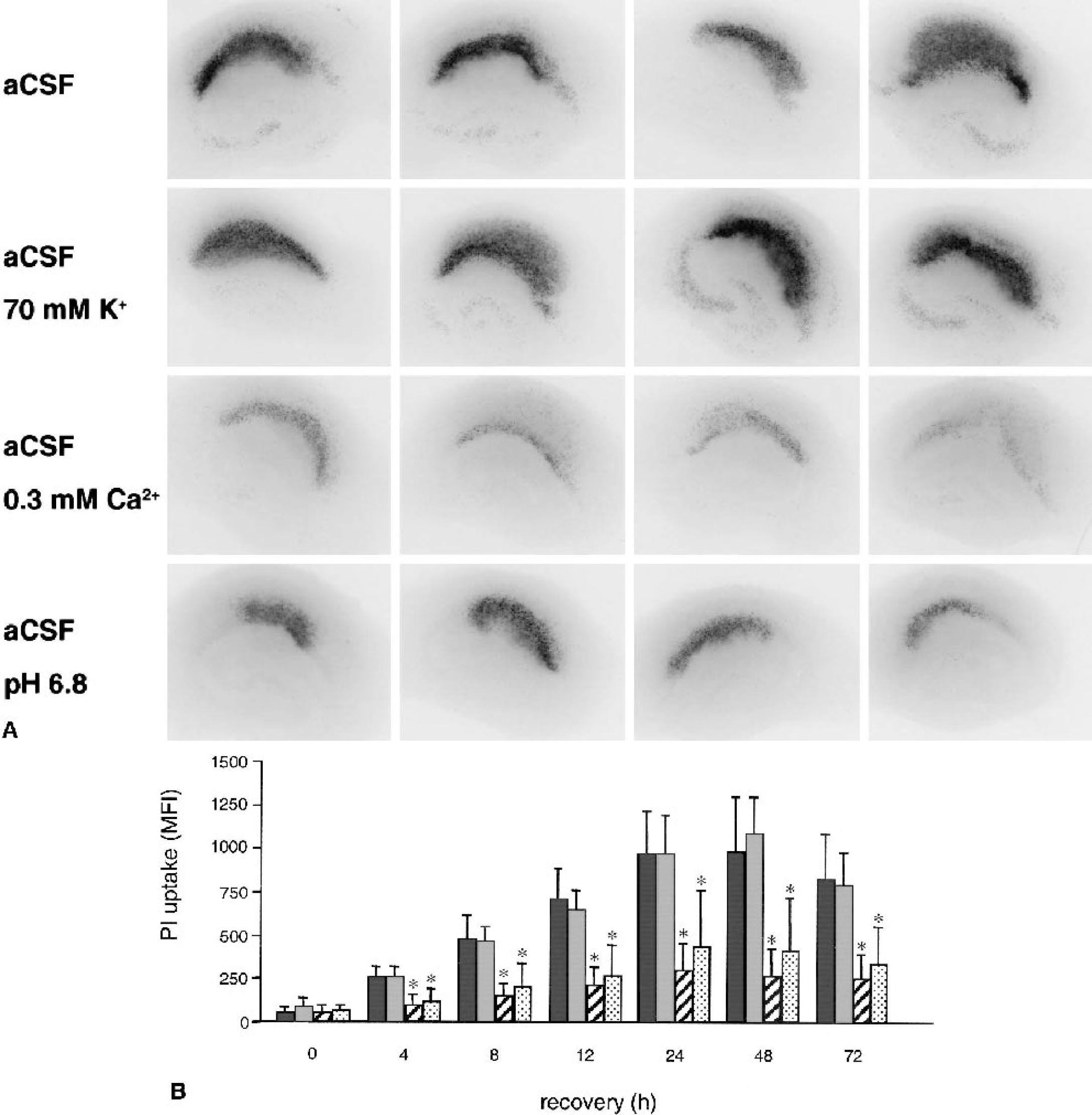
The influence of the ionic composition of the oxygen-glucose deprivation (OGD) medium on cell damage after OGD.
Ischemic cerebrospinal fluid
By changing the OGD medium to 70 mmol/L K+, 0.3 mmol/L Ca2+, and pH 6.8, and in addition decreasing Na+ to 77 mmol/L (Hansen, 1985), the extracellular concentrations in the tissue cultures would be similar to those in the brain during complete ischemia. This glucose-free medium was called ischemic cerebrospinal fluid (iCSF). The development of cell damage after OGD in iCSF was compared with that in aCSF (Fig. 6). Cultures exposed to a 15-minute OGD in iCSF developed a selective cell damage restricted to the CA1 area without damage to CA3 neurons. These cultures also displayed a slower development of cell damage than their aCSF-treated counterparts. In aCSF-treated cultures a significant increase in PI uptake was seen after 4 hours of recovery. Damage developed rapidly and there was no statistically significant increase in PI uptake between 24 and 48 hours of recovery. In the iCSF-treated cultures, on the other hand, a significant increase in PI uptake was not found until after 8 hours of recovery, and PI intensity continued to increase significantly between 24 and 48 hours of recovery.
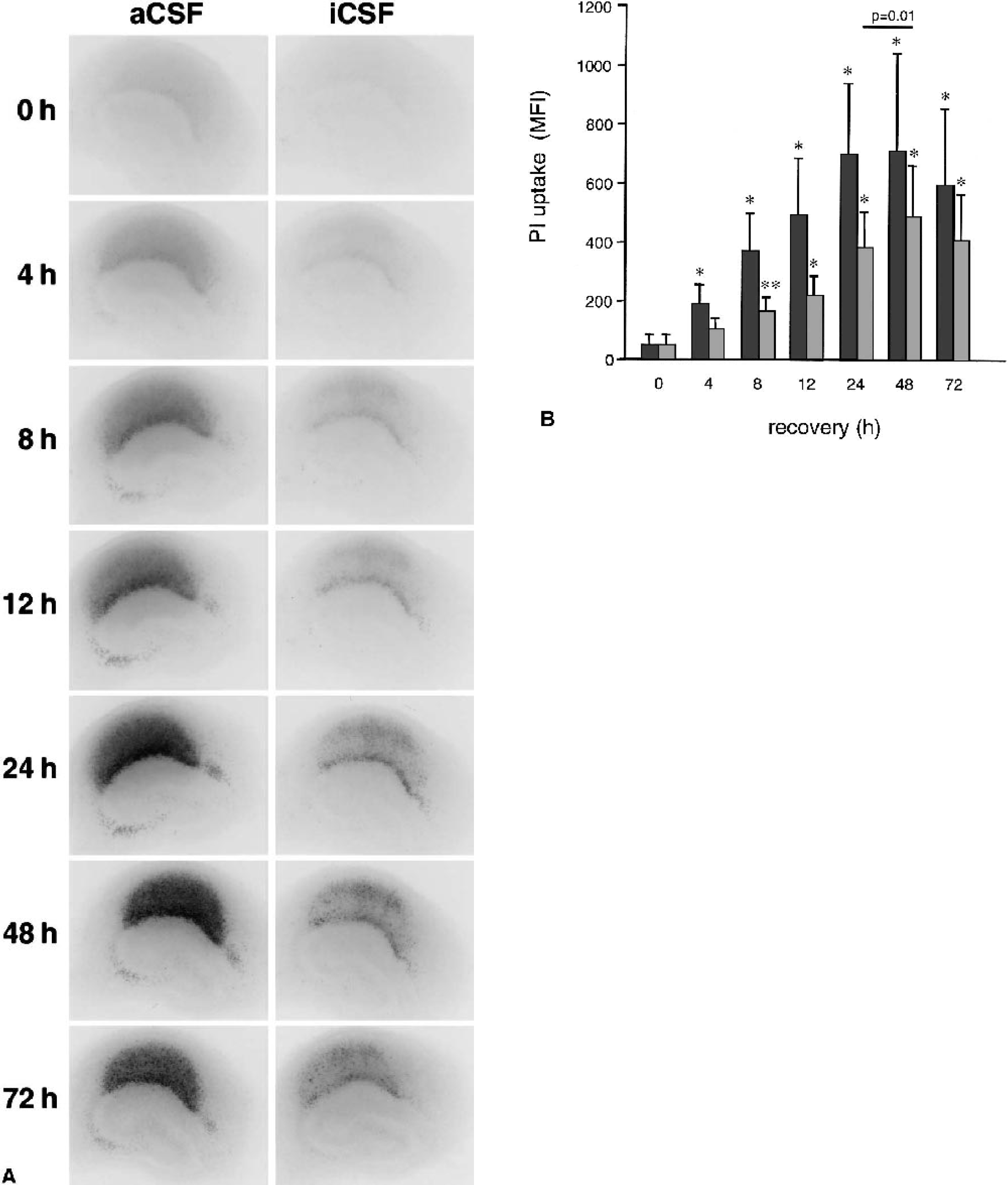
Temporal development of cell damage after oxygen-glucose deprivation (OGD) in artificial cerebrospinal fluid (aCSF) and ischemic CSF (iCSF).
To investigate whether iCSF or the washing procedure in itself could be harmful to neurons, we incubated groups of cultures in iCSF and aCSF for 15 minutes following the same protocol as in the OGD experiments but exchanging the anaerobic incubator for the regular CO2 incubator. None of the groups displayed any elevated PI uptake at 48 hours of recovery compared with culture controls (data not shown).
Cell damage was diminished with antagonist to NMDA and AMPA receptors
To investigate the involvement of glutamate receptors in the damage induced by OGD in iCSF, NMDA receptors were blocked with the noncompetitive antagonist MK-801 and the competitive antagonist D-AP5. Pretreatment with 20 μmol/L MK-801 for 1 hour before a 15-minute OGD in iCSF resulted in significantly (80%) less damage in the CA1 region at 24 hours of recovery compared with OGD-control cultures (Fig. 7A). A similar (75%) protective effect that was seen when 150 μmol/L D-AP5 was added to the culture medium 1 hour before OGD or to the iCSF during OGD was present throughout recovery (Fig. 7B). Involvement of AMPA receptors was investigated with the competitive antagonist NBQX, which was included in the medium from 1 hour before to 24 hours after a 15-minute OGD. This addition resulted in a significant (50%) reduction of PI uptake compared with controls (Fig. 7C).
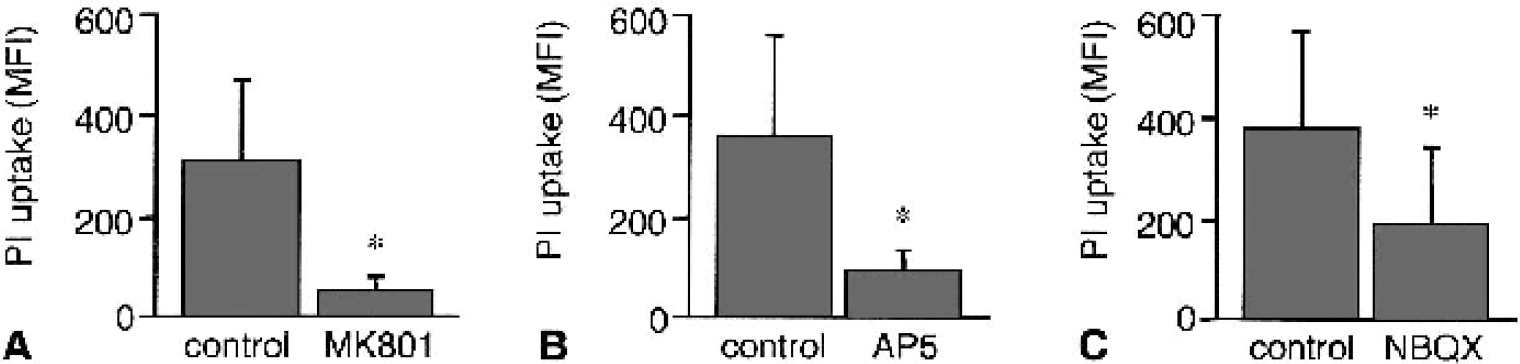
Protective effect of glutamate antagonists. The glutamate antagonists
The presence of glucose during hypoxia in ischemic cerebrospinal fluid exacerbated cell damage
When aCSF was supplemented with 40 mmol/L glucose (the glucose concentration of the culture medium) during a 15-minute hypoxia, no cell damage was observed (Fig. 8). These glucose-supplemented hypoxia-exposed cultures displayed the same level of PI uptake in the CA1 area as control cultures kept in the CO2 incubator throughout the experiment. In contrast, the addition of 40 mmol/L glucose to iCSF during a 15-minute hypoxia had the opposite effect and resulted in significantly more PI uptake in the CA1 area than the iCSF-OGD controls run in parallel. Measurements were made after 48 hours of recovery in all groups.
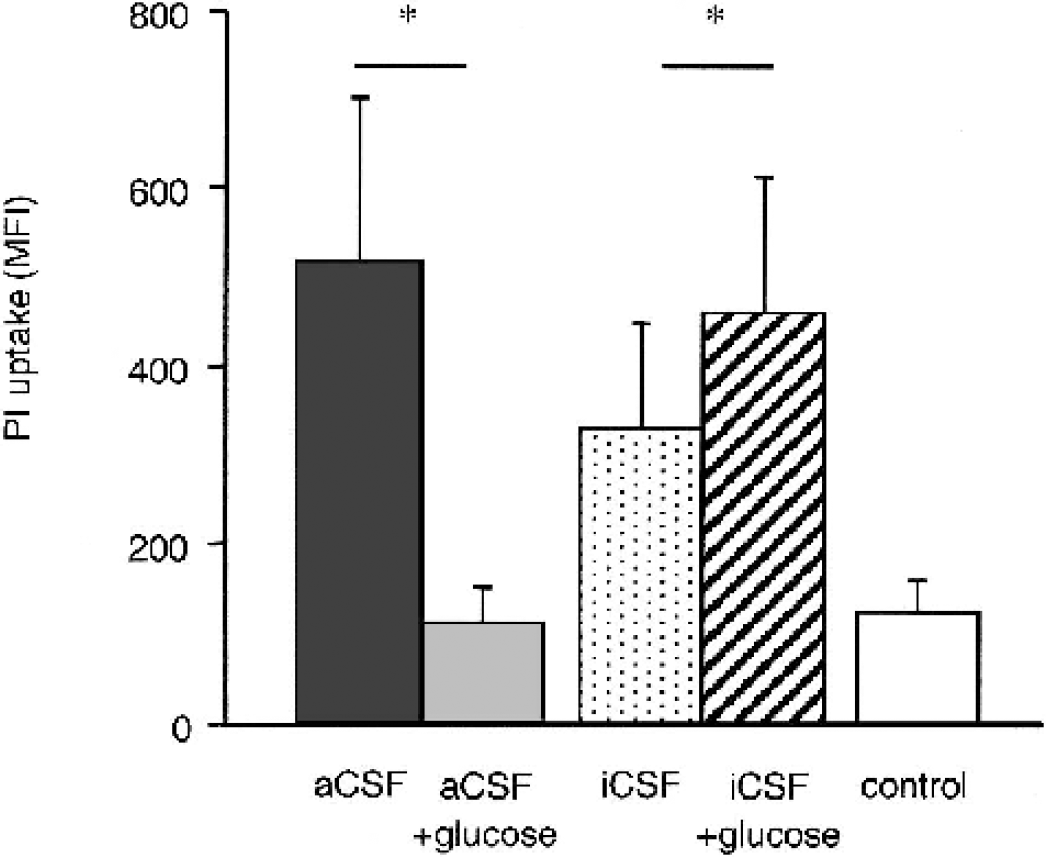
Glucose decreases cell damage after hypoxia in artificial cerebrospinal fluid (aCSF) but increases cell damage after hypoxia in ischemic CSF (iCSF). Addition of 40 mmol/L glucose to the glucose-free aCSF completely prevented cell damage after 15-minute hypoxia. The propidium iodide (PI) intensity in the CA1 area of aCSF with cultures treated with 40 mmol/L glucose (light gray bar) did not differ from controls (white bar) and was significantly (P < 0.0001) less than the intensity of the aCSF-treated cultures (dark gray bar). The addition of 40 mmol/L glucose to iCSF caused significantly (P = 0.0242) worse cell damage in the CA1 area after a 15-minute hypoxia in iCSF alone. Results are derived from three different experiments and calculated by ANOVA with Scheffé post hoc test (n = 18) for each group.
DISCUSSION
Hippocampal organotypic tissue cultures have been widely used to study ischemic, hypoxic, hypoglycemic, oxidative, and excitotoxic damage (Abdel-Hamid and Tymianski, 1997; Barth et al., 1996; Hsu et al., 1994; Newell et al., 1990, 1995; Perez Velazquez et al., 1997; Pringle et al., 1996; Strasser and Fischer, 1995; Vornov et al., 1991, 1994). In these experiments, ischemialike conditions are induced by a combination of OGD achieved by an anoxic atmosphere in combination with a glucose-free medium in a manner similar to that described for dissociated cell cultures (Goldberg and Choi, 1993; Rothman 1983), or by inhibition of oxidative phosphorylation using cyanide in combination with 2-deoxy-glucose (Bernaudin et al., 1998; Vornov et al., 1994).
Here we describe a model for OGD in mouse hippocampal organotypic tissue cultures that, in many aspects, mimic the tissue reaction to brain ischemia in vivo. The results show that the composition of the OGD medium is of critical importance for the development of cell death after in vitro ischemia. By adapting the medium used during OGD to the pathophysiologic concentrations of potassium, calcium, and hydrogen ions measured during in vivo global ischemia, the selectivity and temporal profile of damage seen in vivo could be mimicked.
For assessment of damage, we used PI uptake as cell death indicator. In concordance with previous reports, we did not observe any harmful effects of PI on cell survival. Propidium iodide fluorescence has been shown to correlate linearly with histologically defined neuronal damage as well as lactate dehydrogenase release (Abdel-Hamid and Tymianski, 1997; Laake et al., 1999; Macklis and Madison 1990; Newell et al., 1995; Noraberg et al., 1999; Pringle et al., 1996; Strasser and Fischer, 1995; Vornov et al., 1994; Wilde et al., 1997). Also in our experiments, PI staining correlated with histologic evaluation of neuronal damage. In concordance with (Macklis and Madison, 1990) we observed that the PI uptake during the early recovery phase was diffuse and became pinpoint later during recovery.
Cell death in the hippocampus in vivo is characterized by a selective neuronal damage to the CA1 region after moderate ischemia periods with recruitment of damage to the CA3 region after prolonged ischemia and delayed neuronal death. Selective vulnerability and delayed death of CA1 neurons after global ischemia have been described in gerbils (Ito et al., 1975) and rats (Kirino, 1982; Pulsinelli et al., 1982). Also in mouse, 10 minutes of bilateral common carotid artery occlusion in combination with systemic hypotension resulted in delayed CA1 cell death occurring between 1 and 3 days of reperfusion (Sheng et al., 1999). Thus, the development of cell damage after global ischemia seems to have a similar course of events in rat and mouse.
A selective and delayed cell death in the CA1 region, as seen in vivo, has so far not been reported in hippocampal cultures. Using rat hippocampal organotypic tissue cultures several authors have reported a relative selectivity in cell death in CA1 after OGD (Laake et al., 1999; Newell et al., 1990; Pringle et al., 1997; Strasser and Fischer, 1995). For example, cell damage in the CA1 region was twice that seen in the CA3 region of hippocampal organotypic tissue cultures after 30-minute OGD (Strasser and Fischer, 1995). The development of cell death in cultures is generally faster than in vivo. A significant elevation of lactate dehydrogenase in the medium 2 hours after OGD, which was maximal after 12 to 16 hours, was found in neocortical cell cultures (Goldberg and Choi, 1993). In rat hippocampal organotypic tissue cultures, PI fluorescence increased in the CA1 area 6 hours after 60 minutes of OGD and continued to increase for up to 24 hours of recovery (Pringle et al., 1997).
Our initial experiments were performed with aCSF as OGD medium. We found that cell death developed rapidly and that there was no clear selective CA1 damage. When the cultures were exposed to OGD for 15 minutes, cell death was seen in CA3 cells and dying cells were seen in CA1 after 4 hours of recovery. We recognized that an important difference between the in vivo and in vitro experiments is the composition of the extracellular fluid (Silver and Erecinska, 1992). In most models of in vitro ischemia, glucose-free aCSF or balanced salt solutions are used as the medium during OGD. Because the size of the extracellular space in a culture (i.e., the volume of the medium) is several magnitudes larger than that in vivo, the redistribution of ions during OGD due to membrane depolarisation will have a negligible effect on the extracellular ion concentrations. The intracellular ion concentrations during OGD will therefore be highly dependent on the ion content of the medium. The ion composition of aCSF is similar to the CSF of normal brains and markedly different from the composition of the extracellular fluid in the brain during an ischemic insult. Thus, to simulate the conditions during in vivo ischemia more closely, we examined the effect of changing the concentrations of calcium, potassium, and hydrogen ions.
The loss of ATP during ischemia results in a rundown of transmembrane ion gradients, leading to a rapid membrane depolarization (occurring within 1 or 2 minutes after onset of ischemia) due to the reversal of the sodium-dependent ion transporters and the opening of voltage-and agonist-dependent ion channels. Using potassium-selective electrodes, the potassium concentration in the extracellular space was found to increase to approximately 70 mmol/L during ischemia (Hansen, 1985; Harris et al., 1981). When our cultures were subjected to glucose-free aCSF with a potassium concentration of 70 mmol/L, a steep membrane depolarization occurred, similar to that seen in vivo (data not shown). Still, under normoxic conditions incubation in 70 mmol/L potassium did not cause damage to the cultures (data not shown). Cell damage in cultures exposed to 15-minute OGD in aCSF with 70 mmol/L potassium was similar as that in cultures exposed to OGD in aCSF (2.5 mmol/L potassium). Obviously, high potassium levels during OGD did not aggravate cell damage.
Increased intracellular calcium ion levels play an important role in ischemic neuronal injury (Kristian and Siesjö, 1998). When the cell is depolarized and the calcium extrusion and sequestration mechanisms fail, the intracellular calcium concentration rises (Silver and Erecinska, 1992). Using calcium selective electrodes, the extracellular calcium concentration was found to fall to 0.1 mmol/L (Benveniste et al., 1988) or 0.2 or 0.3 mmol/L (Harris et al., 1981) during complete global ischemia. When we performed OGD in aCSF with 0.3 mmol/L calcium, damage was significantly reduced compared with that seen with aCSF only (2 mmol/L calcium). Also, CA3 cells were not damaged. This neuroprotective effect of decreased levels of calcium during OGD has previously been shown in cell cultures (Rothman, 1983) and in hippocampal organotypic tissue cultures (Newell et al., 1995).
During ischemia, the anaerobic glycolysis leads to lactic acid production and to a decrease in extracellular pH from 6.8 to 6.2. The magnitude of the drop in pH depends on the preischemic plasma levels of glucose (Li et al., 1995). Although acidosis and hyperglycemia increase damage in vivo, in all in vitro systems reported so far, lowering the medium pH to 6.8 during OGD significantly reduces rather than increases damage (Giffard et al., 1990). This protective effect has been attributed to the fact that low pH inhibits NMDA receptors and thus protects against glutamate toxicity (Giffard et al., 1990; Tang et al., 1990). By lowering the pH of the incubation medium to 6.8, damage was significantly reduced in the CA1 and in the CA3 regions in our system. Interestingly, the area displaying damage within the CA1 region was also reduced.
It was evident that, by changing the ion composition of the medium to that of the extracellular fluid in the brain during ischemia, the extent and development of cell death markedly changed. We therefore defined a medium, iCSF, that consisted of 70 mmol/L potassium, 0.3 mmol/L calcium, a pH of 6.8, and 77 mmol/L sodium ions, based on available data in the literature (Hansen, 1985; Hansen and Nedergaard, 1988; Harris et al., 1981; Siesjo 1992; Silver and Erecinska, 1992). In this medium, 15-minute OGD caused selective CA1 damage with no damage to CA3 cells. Also, damage was delayed compared with when OGD was induced in aCSF and appeared at 8 hours of recovery. In the mouse brain, damage to CA1 pyramidal cells after 10-minute global ischemia is induced at 24 hours of recovery (M.-L. Smith, Ph.D., personal communication, 2002); that is, much earlier than in rat hippocampus. Furthermore, the 15-minute duration of OGD used to induce selective neuronal damage is much shorter than that reported earlier, and is comparable to the time required to induce damage in vivo.
Glutamate receptor blockade of both NMDA (using MK-801 and AP5) and non-NMDA (using NBQX) subtypes have repeatedly been shown to protect neurons in different ischemia models, both in in vivo (focal ischemia: Park et al., 1988) and in in vitro models using dissociated neuronal cultures (Goldberg and Choi, 1993; Kaku et al., 1993), as well as in organotypic hippocampal cultures (Breder et al., 2000; Newell et al., 1995; Pringle et al., 1997; Vornov et al., 1994). Also, in a mouse model of global ischemia, MK-801 protected CA1 cells when given after the ischemic insult (M.-L. Smith, Ph.D., personal communication, 2002). Here we show that damage after 15 minutes of OGD in iCSF is mitigated by NMDA and AMPA receptor blockade, demonstrating that glutamate receptor activation contributes to the cell death process.
An important pathophysiologic feature of ischemic brain damage is the aggravation of cell death by hyperglycemia. In rat models of global ischemia, hyperglycemia—when established before the insult—worsened the injury (Myers and Yamaguchi, 1977), possibly through an acidosis-related mechanism (Siesjö, 1985). This aggravating effect of hyperglycemia during ischemia in vivo has so far not been reproduced in vitro. On the contrary, murine neocortical cell cultures survive 8 hours of anoxia in the presence of 20 mmol/L glucose, but develop extensive damage when exposed to 30-minute OGD (Goldberg and Choi, 1993). Also, in hippocampal organotypic rat cultures, 60 minutes of OGD led to significant neuronal damage that was abolished when glucose was included in the medium (Pringle et al., 1997). In concordance with these results we found that the presence of 40 mmol/L glucose during anoxia in aCSF completely prevented cell death. Conversely, glucose significantly exacerbated neuronal death when present during anoxia in iCSF. This is, to our knowledge, the first demonstration that glucose may worsen hypoxic cell death in vitro. Clearly this model is useful for studies of the pathophysiologic mechanisms behind the aggravating effect of hyperglycemia during ischemia, which will be the subject of further investigations.
In summary, we have established a mouse hippocampal organotypic tissue culture model of OGD-induced cell death that may be useful in the analysis of the mechanisms of cell death after brain ischemia and for the screening of antiischemic drugs. By adopting the in vivo intraischemic extracellular concentrations of calcium, potassium, sodium, and hydrogen ions to the OGD medium, the model shows conspicuous similarities with the temporal and special development of cell death in vivo: selective and delayed CA1 damage, a damage mitigated by blockade of the NMDA and AMPA receptors, and a striking augmentation of damage by high levels of glucose.
Cell death induced by chemical anoxia and glucose deprivation has previously been studied in rat organotypic hippocampal tissue cultures. The present model is the first using OGD on mouse tissue cultures, and will therefore be useful in studies on hippocampi from genetically modified mice.
Footnotes
Acknowledgements
The authors thank Luc Stoppini and Stefan Seth for valuable discussions, suggestions, and expert technical help, and Blanka Boberg and Ulrika Sparrhult-Björk for excellent technical assistance.