
Abstract
In in vivo positron emission tomography (PET) studies, dopamine that is released secondary to amphetamine administration appears unable to achieve a receptor occupancy that is significantly higher than 50% (“ceiling effect”). Also with exogenous agonists no studies have reported a higher than 50% occupancy. To investigate the feasibility of exceeding 50% occupancy in vivo with a dopamine receptor agonist we administered D2/D3 agonist (+)−PD 128907 over an extensive dose range. Two anesthetised Macaca mulatta males were used in a bolus-infusion protocol for [11C]raclopride. (+)- PD 128907 was administered as an intravenous challenge during separate PET scans in a dose range of 10 to 10000 nmol/kg. Occupancy by (+)−PD 128907 was estimated by comparing the binding before and after challenge. In a striatal region of interest, receptor occupancy by (+)−PD 128907 increased in an orderly dose-dependent manner to a maximum of at least 85%. This is the first indication that virtually all dopamine D2/D3 receptors in the striatum are in principle accessible to agonist binding. In the case of dopamine a number of protective mechanisms may be responsible for the ceiling effect.
The PET tracer [11C]raclopride is a benzamide antagonist for dopamine D2 and D3 receptors. Like other benzamide and catecholamine tracers, [11C]raclopride binding is sensitive to occupancy by other receptor ligands in a way that is consistent with the occupancy model i.e. binding is reduced when competing ligands such as dopamine are present (Laruelle 2000). For this reason [11C]raclopride is often applied to study dynamic changes in brain dopamine concentrations. Many studies have used dopaminergic manipulations that increase dopamine concentration. The amphetamines d-amphetamine, d-methamphetamine and d-methylphenidate cause an increase in dopamine concentration in the brain and a reduction in [11C]raclopride binding (Dewey et al., 1993; Volkow et al., 1994; Tsukada et al., 1999).
The ceiling effect entails that even high dosages of amphetamines do not reduce [11C]raclopride binding by more than 50% (Laruelle 2000). Laruelle has suggested three possible contributing phenomena for this observation: i) 50% of receptors being extrasynaptic, ii) the low efficiency of dopamine, being an agonist, in occupying low affinity state receptors and iii) basal occupancy by dopamine (Laruelle 2000).
In vitro, the ceiling effect does not exist as such. E.g. the dopamine D2/D3 receptor agonist (+)−PD 128907 completely displaces the dopamine antagonist [3H]spiperone from dopamine D2 and D3 receptors on membrane fractions, despite the presence of the nonhydrolysable GTP analogue GppNHp in the medium, which favors the low affinity state. In this case the displacement curve is ‘shallow’ with an apparent Hill coefficient of less than unity (Pugsley et al., 1995).
In vivo binding behavior of exogenous agonists could indicate whether something fundamental is limiting agonist binding to 50% and clarify the relative contribution of phenomena i) and ii). If phenomenon i) plays a key role in the ceiling effect, the ceiling effect should not apply to exogenous dopamine receptor agonists since they are expected to reach similar intra- and extrasynaptic concentrations. Binding is expected to be complete and without evidence for multiple sites. If phenomenon ii) was to play the main role in the ceiling effect, exogenous dopamine agonists are also expected to reach 100% occupancy, but with a “shallow” or two-step association curve.
Experiments with exogenous agonists have not found occupancy of dopamine receptors in excess of 50% as measured with benzamide radiotracers for PET or SPECT. E.g. the binding of [123I]-iodobenzamide was reduced by ≈5% in parkinsonian patients treated with dopamine agonists pramipexole or lisuride (Schwarz et al., 1996) and by 45% in parkinsonian monkeys treated with the dopamine agonist LY 171555 (Vermeulen et al., 1994).
However, since these studies have not used extensive dose ranges, it is unclear what type of association behavior occurs between dopamine agonists and dopamine receptors in vivo. To obtain this information we administered escalating dosages of D2/D3 agonist (+)−PD 128907 (Dijkstra et al., 1988) while measuring free D2/D3 receptors with [11C]raclopride and PET.
METHODS
Subjects
Two adult male rhesus monkeys (5.5 to 8 kg) were used. (±)-Ketamine was administered for handling, transportation and anesthetic induction. Two intravenous cannulae were inserted, one for administration of tracer and (+)−PD 128907, the other for blood sampling. Atropine (0.06 to 0.09 mg/kg intramuscular) and sodium pentobarbital (8 to 11 mg/kg intravenous) were administered, the trachea was intubated for ventilation with isoflurane (1 to 2% (v/v)) in a carrier gas mixture of O2:N2O 50:50. Heart rate, electrocardiogram, respiration rate and O2 saturation were monitored throughout the experiment.
Drugs and tracer administration
The hydrochloride salt of (+)−PD 128907 was synthesized as described before (Dijkstra et al., 1988). Unlabeled raclopride and domperidone were obtained from RBI (Natick, U.S.A.).
The tracer [11C]raclopride was synthesized by [11C] methylation of the hydroxy-precursor (Farde et al., 1988). A total of 63 to 203 MBq of activity was dissolved in 20 mL sterile 0.9% saline, and taken to the subject in the PET scanner.
At t = 0 minutes, over the first 60 seconds of the acquisition, a tracer volume corresponding to 83 minutes of infusion (60 for the studies at 0 nmol/kg) was injected. Immediately after completion of the bolus the infusion was started.
At t = 15 minutes, domperidone (200 μg/kg, intravenous) was administered to block peripheral dopamine D2/D3 receptors. This treatment was included after noting cardiovascular effects of (+)−PD 128907 at the highest dose. The studies at 0 and 10000 nmol/kg were performed without domperidone pretreatment.
At t = 45 minutes, an intravenous challenge of (+)−PD 128907 was injected over 60 seconds. Monkey 1 received 0 (three times) and 10000 nmol/kg, monkey 2 received 1.0; 3.2; 10; 31.6; 100; 316; 562; 1000 and 3162 nmol/kg. The dose of 31.6 was repeated, since on the first occasion the binding of [11C]raclopride was still rising at the time of challenge. All studies were included in the displacement curve.
Scanning procedures
The monkey was positioned in a stereotaxic frame (all studies except the very first one at 0 nmol/kg) in an ECAT HR+ PET scanner (Siemens, Erlangen, Germany). The field of view included the entire brain and part of the thorax.
A 20-minute transmission scan was performed for attenuation correction. There was always a minimum of 13 days between two consecutive scans in the same monkey.
After the experiments at 0 and 10000 nmol/kg which used 6 × 2.5, 6 × 5, 6 × 2.5 and 6times5 minutes frame durations, scanning duration and temporal resolution were increased to 48 × 2.5 minutes. In the cases where frame duration was 5 minutes the time activity curves and binding ratio curves were interpolated. Reconstruction was performed to a 128 × 128 × 63 matrix with a plane separation of 0.2425 cm and a bin size of 0.2250 cm.
Data analysis
A region of interest analysis was performed with Clinical Applications Programming Package 5 (Siemens, Erlangen, Germany) for the striatum and the cerebellum. Binding ratio was defined as (striatal activity - cerebellar activity) / cerebellar activity. Under equilibrium conditions the binding ratio equals the binding potential.
The binding ratio in absence of (+)−PD 128907 was calculated for the striatal ROIs by averaging the binding ratio over nine frames before the 45 minute point.
For every PET experiment, the minimum in the binding ratio due to displacement was located by a second degree polynomial fit on the curve after (+)−PD 128907 administration. This was expressed relative to plateau and plotted against dose of (+)−PD 128907. The resulting curve was fitted with a logistic 4 parameter fit by SigmaPlot (v7.0, SPSS Inc., Chicago, U.S.A.). The parameters were: Bmin, Bmax, ED50 and nH. The parameter nH is the Hill coefficient which, if significantly different from unity, indicates a deviation from single site, non-cooperative binding behavior. Its absolute value cannot exceed the number of binding sites for a ligand (Monod et al., 1965).
RESULTS
Over the separate experiments, increasing dosages of (+)−PD 128907 were injected to occupy dopamine D2/D3 receptors as measured with infused [11C]raclopride.
Figure 1 shows the curves for binding ratio for [11C]raclopride in time. At the three highest dosages of (+)−PD 128907, there was an evident reduction in binding ratio, that was larger when the (+)−PD 128907 dose was larger. After 1000 nmol/kg there was partial recovery towards the baseline binding ratio, but at the highest two dosages this did not occur. At the highest dose of (+)−PD 128907 the binding ratio was reduced almost to zero.
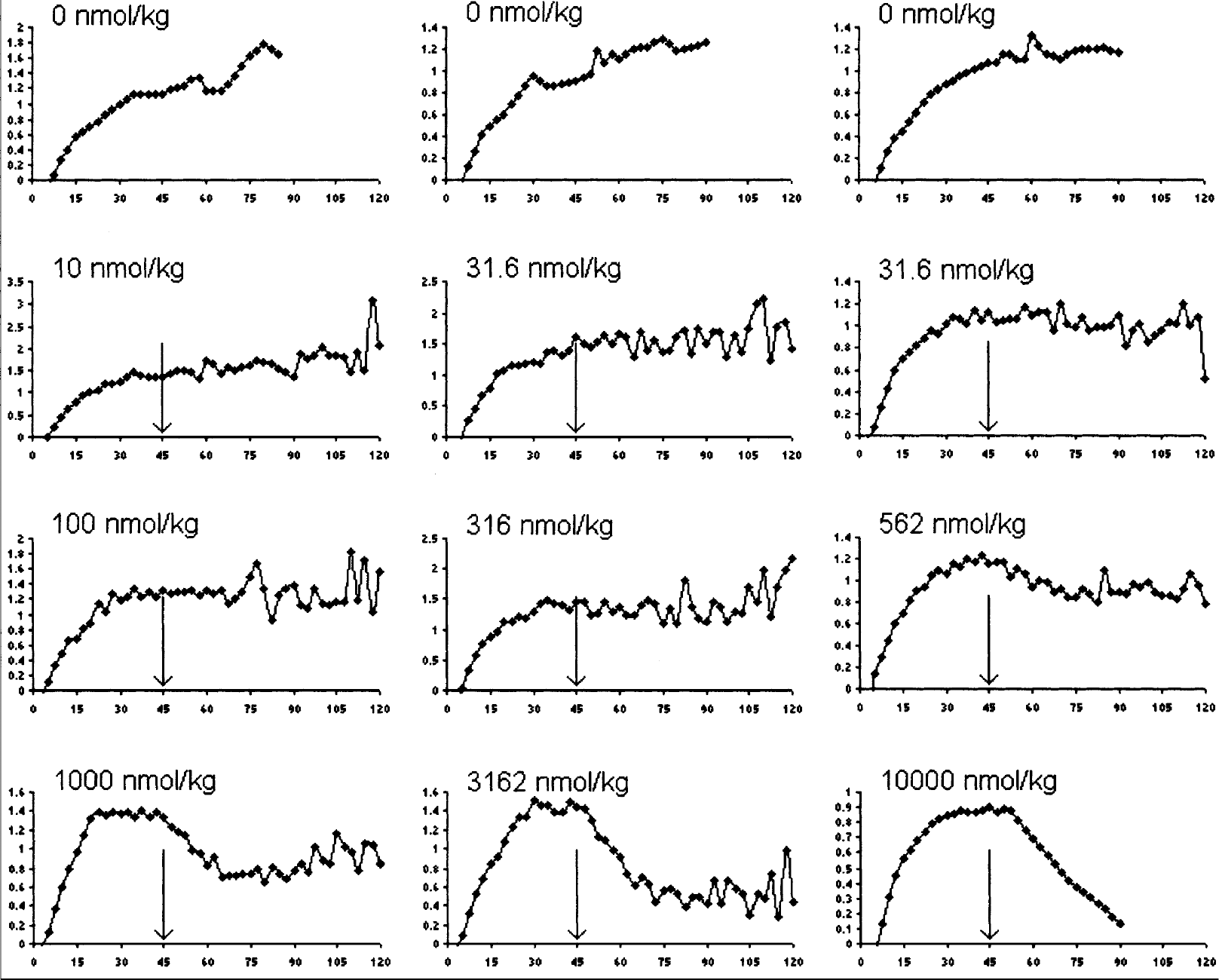
Binding ratio in time (min) for different dosages of (+)−PD 128907.
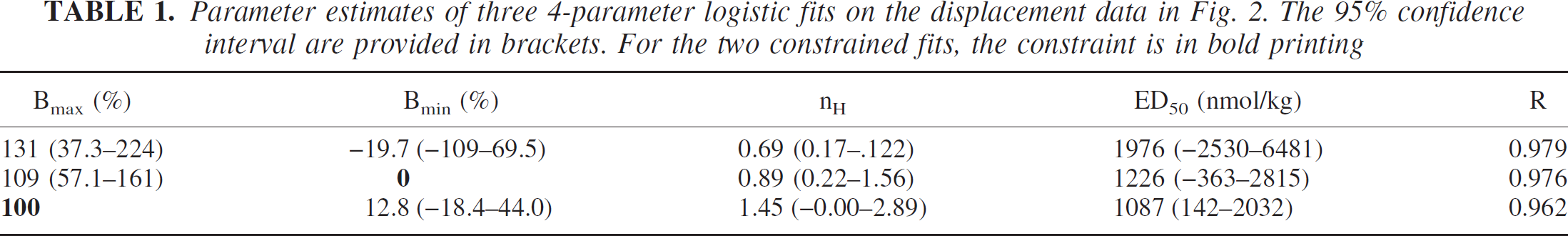
The fitted minimum values of the curves in Fig. 1 are plotted against dose of (+)−PD 128907 in Fig. 2. This is thus the minimum relative binding ratio due to (+)−PD 128907 injection against the dose. There clearly is an orderly dose dependent decrease in minimum binding ratio for [11C]raclopride. When the data were fitted with three different 4-parameter logistic fits (unconstrained, constrained to Bmin = 0%, constrained to Bmax = 100%), all three fits described the data well. Judging by the correlation coefficient R, none of the fits was superior to the others. Table 1 provides the parameter estimates from the three fits. The data did not support rejection of the hypotheses Bmax = 100%, Bmin = 0% and nH = 1.000.
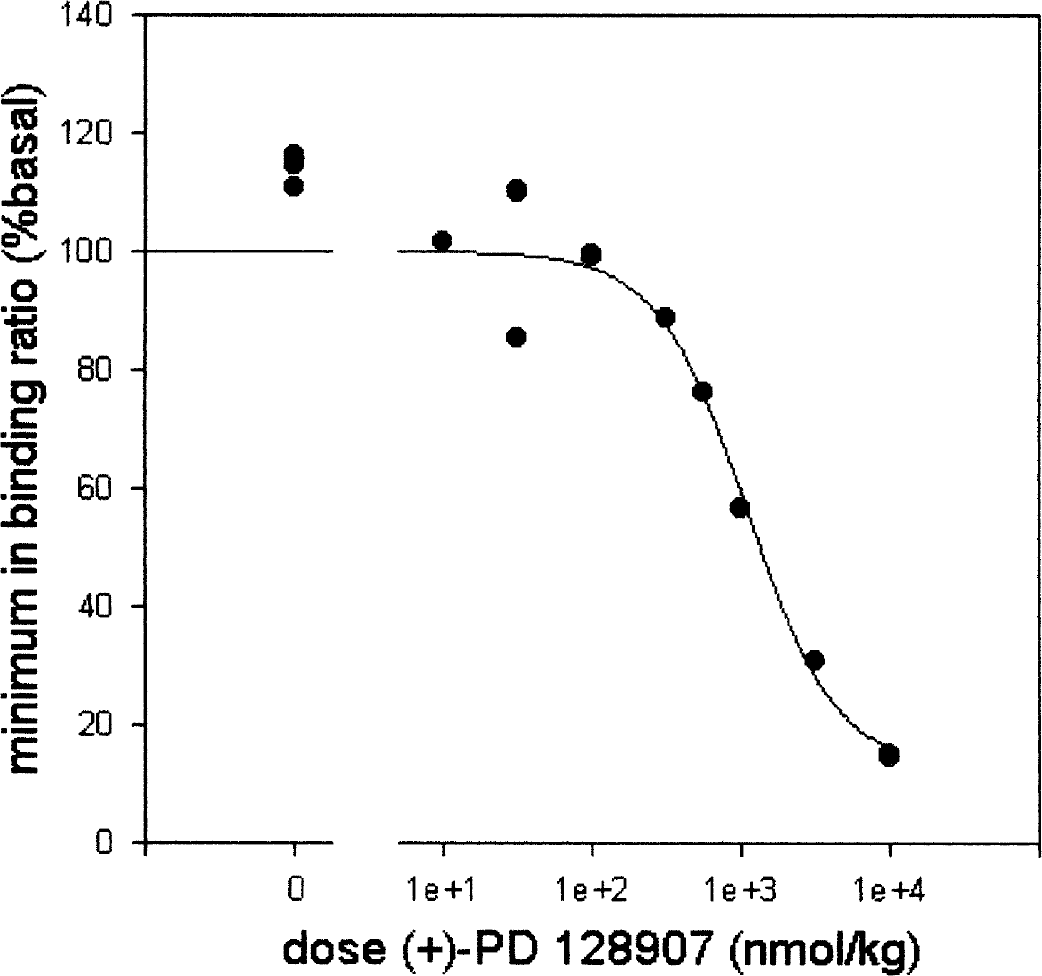
Displacement of [11C]raclopride by (+)−PD 128907. The minimum relative binding ratio for [11C]raclopride is plotted against dose of (+)−PD 128907. The line represents a 4-parameter logistic fit constrained to a Bmax of 100%.
Parameter estimates of three 4-parameter logistic fits on the displacement data in Fig. 2. The 95% confidence interval are provided in brackets. For the two constrained fits, the constraint is in bold printing
DISCUSSION
The highest dose of the dopamine agonist (+)−PD 128907 reduced specific binding of [11C]raclopride in the striatum by at least 85%. The shape of the binding ratio curve after 10000 nmol/kg (+)−PD 128907 (Fig. 1 bottom right) suggests that measuring for longer would show an even larger decrease. This is a much larger effect than that reported with dopamine releasers, so that the “ceiling effect” of 50% does not apply to (+)−PD 128907. Provided that (+)−PD 128907 acts in vivo as an agonist and [11C]raclopride as an antagonist at dopamine D2/D3 receptors, these data prove that no fundamental mechanism is restricting agonist binding to ≤ 50%.
The sigmoidal curve in Fig. 2 did not display overt two-step behavior, nor was the apparent Hill slope (nH) significantly different from unity. This means that the present data (from twelve PET scans) do not have sufficient statistical power to prove heterogeneity of the receptor population that [11C]raclopride and (+)−PD 128907 bind to. Hence there is no evidence in favor of a multiple sites model representing e.g. high and low affinity receptors or the D2 and D3 subtype (both [11C]raclopride and (+)−PD 128907 have relatively high affinity (Kd≈1nM) for dopamine D3 receptors).
Since the ceiling effect, as seen with dopamine release secondary to amphetamine, does not apply to (+)−PD 128907, it could be caused by extrasynaptic dopamine levels not rising sufficiently to occupy extrasynaptic receptors which do remain available for antagonist tracer binding. These dopamine levels are limited by reuptake, degradation and the available intracellular dopamine pool, and it appears likely that the brain has efficient mechanisms to prevent maximal agonistic stimulation. Since (+)−PD 128907, like [11C]raclopride but unlike dopamine, is expected to have full access to extrasynaptic receptors the former two compounds compete for the same receptors. Another possibility could be that amphetamine is in fact able to cause 100% occupancy by dopamine of D2/D3 receptors, but does so only at a toxic dose level. If one constructs a displacement curve for published primate studies with the amphetamines it becomes clear that a ≈50% reduction in [11C]raclopride binding occurs over a dose range of about one order of magnitude. This is not inconsistent with a maximal effect size of 100%, although not enough is known about this relationship to make a reliable extrapolation.
In the present study the mean basal binding ratio was 1.1, which is lower than in similar studies reported in the literature, e.g. (Ito et al., 1998; Tsukada et al., 2002). This may be partly due to the partial volume effect and a relatively large size of the regions of interest. Anesthesia may also play a role since this is known to lower the binding of [11C]raclopride (Tsukada et al., 2002). In particular ketamine, which our animals received twice, could be the cause of this since it has been found to reduce [11C]raclopride binding (Vollenweider et al., 2000). However, none of these issues influence the main finding that the agonist (+)−PD 128907 was able to occupy a very large portion of the receptor ensemble.
After 1000 nmol/kg of (+)−PD 128907 the time activity curve and binding ratio curve (Fig. 1) recovered towards basal, presumably as the compound was cleared from the blood and brain. This recovery was much faster than that seen after amphetamine administration, where tracer binding remains reduced for hours (Laruelle et al., 1997; Laruelle 2000). In the case of amphetamine, agonist induced internalization of dopamine receptors (Laruelle 2000) together with an inability of raclopride to reach these internalized receptors (Sun et al., 2003) has been postulated to explain the lasting reduction.
In summary, the ceiling effect of 50% dopamine receptor occupancy does not apply to the exogenous agonist (+)−PD 128907. Thus there is no reason to assume that dopamine can not in principle bind to low affinity D2/D3 receptor sites. The ceiling effect as noted with amphetamines could be caused by (extracellular) dopamine concentrations being limited by available dopamine pools, uptake and degradation mechanisms and toxicity issues.
Footnotes
Acknowledgments
The authors are grateful to M. Faassen, T. Peters, A. Hanssen, T. Wegman and the medical nuclear workers for able technical support and Dr. W. Timmerman for valuable discussions.