
Abstract
The ischemic penumbra has been documented in the laboratory animal as severely hypoperfused, nonfunctional, but still viable brain tissue surrounding the irreversibly damaged ischemic core. Saving the penumbra is the main target of acute stroke therapy, and is the theoretical basis behind the reperfusion concept. In experimental focal ischemia, early reperfusion has been reported to both prevent infarct growth and aggravate edema formation and hemorrhage, depending on the severity and duration of prior ischemia and the efficiency of reperfusion, whereas neuronal damage with or without enlarged infarction also may result from reperfusion (so-called reperfusion injury). Activated neutrophils contribute to vascular reperfusion damage, yet posthypoxic cellular injury occurs in the absence of inflammatory species. Protein synthesis inhibition occurs in neurons during reperfusion after ischemia, underlying the role that these pathways play in prosurvival and proapoptotic processes that may be differentially expressed in vulnerable and resistant regions of the reperfused brain tissue. Ischemia-induced decreases in the mitochondrial capacity for respiratory activity probably contribute to the ongoing impairment of energy metabolism during reperfusion and possibly also the magnitude of changes seen during ischemia. From these experimental data, the concept of single-drug intervention cannot be effective. Further experimental research is needed, especially of the study of biochemical markers of the injury process to establish the role of several drugs.
Keywords
An acute occlusion of cerebral arteries, if not reversed within a defined short period, usually results in cerebral ischemia with subsequent cell death within the perfusion territory of the affected vessels. The infarction volume is an important determinant of long-term neurologic outcome and, thus, there has been a considerable interest in developing new therapeutic options to potentially ameliorate this brain tissue damage. In the treatment of acute cerebral ischemia, two primary therapeutic strategies have to be applied: (1) limitation of cerebral ischemia by early reperfusion after cerebral ischemia (the vascular approach), and (2) interference with the pathobio-chemical cascade leading to ischemic damage (the cellular approach) (Heiss et al., 1999). To reduce potential neurologic morbidity, there has to be a sufficient understanding of the pathophysiologic mechanisms involved in ischemic damage and repair to design clinically effective therapeutical options. A necessary prerequisite of every therapeutic strategy is the existence of functionally impaired but viable and potentially salvageable brain tissue (i.e., a penumbra). The potential time window for the various treatment options to be efficacious is likely to be completely different: of rather short duration for effective approaches, of longer duration for neuroprotection, and particularly prolonged duration for antioxidant and antiapoptotic approaches. However, delayed reperfusion can precipitate a progressive destruction of reversibly damaged neuronal cells, leading to paradoxical tissue dysfunction or cellular necrosis. Although early reperfusion is accepted as a standard therapeutic option, it remains a controversial topic when linked to different experimental paradigms (e.g., functional outcome, lesion size, markers of inflammation, apoptosis). Improved understanding of the early course of focal ischemia has helped to close the gap between the molecular and biochemical concept of ischemic damage and the potential usefulness of early reperfusion.
The authors review the current literature on the pathophysiologic mechanisms of reperfusion after cerebral ischemia. The pathophysiology due to cerebral ischemia per se is only addressed when relevant to reperfusion in the cellular or molecular context.
MAJOR PATHOPHYSIOLOGIC PHENOMENA
Within a certain period, the reversal of an acute disruption in blood supply to a specific cerebrovascular territory, either spontaneously or as a result of thrombolytic treatment, can markedly reduce the subsequent infarction volume (Fig. 1). There exist different experimental data for the extent of ischemic damage that is observed when reperfusion is initiated during the first few hours of focal ischemia (Heiss et al., 1997) (Table 1). Nonetheless, in rodent models, the initiation of reperfusion within the first 1 to 2 hours after focal ischemia generally results in smaller infarct volume when assessed several hours or days later, although ischemic damage in selective vulnerable neuronal populations can subsequently develop many days later in affected regions of brain tissue. Periods of focal ischemia exceeding 1.5 to 2 hours in different animal models lead to similar cellular damage as produced by permanent focal cerebral ischemia (Heiss et al., 2001). As a consequence, the theory of a “therapeutic window”—the duration of time during which effective CBF restoration and intervention to prevent the consequences of biochemical alterations can be successful in ensuring the viability of brain tissue—has been postulated (Schaller and Graf, 2002). Interestingly, reperfusion after longer periods of focal ischemia (3 to 6 hours) has also been reported—in only few experimental studies—to reduce infarct size compared with permanent ischemia (Astrup et al., 1977). These findings underline the possibility that the time window for therapeutic reversal of vascular occlusion in humans may also be longer than currently predicted (Schaller et al., 2003). However, the effects of some pharmacologic therapeutic interventions, which have been primarily examined in focal cerebral ischemia, indicate that irreversible cellular damage is not determined during the ischemic period, but can develop during reperfusion, especially during delayed reperfusion. Thus, therapeutic interventions initiated up to several hours after 2 to 3 hours of induced focal ischemia may be able to substantially reduce infarction volume, not only in the perifocal regions, but also in brain tissue that has formed parts of the severely disturbed ischemic core (Arai et al., 1986). However, reperfusion can result in ischemic damage in excess of that which might transpire if reperfusion had not occurred.
Alterations in energy-related metabolites in brain tissue in response to short-term global ischemia or focal ischemia∗

Zero (0) indicates no significant difference compared with nonischemic tissue; (−), small but significant decrease (to >70% of nonischemic tissue); (−), moderate reduction (30–70%); (—), severe decrease (to 30% of nonischemic tissue); (+), moderate increase (to less than four times nonischemic tissue); (++), large increase (to more than four times nonischemic tissue). The use of combined symbols reflects differences between studies.
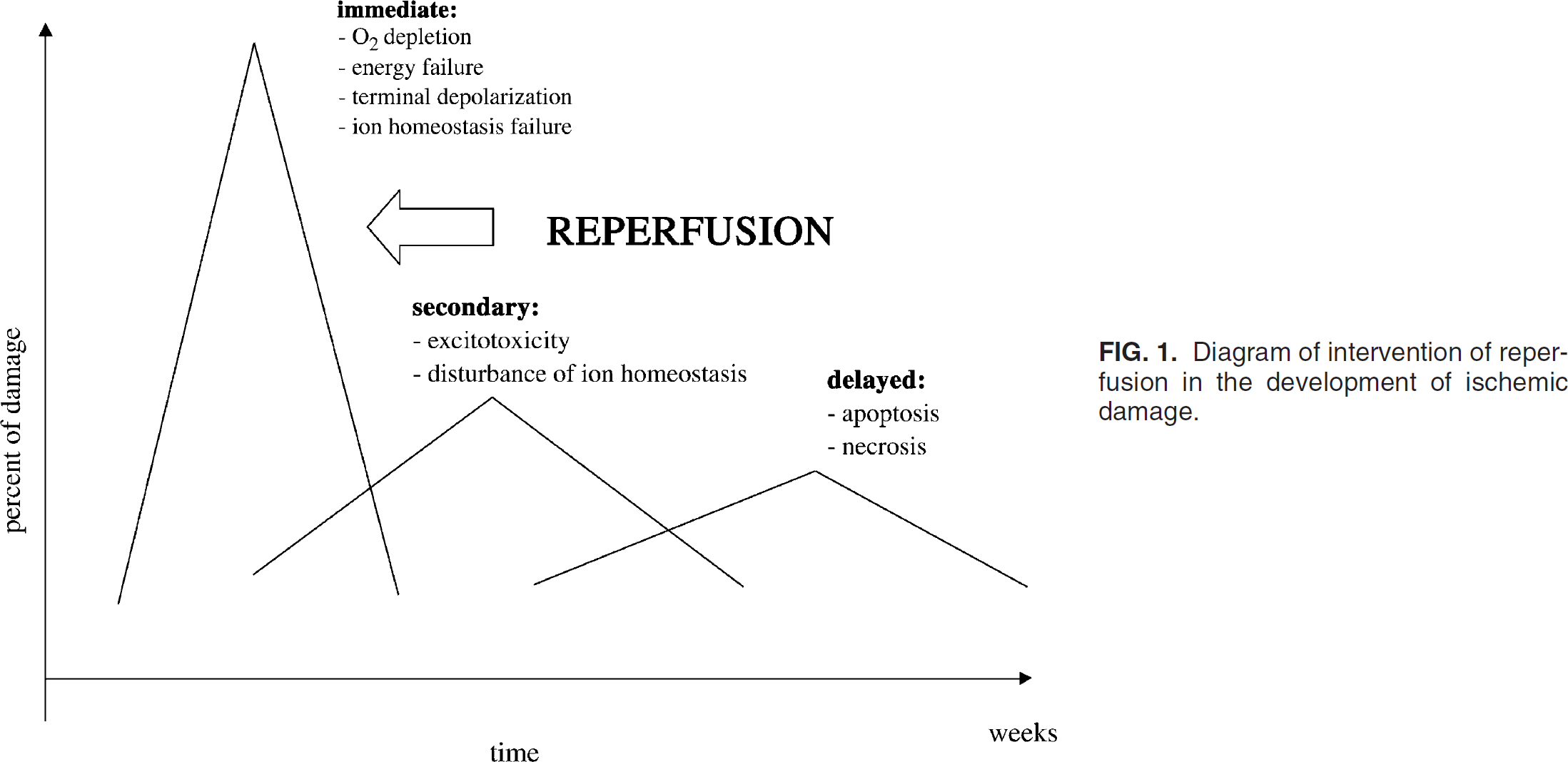
Diagram of intervention of reperfusion in the development of ischemic damage.
Resistance to infarction
Reversible middle cerebral artery occlusion (MCAO) in animal models and the observations in reperfused stroke patients suggest that the severity of cerebral ischemia—measurable as residual CBF rate—may be a principal determinant of the subsequent course and extent of ischemic damage. If reperfusion is accomplished in oligemic tissue with a flow rate above 12 mL · 100 g− 1 · min− 1, the threshold for morphologic integrity, subsequent ischemic damage may be minimal. If reperfusion is induced in already irreversibly damaged areas bordering the infarct core, the increased vascular permeability during reperfusion leads to brain edema formation and parenchymal hemorrhage. In addition, it may enhance ischemic damage by inducing different pathophysiologic mechanisms (e.g., massive increase in the release of excitatory amino acids, production of reactive oxygen species [ROS], excessive Ca2+ influx into cells). Experimental findings stress the importance of a sufficient residual CBF level for the further course of reperfusion. This hypothesis is supported by experimental facts showing that early reperfusion preserves penumbral brain tissue whereas late reperfusion increases ischemic damage, and that good collateral CBF is essential for spontaneous recovery as well as for the beneficial effects of thrombolytic therapy (Astrup et al., 1977). Resistance to cerebral ischemia can also be affected by various inherent or external factors: hypertensive animals are less resistant than normotensive animals (Jacewicz et al., 1992), and resistance is dependent on the number of ischemic episodes and body temperature (Schaller and Graf, 2002, 2003;Schaller et al., 2003). It must also be kept in mind that delayed neuronal death after reperfusion may result from selective vulnerability of neurons.
Cellular aspects of reperfusion
The recovery of energy metabolism during the initial 20 minutes of reperfusion (after 10-minute forebrain ischemia) is accompanied by a restoration of the distribution of ions to near their preischemic state. Although Ca2+ changes are largely reversed in the initial reperfusion period, complete recovery may require 1 to 2 hours. When reperfusion was extended to 4 hours after 2 hours of transient focal cerebral ischemia, a deterioration of all energy metabolites (decreased ATP, phosphocreatine, acetylated energy charge and increased lactate) was seen within the previously ischemic core (Folbergrova et al., 1995), presumably associated with the progression towards irreversible cell dysfunction and death. In perifocal tissue, a comparable pattern of partial recovery during initial reperfusion with subsequent delayed deterioration was also shown, but this was again less pronounced than in the previously ischemic core. At 2 hours of reperfusion, phosphocreatine and the adenylate energy charge were essentially fully recovered and ATP amounted to 70% of nonischemic values. A moderate deterioration in these metabolites was again observed with prolonged reperfusion. However, focal ischemia data have identified ATP changes in focal and perifocal brain tissue at both 1 hour (Welsh et al., 1991) and 4 hours of reperfusion (Selman et al., 1990) that are consistent with the major findings described earlier. In one of these studies (Welsh et al., 1991), there was substantial reversal of [lactate] during reperfusion, suggesting a lesser reliance on glycolysis for ATP generation than that seen by Folbergrova et al. (1995). This could reflect differences in the nature and severity of cerebral ischemia between the animal models used, although both produced large middle cerebral artery (MCA) infarction. Decreases in glucose use in both focal and perifocal brain tissue regions have been observed during the early phase of reperfusion after 2 hours of focal ischemia (Belayev et al., 1997). Thus, increased glycolytic activity was also apparently not a major contributor to postischemic ATP production. Overall, these findings provide clear evidence for marked imbalances between energy use and production during focal ischemia as well as during reperfusion that are sufficient to initiate extensive ischemic damage.
Hyperglycemia and reperfusion
The process by which hyperglycemia worsens morphologic outcome is uncertain. In animal models, preexisting short-term hyperglycemia that was not reversed after more extended periods of MCAO (60 to 90 minutes) also significantly increased infarction volume relative to nonhyperglycemic animals (Gisselsson et al., 1999). In more severe or prolonged reperfusion periods (60 to 240 minutes), CBF during reperfusion was seriously impaired, and this significantly correlated with increased infarct volume (Palmon et al., 1995). Acute hyperglycemia achieved by injection of 50% D-50 glucose solution also dramatically increased the risk of hemorrhagic transformation while reducing CBF during reperfusion (Kawai et al., 1997). These findings suggest that a vascular mechanism may be a critical component for worsened outcome under hyperglycemic conditions, whereas metabolic impairment may be a more salient factor. Potential mechanisms include hyperglycemia-induced damage to endothelium, increased expression of adhesion molecules, and/or glycosylation of critical proteins that generate vasodilating or antithrombotic substances such as nitric oxide (NO) (Amore et al., 1997). The role of NO resulting from neuronal constitutive (nNOS) or inducible (iNOS) NO synthase in mediating the deleterious effects of hyperglycemia on cerebral ischemia is confirmed by the observation that pretreatment with the nonselective NO synthase inhibitor, N-nitro-L-arginine methyl ester, provides neuroprotection in reversible MCAO under hyperglycemic conditions and prevents the vascular compromise. In addition, hyperglycemia affects recovery of metabolic function after cerebral ischemia. Several potential cellular factors have been associated with worsened outcome: (1) deteriorating acidosis, which directly injures brain tissue; (2) prolongation of postischemic alkalosis, which interferes with mitochondrial function; (3) delayed postischemic hyperthermia, which promotes deleterious enzymatic reactions such as the formation of peroxynitrate; and (4) promotion of hydroxyl (OH) radical formation. A common end-point in hyperglycemic cellular changes is ROS production. The formation of advanced glycation end products (AGE) (as a function of elevated nonenzymatic glycation of proteins, lipids, and nucleic acids) is accompanied by oxidative, radical-generating reactions and thus represents a major source of ROS under chronic hyperglycemic conditions. AGE have been proposed as a potential link between the harmful effects of hyperglycemia and inflammation. AGE are formed by nonenzymatic glycosylation of proteins and protein precursors. Increased AGE can appear intracellularly after episodes of hyperglycemia lasting only several hours in endothelial cell cultures. The formation of AGE is accompanied by the generation of ROS (Schmidt et al., 1994) that may contribute to their damaging effects. Additional mechanism of injury due to AGE includes the action of receptors for AGE (RAGE) that are found on many different tissues (Brett et al., 1993), such as the CNS as well as in vascular and arteriolar endothelial cells (Weber et al., 1998). AGE—RAGE interaction is linked to nuclear factor κB (NF-κB) expression, apparently through ras but not rac or Cdc42 (Huttunen et al., 1999), and greatly increases microvascular injury (Selman et al., 1990). Administration of AGE-linked albumin before 30 minutes of focal ischemia followed by partial reperfusion worsened infarct volume (Zimmermann et al., 1995), but AGE-linked albumin administered systemically did not reduce CBF, actions resembling those seen in the briefer ischemia/reperfusion hyperglycemic model.
In addition, hyperglycemia may affect vascular tone by altering patterns of eicosanoid production. Hyperglycemia activates several signaling pathways that could have considerable effects on vascular function. For example, hyperglycemia stimulates lipooxygenase and cyclooxygenase pathways in vascular smooth muscle leading to enhanced formation of vasoconstrictive prostaglandins such as thromboxane A2 (Amore et al., 1997). This action could explain the dramatic reduction in postreperfusion CBF and may provide a target for potential therapeutic intervention, including cyclooxygenase inhibition. Beneficial effects of such medications on vascular tone could be outweighed by their antiplatelet actions.
Recent infection/inflammation and reperfusion
The influence of interleukin-1β(IL-1β)—a specific inflammatory cytokine—on the development of reperfusion induced ischemic damage has been studied. Pre-treatment with IL-1β applied to the cortical surface after MCAO reduced CBF, increased neutrophil recruitment, and increased O2− production during reperfusion (Fabian et al., 2000). These events occurred after 60 to 90 minutes of cerebral ischemia, a period that produced no measurable O2− formation in control (non—IL-1β pretreated) rats. Additionally, exposure to IL-1β accelerated the poor-reflow phenomenon (Fabian et al., 2000) and reduced the cerebral ischemia time necessary to unleash O2− on reperfusion, an effect that could accelerate ROS-mediated reperfusion damage to the vasculature and parenchyma. The potential contribution of acute inflammation in the pathogenesis of cerebral ischemia is supported by the fact that stroke onset in the spontaneously hypertensive stroke-prone rat, precipitated by salt loading, is proceeded by an increase in circulating acute-phase inflammatory factors.
Preexisting inflammation may also have beneficial effects. Pretreatment with lipopolysaccharide (used to model a systemic response to infection) activates multiple inflammatory cytokines as part of the inflammatory response, and induces a neuroprotective effect (Matsuo et al., 1994), despite an increase in neutrophil accumulation. The lipopolysaccharide-induced inflammatory response thus appears to have a complex relationship in ischemic brain tissue, with the potential for inflammatory priming to induce both deleterious and beneficial factors. This may explain why patients with elevated IL-6 levels at the onset of stroke, perhaps reflecting preexisting infection, had poorer neurologic outcome after both ischemic and hemorrhagic stroke (Vila et al., 2001). Therefore, it is conceivable to predict reduced benefit from thrombolysis for stroke with an elevated C-reactive protein, but this has yet to be determined.
Mitochondrial respiratory function and reperfusion
The ongoing metabolite changes commonly seen in affected brain tissue during reperfusion are apparently not due to altered delivery of O2 and must signal either the presence of other changes that impair ATP generation or a large increase in cellular energy demand. Reduced mitochondrial ATP production could arise by a number of mechanisms including direct deleterious modifications in the properties of constituent proteins or lipids, a decreased availability of metabolites such as adenine or pyridine nucleotides that are necessary for normal oxidative phosphorylation, or an accumulation of substances able to inhibit this process.
Effects of reperfusion on mitochondrial function.
A number of mitochondrial enzymes, including cytochrome oxidase and manganese superoxide dismutase (MnSOD), have decreased activity during cerebral ischemia, with predicted effects on O2 metabolism. Cellular cerebral ischemia inhibits expression of the multisubunit CC-oxidase (complex IV) and the final intramitochondrial site of oxidative phosphorylation. Loss of cytochrome oxidase activity leads to cellular damage during reperfusion, because absence of the final electron acceptor increases ROS production by more proximal complexes (Lin et al., 2000). Therefore, the ROS effects should be greatest at the level of the mitochondrial membrane constituents including the complexes of the respiratory chain and lipid constituents particularly rich in unsaturated fatty acids. Increased ROS production in ischemia-injured mitochondria will lead to cytoskeletal changes and likely influence permeability in endothelium and epithelium. ATP-depleted endothelial cells display shortening and disassembly of F-actin filaments, which lead to increased endothelial permeability. In addition, mitochondrial ROS production may cause membrane protein aggregation, alterations in protein polarization (altered apical-basolateral orientation), protein degradation, and changes in molecular chaperons or growth factors.
Mitochondrial complex-I dysfunction occurs after reperfusion of ischemic mitochondria. Complex-I defects increase cellular production of ROS, which may influence MnSOD expression. MnSOD eliminates ROS generated at the Qi site, but in the absence of MnSOD, O2− from that site is free to react. When complex-I dys-function occurs as a result of ROS produced during reperfusion, substrates that bypass complex-I can provide sufficient energy to maintain viability of reoxygenated cells.
Transient but large increases in cellular and mitochondrial [Ca2+] occur during reperfusion. Elevated [Ca2+]i cause mitochondrial depolarization. Release of Ca2+ from storage sites stimulates Ca2+-dependent proteases, nucleases, and phospholipases that trigger apoptosis. Ca2+ leaves mitochondria during reoxygenation through pore formation, reversal of uniport influx carrier, the Ca2+/H+ antiport system, or channel-mediated Ca2+ pathways. Increased [Ca2+]i also appears to be related to the oxidation of adenine nucleotides or to thiol oxidation, because overexpression of the antiapoptotic protein Bcl-2, which has antioxidant properties in mitochondrial membranes, inhibits Ca2+ efflux due to oxidants (Esposti et al., 1999).
In some studies, reperfusion after 2 hours of focal ischemia produced a partial recovery of respiratory capacity during the first hour and then a secondary deterioration at 2 to 4 hours in core tissue. Similar but less marked alterations were seen in perifocal tissue resembling the pattern of postischemic alterations in energy metabolites (Folbergrova et al., 1995), suggesting that the impairment of mitochondrial function is a key determinant of this altered metabolic response. Furthermore, secondary decline in mitochondrial respiratory capacity was greatly ameliorated by postischemic treatment with the spin-trap compound, α-phenyl-N-tert-butyl nitrone (Araki et al., 1992), or the immunosuppressant, FK-506. Both of these treatments significantly reduce the extent of infarction that subsequently develops in brain tissue. Thus, these findings suggest that continued restriction of mitochondrial capacity during reperfusion, or perhaps the secondary deterioration of mitochondrial activity, is one determinant of the severity of ischemic damage.
The effect of reperfusion on mitochondrial permeability transition.
The O2 increase in brain tissue during early reperfusion supports restitution of electron transport chain activity and increased membrane potential. After severe global ischemia, there is apparently a moderate and short-lived increase in mitochondrial [Ca2+], presumably resulting from the return of mitochondrial function during the brief period when [Ca2+]i remains substantially elevated. To our knowledge, there are no data examining mitochondrial Ca2+ during reperfusion after focal ischemia. [Ca2+]i recovers only partially, even with extended periods of reperfusion (Kristin et al., 1998), and raises the possibility that increases in mitochondrial [Ca2+] could develop. Such a change is coupled with the development of a more oxidized intra-cellular environment and a likely increased generation of prooxidants (Arnold et al., 2002), creating conditions expected to favor opening of the transition pore.
The strongest evidence that the permeability transition pore is induced during reperfusion (after focal ischemia) is the protection achieved using postischemic treatment with specific agents to block induction of the transition pore. One such agent, cyclosporin A, has been shown to produce dramatic neuroprotection: intracarotid injection of this compound after 2 hours of focal ischemia reduced the infarct volume by approximately 90% (Yoshimot and Siesjö, 1999). Furthermore, this treatment provided substantial neuroprotection when initiated up to 3 hours after initiation of cerebral ischemia. Large reductions in infarct volume (approximately 65%) have also been seen after oral administration of cyclosporin A for several days before the induction of cerebral ischemia or an intraperitoneal injection immediately on reperfusion.
Alterations in system regulating protein synthesis and reperfusion
Protein synthesis is substantially suppressed in vulnerable neurons during reperfusion. Although the patterns of postcerebral ischemia protein synthesis suppression in brain tissue are well described, the exact pathophysiologic mechanisms that cause it were completely unknown until recently. The integrity of DNA, transcription machinery, mRNA processing and transport, and translational competence of purified ribosomes are preserved in early reperfusion, and there is adequate recovery of high-energy phosphates to support peptide synthesis; thus, these components are not responsible for the inhibition of protein synthesis. Although, there has not been a systematic study of these effects of reperfusion on intracellular levels of all amino acids, the levels of those that have been examined are not substantially reduced (Raley-Susman and Lipton, 1990). Additional investigation is needed because levels of aminoacyl-tRNA that do fall to 60% to 70% of control values (Kleihues and Hossmann, 1971) can be involved in kinase activation directed at inhibition of translation initiation by modification of eukaryotic initiation factors (eIFs).
Protein synthesis may be suppressed by inhibition of translation initiation during early reperfusion. Ribosomal sedimentation profiles obtained from reperfused brain tissue show a preponderance of monomeric ribosomal subunits (Kleihues and Hossmann, 1971), suggesting a block in the formation of the initiation complex (Gaitero et al., 1988). In in vitro translation by brain homogenates, the addition of a chain initiation inhibitor (polyinosinic acid) decreased protein synthesis 63% in samples from control animals as expected. However, polyinosinic acid had no effect on in vitro translation with the postmitochondrial supernatant obtained after 25 minutes of reperfusion, indicating that translation initiation was already maximally inhibited.
Formation of the translation initiation complex.
The initiation complex is an intricate process involving over 140 proteins, including at least nine eIFs. Formation of the initiation complex requires free 40S ribosomal subunits; however, under normal physiologic conditions, formation of inactive 80S ribosomes is favored. Studies of reperfusion have identified two important changes in eIFs: (1) substantial proteolytic degradation of eIF4G mediated by γ-calpain during both cerebral ischemia and reperfusion, and (2) a rapid 20-fold increase in eIF2α (P) during reperfusion. Autoproteolytic activation of γ-cal-pain occurs during cerebral ischemia in association with degradation of eIF4G that is blocked by calpain inhibitors (Neumar et al., 1997).
In nonischemic brain homogenates, 1% of total eIF2α is in the phosphorylated form eIF2α (P); however, after 10-minute cardiac arrest and 90-minute reperfusion (but not after ischemia alone), eIF2α (P) is increased to 23% of total eIF2α(Selman et al., 1990). This alteration occurs rapidly: at 10 minutes reperfusion eIF2α(P) is already increased 18-fold, and there is prominent eIF2α(P) immunostaining in the cytoplasm of neuronal tissue in selectively vulnerable areas (i.e., the hippocampus and cortex). At 1 hour of reperfusion, eIF2α(P) remains only in selectively vulnerable neurons and is now prominent both in their cytoplasm on ribosomes and in their nuclei in association with both the nucleolus and chromatin (Goldstein et al., 1999). The nuclear translocation probably reflects the nuclear localization motif (RRRIR) immediately adjacent to the ser-51 phosphorylation site on eIF2α, and α-subunit phosphorylated (but not unmodified) eIF2 binds DNA in electromobility shift assays (Goldstein et al., 1999) and may have important consequences for transcription. Immunohistochemical staining of eIF2α(P) and inhibition of in vivo protein synthesis (by autoradiography of 35S-labeld amino acid incorporation) comap in the reperfused brain (Sullivan et al., 1999). These results provide a mechanistic basis for the inhibition of protein synthesis in selectively vulnerable neurons during reperfusion, and together with other evidence for a role for eIF2α(P) in the mechanisms of apoptosis (discussed later). This suggest that the inhibition of protein synthesis in selectively vulnerable neurons during reperfusion may be a fundamental phenomenon in the causal pathway leading to the death of vulnerable neurons.
Implications of eIF modifications for message selection.
The response of translation initiation to heat shock and to reperfusion in selectively vulnerable neurons shares some common elements, including phosphorylation of eIF2α. However, the response to heat shock is not completely analogous to that seen after cerebral ischemia. Although dephosphorylation of eIF4E is seen in heat shock, dephosphorylation of eIF4E cannot be found after reperfusion. Antisense-induced depletion of eIF4E and eIF4G substantially inhibits protein synthesis, but about 20 proteins, including HSP-70, continue to be synthesized in this situation. Proteolytic loss of eIF4 components is therefore inadequate to explain the blocked translation of HSP-70 in selectively vulnerable neurons, which in fact is almost certainly a consequence of the large increase in eIF2α(P).
Translations of proteins encoded by 3′-distal open reading frames on mRNAs with internal ribosome entry site and long 5′ leaders containing other open reading frames might actually be forwarded in the circumstance of degradation of eIF4G and increased eIF2α(P). Several examples of such mRNAs are associated with reperfusion. The mRNA for eIF4G itself has a long 5′ leader and contains multiple programmed ribosomal frame shift and an internal ribosome entry site. Transfer of this 5′ leader onto reporter genes can increase their translation over 100-fold. Thus, the levels of eIF4G should be able to recover during reperfusion. Other important mRNAs that have recently been found to share these characteristics include those for apoptosis-activating factor-1 (APAF-1), vascular endothelial growth factor, and fibroblast growth factor-2. Apoptosis, in particular, is a crucial area in which these issues are likely to be important. The previously mentioned considerations suggest that careful examination of the products of residual translation in selectively vulnerable neurons during reperfusion may resolve the apparent conflict between the biomolecular inhibition of overall protein synthesis in selectively vulnerable neurons during reperfusion, and suggests that administration of cyclohexamide (an inhibitor of peptide elongations) reduces what is thought to be apoptotic degeneration (Martin et al., 1988).
EIF2α kinases and phosphatases.
There are four mammalian eIF2α kinases: (1) GCN2, (2) the hemin-regulated inhibitor in reticulocytes (HRI), (3) “RNA-activated” protein kinase (PKR), and (4) PERK (PKR-like ER kinase). There are at least three potential reperfusion-induced activation mechanism for GCN2. GCN2s bind unchanged tRNAs via its histidyl tRNA synthase-related region. Although there are no published observations on the effects of reperfusion on levels of aminoacylated histidine tRNA, histidine loss occurs in association with cerebral ischemia (Feig and Loeb, 1993), and histidine administration offers some neuro-protection (Kawamoto et al., 1997). Second a cytoplasmic unfolded protein response, consistent with HSp transcription, could induce dissociation of HSP90 from GCN2 and activation of the enzyme. Finally, GCN2 is also activated by low adenine levels, known to occur in response to reperfusion (Newman et al., 1998).
HRI activity is stimulated by heavy metals (e.g., iron), oxidized glutathione, and polyunsaturated fatty acids (PUFA) and lipoperoxides (DeHerreros et al., 1985), suggesting a potential causal link between radical-mediated iron delocalization, lipid peroxidation, and reperfusion-dependent suppression of protein synthesis. However, HRI-knockout mice show no reduction in brain eIF2α phosphorylation during reperfusion.
PKR levels are increased in differentiated cells (Proud, 1995), and a number of endogenous inhibitors and activators of PKR have been identified and have been correlated with changes in gene expression; it is particularly interesting that activated PKR appears to be required for induced transcription of the FAS-receptor gene involved in apoptosis. PKR is found tightly associated with ribosomes and on the rough endoplasmic reticulum with a small fraction present in the nucleolus. PKR transcription is induced by interferon, and PKR activity is antagonized by ras-activated cell signaling processes, such as those known to be under the control of the insulin receptor. However, PKR knockout mice show no reduction in the accumulation of eIF2α (P) induced by reperfusion (DeGracia et al., 1998).
PERK is an evolutionary recombinant protein with homology to IRE-1 unfolded protein detector in its N-terminal endoplasmic reticulum resident domain and homology to GNC2 in its cytosolic eIF2α kinase domain (Kaufman, 1999). PERK activation involves autophosphorylation, and the active form displays retarded electrophoretic mobility (Harding et al., 1999). PERK is present and activated in response to thapsigargin (a selective inhibitor of the endoplasmic reticulum Ca2+ pump) or arachidonate in neuronally differentiated NB104 cells (Kaufman, 1999).
Regulation of the availability of eIF2 or eIF2α kinases.
A final mechanism that might be involved in eIF2α phosphorylation during postischemic reperfusion is regulation of the availability of eIF2 as substrate for eIF2α kinases. A peptide with a molecular weight of 67 kd (p67) has been identified that, when glycosylated, binds to eIF2 and protects eIF2α from phosphorylation. Preincubation of brain homogenates with an antibody against p67 enhances subsequent phosphorylation of eIF2α in vitro. Different cell lines contain different absolute [p67], and cells with high [p67] exhibit enhanced resistance to viral-induced eIF2α phosphorylation (Bose et al. 1997). Viral-induced phosphorylation of eIF2α and shut-off of host-cell protein synthesis is associated with rapid activation of a 60-kd enzyme that deglycosylates p67, which makes p67 unable to prevent phosphorylation of eIF2α, but does not lead to its degradation (Chakraborty et al., 1994). These findings suggest that p67 deglycosylation during reperfusion and/or the level of expression of p67 by various groups of neurons could affect their susceptibility to eIF2α phosphorylation. However, neither the glycosylation state of p67 nor its location in brain tissue is affected by reperfusion (Charkraborty et al., 1994). Although altered p67 glycosylation does not appear to be involved in the causal pathway leading to phosphorylation of eIF2α during reperfusion, the persistence of glycosylated p67 may be important in limiting the extent of eIF2α phosphorylation and supporting the potential for recovery of translation competent vulnerable neurons.
The importance of recovery of protein synthesis during early reperfusion.
Late reperfusion may be a fundamental mechanism that can perpetuate the suppression of protein synthesis. By 8 hours of reperfusion after a 15-minute cardiac arrest in dogs, ionic concentration gradient of Na+, K+, Ca2+, and Mg2+ is lost, with associated concomitant 30% decrease of lipid double bond content and ultrastructural evidence of large breaches in the plasmalemma in selectively vulnerable neurons. Effective therapy would probably have to initiate recovery of protein synthesis before 4 to 8 hours reperfusion to enable the vulnerable neurons to respond in ways that would prevent the later deterioration.
Reactive oxygen species in intracellular signaling and reperfusion
Reactive oxygen species act as signaling molecules in various cell types, participating in or modifying physiologic events related to receptor-ligand binding and transcriptional activation (Hanson and Leidold, 1998). Specific intracellular signaling pathways have been implicated in cell death after reperfusion, although the specific pathways leading to apoptosis/necrosis might vary among different cell types. Several PK-signaling pathways involved in cellular reperfusion injury were detailed earlier. ROS interact with a number of specific molecular targets in reperfused cells, including extracellular signal-regulated PK and mitogen activated protein kinase (MAPK) that mediate proliferation, stress-activated PK implicated in apoptosis, NF-κB, and several caspases (Whitmarsh and Davis, 1996). The GPTase binding protein Rac1 regulates some PK (e.g., stress-activated protein kinase, p38 MAPK, and c-jun NH2-terminal kinases). These PKs may be regulated further by ROS generated by NAD(P)H oxidase, which requires Rac1 for activity (Irani, 2000). Inhibition of rac1 lessens both ROS generation and reperfusion injury (Kim et al., 1998).
ROS also regulate or participate in growth, apoptosis, and the adaptive response to injury or stress. Platelets exposed to reperfusion generate O2− and OH, which activate arachidonic acid (AA) metabolism via phospholipases A2 and C (Leo et al., 1997). Platelet activation is associated with inositol 1,3,4-triphophate and thromboxane A2 production, and platelet activation is inhibited by antioxidants.
Stress kinases, JNK, and activator protein-1.
Several studies have shown modifications in the expression of stress kinases after reperfusion. ROS production during reperfusion appears to stimulate stress-activated PK, including c-jun NH2-terminal kinases (JNK) and p38 MAPK in the penumbra. However, the majority of studies are focused on particular PK or on certain substrates, and it is difficult to establish a coordinated view of the underlying pathophysiologic mechanisms. Moreover, some aspects of PK response are poorly documented, particularly those occurring at early stages after cerebral ischemia.
JNK and p38 participate in a variety of metabolic pathways including survival. JNK-1 activity translocated to the nucleus increases during reperfusion, at which time JNK-1 is serine phosphorylated. An upstream kinase, stress-activated protein kinase/ERK kinase-1 (SEK1), activates JNK-1 in the nucleus. Another upstream kinase for the MAPK, MAPK/ERK kinase 1 (MEK1), in contrast, remains localized to the cytosol. Increased JNK-1 activity after ischemia depends on its translocation to the nucleus and activation of upstream PK (probably SEK1) during reperfusion. Supporting evidence for activation of PK pathways by ROS during reperfusion includes inhibition of reperfusion-induced apoptosis by an antisense oligonucleotide targeted to JNK-1. Activator protein-1 (AP-1) binding increased markedly during reperfusion, as did poly-(ADP-ribose)-polymerase cleavage and capsase-3 activation. Induction of c-jun/c-fos (AP-1) expression during ischemia was followed by poly-(ADP-ribose)-polymerase activation and histone H1 ADP-ribosylation. Activation of the AP-1 pathway was essential for initiation of programmed cell death (Chen et al., 1998). C-jun is involved in neuronal survival and neuronal regeneration, in addition to cell death. It could be shown that c-fos and c-jun are markers of cell sensitivity after MCAO. However, early c-jun-Pser63 expression, correlating with early stress-activated protein kinase/JNK-P nuclear expression, is associated with cell death in the infarct core. In addition, strong c-jun expression also develops in the cytoplasm of neurons in the penumbra at 4 hours after reperfusion (Laderoute and Webster, 1997). Therefore, the outcome of a cell expressing c-jun phosphorylated at ser63 depends on the subcellular localization of c-jun and, probably, on the interactions of c-jun with other factors. Taken together, the present findings indicate that early increased activation of several transcription factors is associated with cell death in the infarct core. However, different signals converge in the cytoplasm of neurons located at infarct borders after reperfusion, revealing the struggle of death promoters and life facilitators at the penumbra.
Protein kinase c.
Members of protein kinase C (PKC) family are key enzymes in the mediation of ischemic signals in the nerve cell. PKC acts upstream of MAPK and induces its activity through MEK activation during reperfusion. During reperfusion, [ATP] is restored and protein phosphorylation, due to specific localization of actually translocated PK, might be accelerated. Phosphorylation and MAPK activation during reperfusion can be blocked both by PKC inhibition and phosphoinositide 3-kinase inhibitors. However, permeability changes due to cerebral ischemia and low glucose could be blocked by chelating Ca2+, PKC, and PKG inhibition or by inhibition of p38 MAPK-1. Ca2+ induced dissociation of cadherinactin and occluding-actin junctions might mediate these increases in endothelial permeability, through pathways that involve PKC, PKG, and MAPK. ROS may act as intracellular signals, or they may interact with specific molecules in intracellular signaling cascades. Stress-activated PKs, particularly JNK and p38 MAPK, have been most strongly associated with reperfusion injury. PKC appears to be responsible for induction of MAPK during reperfusion and may represent an important mediator of reperfusion-induced brain injury.
Nuclear factor-κB.
Nuclear factor-κB activation is involved in reperfusion injury, especially in the vascular endothelium, where NF-κB activation leads to neutrophil adhesions in vivo. NF-κB nuclear binding increased in microvascular endothelial cells during reperfusion, but not during cerebral ischemia. NF-κB binding and intracellular adhesion mocecule-1 (ICAM-1) surface expression were induced, concurrent with ROS production. An NO donor molecule, A-nitroso-acetyl-penicillamine (SNAP), blocked NF-κB activation and inhibited ICAM-1. Blockade of NO synthase by L-nitroarginine increased ICAM-1 expression after reperfusion. NO thus appears to inhibit ICAM-1 activation by decreasing the level of oxidant stress responsible for NF-κB activation.
Nitric oxide can block NF-κB activation by reperfusion, and NF-κB appears to be required for development of tolerance to reperfusion. NF-κB is activated when an inhibitory factor, Ik-B-α, is degraded, thus allowing NF-κB subunits to dimerize, transmigrate to the nucleus, and bind to several transcriptions promoting regions of the genome. Ik-B-α is primed for degradation by phosphorylation by the Iκ B kinase (IKK), followed by ubiquination. In turn, IKK is activated by phosphorylation by several PK, including those activated by IL-1, tumor necrosis factor (TNF) α, IL-6 and IKK itself. NF-κB stimulates the increased transcription of inflammatory cytokine mRNAs including IL-1, IL-6, TNF-α and others (Caroll et al., 1998). Thus, on reperfusion, oxidative stress increases activation of NF-κB, which results in increased expression of proinflammatory cytokines that further activate NF-κB through IKK signaling pathways (Carden and Granger, 2000). Moreover, proinflammatory cytokine transcription stimulates increased expression of endothelial adhesion molecules such as the selectins and ICAM-1. Inflammatory cells generate ROS that increase oxidative stress and are likely to further activate NF-κB and AP-2 as key regulatory transcription factors that would be expected to have additional deleterious effects on vascular endothelium. Inhibition of the proinflammatory cytokine influence of NF-κB activation and translocation to the nucleus have been found to reduce reperfusion injury (Schneider et al., 1999).
Reactive oxygen mechanisms and reperfusion
Increased ROS production has been proposed to explain how reperfusion worsens ischemic damage. O2− generated by xanthine oxidase (XO), was postulated initially to be the ROS responsible, but now the entire spectrum of ROS and reactive nitrogen species, including peroxynitrite (ONOO−) (Beckman, 1991), has been implicated in posthypoxic cellular injury (Table 2). ROS generated by hypoxia or reperfusion are now recognized as interacting with physiologic signal transducers rather than behaving as simple reactants that peroxidize membrane lipids, oxidize DNA, or denature enzyme proteins. ROS can affect signal transduction in posthypoxic cells, and ROS are able to initiate cell death programs in the form of apoptosis or necrosis (Fig. 2). Diverse sources of ROS (e.g., enzymes, mitochondria) exist normally within cells, some of which produce excess ROS during reperfusion. Excess ROS from endogenous sources can account for autocrine and paracrine cellular damage during reperfusion.
Important mechanisms and sequelae of cellular reoxygenation injury

ROS, reactive oxygen species; XD, xanthine dehydrogenase; XO, xanthine oxidase; GSH, glutathione; NF-κB, nuclear factor-κB; PG12, prostacyclin; NOS, nitric oxide synthase; RNS, reactive nitrogen species; PKC, protein kinase C; HSP, heat shock protein.
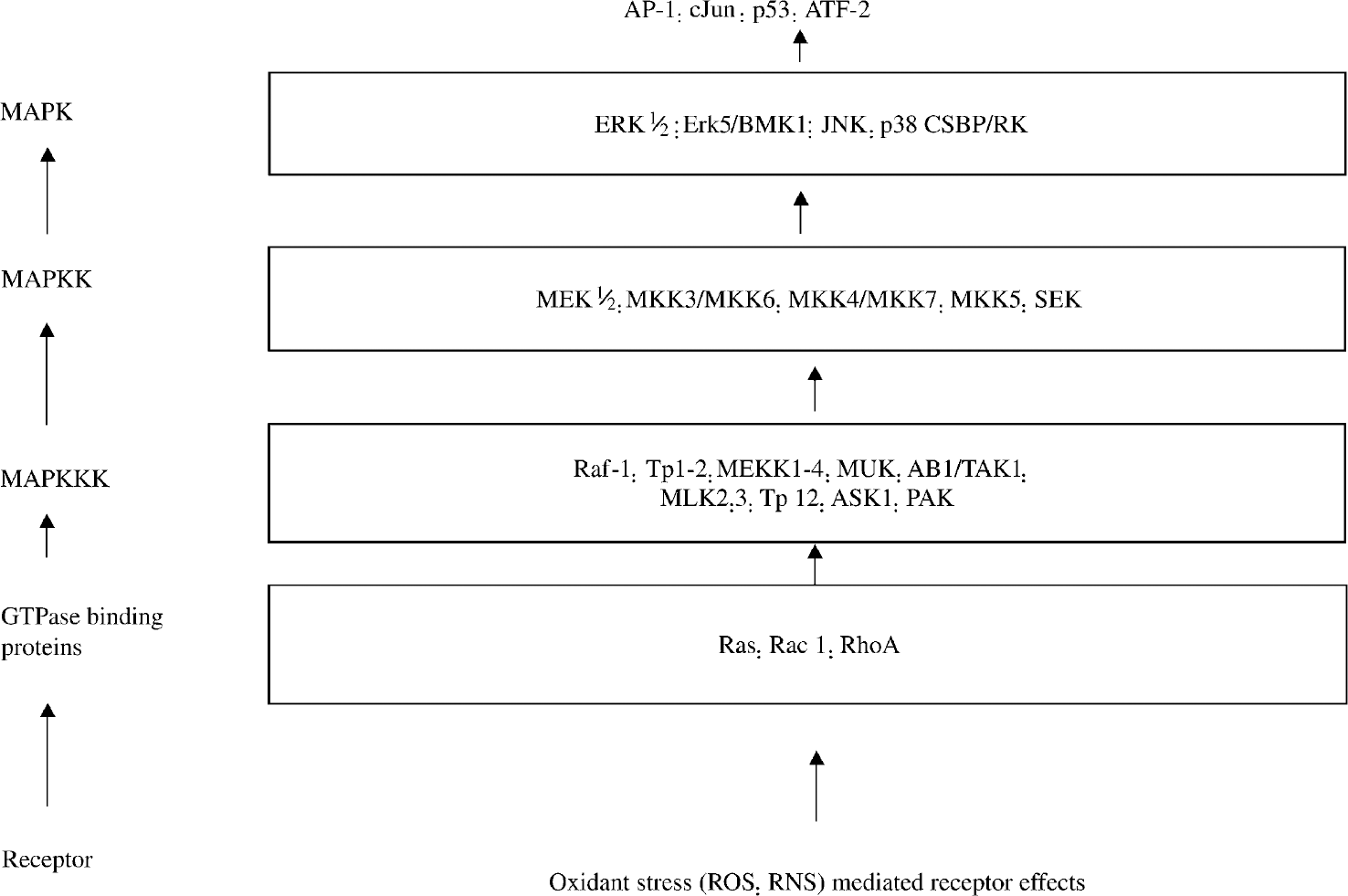
Protein kinase signaling pathways possibly involved in reoxygenation injury. MAPK, mitogen-acitvated protein kinase; MEK, mitogen-acitvated protein kinase/extracellular signal-regulated kinase; ERK, extracellular signal-regulated kinase; JNK, c-jun NH2-terminal kinase; SEK, stress-activated protein kinase/extracellular signal-regulated kinase; ROS, reactive oxygen species; RNS, reactive nitrogen species.
The role of ROS in the pathogenesis of reperfusion damage is well known: however, certain details are emerging that have clinical implications. First, the production of toxic ROS, particularly O2−, after reperfusion is very rapid. ROS may produce additional tissue damage through lipid peroxidation, reaction with nucleotide bases, attack on DNA repair enzymes, activation of other related enzymes, such as poly-(ADP-ribose)-synthase, reaction with proteins and reduction of NO. NO reacts with O2− to form peroxynitrite and is scavenged by hemoglobin that may be released into brain tissue, leading to delayed neuronal death (Hata et al., 2000). Oxygen-dependent NO synthase is activated in ischemic tissue when Ca2+ enters ischemic neurons. NO levels are reduced as peroxynitrite is generated when reperfusion floods ischemic tissue with O2 (Beckman, 1991). NO production may also be reduced by vascular endothelial damage that may occur with cerebral ischemia or a secondary inflammatory response (Akopov et al., 1994). Loss of NO has the potential to lead to vascular contraction and the N2O2 radical is potentially damaging in its own right.
Activation of NF-κB promotes transcription of genes for proteins involved in reduction of apoptosis, such as NGF, Bcl-X1, and apurinic/apyrimidinic endonuclease (APE)/Ref-1, and for production of antioxidants such as MnSOD (Grilli and Memo, 1999). Similarly, paradoxical exacerbation of ischemic damage was seen, when the TNF receptor was knocked out in mice (Bruce et al., 1996). NF-κB is activated by a variety of stimuli that include cytokineses such as TNF and IL-1 and oxidative stress. Perhaps attention to specific downstream effects of blocking oxidative stress (i.e., activation of specific subunits of heterodimer of NF-κB, expression of proinflammatory cytokines, proapoptotic factors) may provided more promise for neuroprotective efficacy than reduction of ROS. The interaction of these events is complex, and the outcome of therapeutic interventions aimed at these elements of cellular injury is uncertain without more rational and specific targeting of these mechanisms and knowledge of the underlying state of the organism with respect to these factors.
Intracellular sources of reactive oxygen species and reperfusion
Xanthine dehydrogenase and xanthine oxidase.
One of the pathways in the ischemia-initiated cascade involves transformation of xanthine dehydrogenase into xanthine oxidase (XO), which intracellular ATP degrades to hypoxanthine. XO and the closely related aldehyde oxide are potentially capable of ROS generation. XO is the more important source of both O2− and H2O2, but XO varies widely in abundance among cell types, organs, and species. XO normally occurs in vivo as a NAD+-dependent dehydrogenase (xanthine dehydrogenase [XD]) incapable of ROS production. XD activity converts by sulfhydryl oxidation or limited proteolysis (conditions that exist during reperfusion) to an oxidase that produces both O2− and H2O2 (Kayyali et al., 2001). XO is phosphorylated in ischemic microvascular endothelial cells through a mechanism involving p38 MAPK and casein kinase II. Phosphorylation appears to be necessary for ischemia-induced enzyme activation (McLeod and Alayash, 1999). However, reperfusion induced brain tissue damage also unquestionably occurs in cells in which XO activity is exceedingly low or absent.
There has been reasonable doubt expressed about the XO system as a main source of ROS, because an intermediate of XD is formed during the oxidation process. Furthermore, allopurinol, an XO inhibitor, has been shown to have HO−-scavenging activity of its own. How-ever, the role of XO in reperfusion damage is still unclear, with doubt about intracellular XO's ability to serve as a source of O2− and whether or not the generated O2− can cause cell damage.
Ferrylhemoglobin.
Hemoglobin and myoglobin, which are released into plasma after cellular stress, can mediate endothelial cell oxidative stress, and a ferrylhemoglobin intermediate has been detected in cellular reperfusion models. Heme-mediated redox reactions have been suggested to contribute to organ dysfunction and/or tissue damage after reperfusion. Incubation of hemoglobin or myoglobin with endothelial cells during reperfusion causes transient oxidation of Hb to the reactive ferryl species (Fe4+). Lipid peroxidation, which results from exposure of endothelial cells to ferrylhemoglobin, increased after reperfusion. Incubation of endothelial cells with Hb also caused a dose-dependent decrease in intracellular glutathione (GSH), confirming an oxidative stress (Privalle et al., 2000). NO acts as an antioxidant to inhibit lipid peroxidation and damage due to ferrylhemoglobin, because arginine inhibited both lipid peroxidation and formation of ferryl intermediate (Ozaki et al., 2000). Hemoproteins oxidized to their ferryl forms could account for heme-mediated injury of endothelial cells that have been stressed by cerebral ischemia, reperfusion, and glutathione depletion. For this reason, data indicate that ferrihemoglobin induces an initial oxidative stress, mainly reflected in the early oxidation of lipids and further decrease in GSH and ATP, but the cell may gradually adapt to this situation by recovering their endogenous ATP and GSH levels.
NADPH oxidase.
Endothelial cells express a membrane NADPH oxidase, similar to the membrane enzyme complex that produces the respiratory burst in granulocytes The membrane oxidase complex is probably an important source of ROS in reoxygenated tissues. ROS produced p47phox, a subunit of the NADPH oxidase, is detected by Western blotting of endothelial proteins. Endothelial cells adapted to flow in vitro for 2 to 7 days showed a nearly twofold increase in ROS production during simulated no-flow ischemia, compared with continuously perfused cells (Ozaki et al., 2000). Absence of flow causes flow-adapted cells to activate both NF-κB and AP-1. Transcription factor activation appears to be dependent on transduction of mechanical signals rather than altered oxygenation, showing that ischemia rather than hypoxia is the more important determinant in flow-adapted cells.
A dominant negative rac1 construct completely inhibited reperfusion NF-κB activation induced by ROS. The dominant negative gene product (N17rac1) inhibits the intracellular ROS burst after reperfusion. N17rac1 expression protected smooth muscle cells, fibroblasts, and endothelial cells, indicating an important role for rac1. A constitutively active rac1 mutant does not increase intracellular ROS. Rac1 GTPase is, therefore, a necessary, but not sufficient component of the pathway leading to ROS during reperfusion.
Multiple sources of ROS exist within cells and may account for increased ROS production during reoxygenation. Although XD was identified early as a ROS source during reperfusion, other intracellular sources including redox cycling of iron and NAD(P)H oxidases have been shown to produce ROS and contribute to oxidant stress during reperfusion.
Leukocytes and reperfusion
The inflammatory cascade, characterized in part by early leukocyte interaction with endothelium, may contribute to additional injury to blood vessels and surrounding brain tissue, extending the area of infarction. Accumulation of neutrophils, together with red blood cells and platelets, can lead to capillary plugging (Engler et al., 1986), and a consecutive reduction in microvascular CBF, resulting in focal areas of stasis. Once infiltrated into brain tissue, neutrophils are capable of releasing various proteases, lipid derived mediators, and ROS that can injure potentially salvageable cells. Adhesion molecules on both endothelial cells and neutrophils are key factors that mediate the sequential events of neutrophil rolling, adherence, activation, and emigration in the tissue. Several classes of cell surface glycoproteins orchestrate the adhesion and migration of neutrophils. These include the leukocyte integrins (CD11/CD18), the immunoglobulin supergene family (ICAM-1; vascular cell adhesion molecule-1), and the selectins (L-,P-, and E-selectin) (Lipsky, 1993). Studies employing monoclonal antibodies directed at specific members of these various pathways have been instrumental in showing the importance of neutrophils in reperfusion injury (Malinski et al., 1993).
Intracellular adhesion mocecule-1.
Several animal studies have reported increased levels of cerebrovasculature ICAM-1 in reperfusion injury. Permanent or transient MCAO gives rise to increased levels of ICAM-1 message and protein (Schroeter et al., 1994). It is of interest that patients with risk factors for vascular disease exhibited increased soluble [ICAM-1] in serum samples. Two studies with mice deficient in ICAM-1 (homozygous null ICAM-1; ICAM-1−/−) support the role of ICAM-1 in the etiology of reperfusion injury. Compared with wild-type mice, ICAM-1−/− mice exhibited reductions in infarct size and neurologic deficit after 22 hours of reperfusion after focal ischemia (MCA, 45 minutes) (Connolly et al., 1996). The ICAM-1−/− groups had an increased survival (35% increase) and had an elevated CBF in the cortical area of the infarcted hemisphere. Similar results were obtained in wild-type animals immunodepleted of neutrophils. After (3 hours) MCAO in ICAM-1−/− mice followed by either 21 or 45 hours reperfusion, the ICAM-1−/− mice had a 5.6-fold (at 21 hours) or 7.8-fold (at 45 hours) reduction in cerebral infarct size compared with the wild-type mice (Soriano et al., 1996).
Selectins
The selectin family of adhesion molecules mediates the initial rolling and tethering of leukocytes to endothelium. Upregulation of selectins has been shown on cerebral vascular endothelium after reperfusion. P-selectins are upregulated on microvascular endothelium in the basal ganglia after MCAO (3 hours) and reperfusion (1 and 4 hours). Another study (Haring et al., 1996) showed that E-selectin was upregulated during MCAO and persisted during reperfusion. These authors also found a delayed (at 24 hours) upregulation of E-selectin in the contralateral (nonischemic) basal ganglia, suggesting a remote induction by humoral factors generated from the ischemia area. E-selectin mRNA has also been reported to be induced in the brain after focal ischemia in animals (Wang and Feuerstein, 1995), and in stroke patients, soluble E-selectin was found to increase transiently in serum samples after acute cerebral ischemia without differences in the levels of L-selectins.
The efficacy of a selectin oligopeptide that contained an amino acid sequence of the common lectin domain of selectins was studied using animal models of either permanent or transient focal ischemia in spontaneously hypertensive rats. The selectin oligopeptide, or an oligo-peptide with a scrambled sequence, was administered just before cerebral ischemia and was not effective in limiting infarct volume in the permanent cerebral ischemia (MCA/common carotid artery) model. However, the selectin oligopeptide caused a significant decrease in the volume of ischemic damage (2 hours of MCAO/common carotid artery occlusion) followed by reperfusion. Administration of an oligopeptide with a scrambled sequence did not confer any protection and authors reported a significant increase in CBF in animals that received the selectin oligopeptide (Wang and Feuerstein, 1995).
P-selectin occurs on stimulated platelets and endothelial cells and this glycoprotein can mediate interaction of these cells with leukocytes. When P-selectin—dependent interaction is inhibited, fibrin deposition was blocked in a primate model of thrombosis (Palabrica et al., 1992). Fibrin deposition is a probable component of the microvascular complications associated ischemia/reperfusion injury. In baboons, fibrin deposition was evident in microvessels from an ischemic zone and deposition increased during reperfusion. Extravascular fibrin was evident at 24 hours of reperfusion, due to extravasation of fibrinogen and reaction with parenchymal tissue factors. Therefore, p-selectin-dependent adhesive events are attractive targets to exploit for developing possible therapeutic strategies in stroke.
Nitric oxide and reperfusion
Nitric oxide is generally considered to play a major role in the development of ischemic damage (Lipton, 1999). NO generation via the neural form of NO synthase seems to be particularly important, based on the protective effects of inhibitors of this enzyme, as well as studies in mice expressing knockout mutations of iso-form of NO synthase. Mitochondria are a target for multiple deleterious effects produced by NO. Low concentration of this substance (in the nanomolar range) can compete with O2 to reversibly inhibit CC-oxidase. Such inhibitory actions of NO could be a further contributor to changes observed in ATP and related metabolites during reperfusion, but this has not been directly tested. Higher [NO] can produce irreversible modifications of proteins or lipids and impairment of mitochondrial respiration. The N2O2 generation, formed from NO reacting with O2−, represents a major mediator of these effects (Canevari et al., 1997). N2O2 can directly produce damage or be converted to other strong oxidants, including the highly reactive OH radical. The targets of N2O2 damage can include the major enzyme complexes of the electron transport chain and ATP synthase, which could directly impair respiration. However, such modifications require [N2O2] in the high micromolar range. Direct assay of the activity of respiratory chain complexes after focal ischemia revealed only a moderate decrease in the activity of CC-oxidase during cerebral ischemia and no changes in any of these complexes on reperfusion (Canevari et al., 1997). Thus, N2O2 accumulations of sufficient magnitude to target these proteins may not be produced even under the abnormal conditions of reperfusion. Possible modifications to other mitochondrial proteins and lipids could contribute to dysfunction but have not as yet been directly investigated. In addition, N2O2 is a stronger inducer of the permeability transition pore (Canevari et al., 1997) and could contribute to injury by activating this response during reperfusion.
Antioxidant defense mechanism and reperfusion
Antioxidants inhibit cellular reperfusion injury.
Endogenous antioxidant systems are critically important in limiting reperfusion-induced cellular damage. The preponderance of experimental data shows that exogenous antioxidant enzymes and low-molecular-weight ROS scavengers inhibit reperfusion injury in cellular models. However, studies using scavengers do not by themselves identify specific ROS or reactive nitrogen species responsible for reperfusion damage, and they do not exclude other mechanisms. The level and duration of ischemia are themselves important in regulating levels of both the antioxidant enzymes and low-molecular-weight ROS scavengers, including glutathione, which ultimately might determine the extent of reperfusion damage (Horakova et al., 1997).
Reperfusion (after 12-minute ischemia) of hippocampal slices in vitro increased lactic dehydrogenase release and lipid peroxidation significantly (Cadenas and Davies, 2000). Exogenous SOD and catalase inhibited injury, showing involvement of ROS (Cadenas and Davies, 2000), which appeared to arise from prostaglandin synthesis, XO, and mitochondria, on the basis of the inhibitor profile. Endogenous low-molecular-weight antioxidants are important in protection from reperfusion damage. Thioredoxin proteins act as disulfide oxidoreductases and electron donors for thioredoxin peroxidases. Thioredoxin shows functions as disulfide bond reductants, similar in mechanism to N-acetylcysteine. Oxidized thioredoxin is reduced by thioredoxin reductases requiring NADPH.
Effects of ischemia on antioxidant enzymes.
Brain capillary endothelial cells similarly respond to cerebral ischemia preexposure by decreasing activities of glutathione peroxidase, glutathione reductase, catalase, and SOD, as well as total cellular GSH content (Li et al., 2002). Such changes, which represent possibly adaptive downregulation of antioxidant defenses by unspecified mechanisms in cerebral ischemia, would predispose to increased ROS production by reperfused mitochondria.
Hypoxia-induced decreases in cellular antioxidant enzymes, especially MnSOD, have potentially important biologic consequences because they could lead indirectly to cellular reperfusion injury by exacerbating oxidant stress. The general response to cerebral ischemia, therefore, appears to be decreased activity or expression of antioxidant defenses. But the mechanism by which anti-oxidant enzyme regulates expression by cerebral ischemia is not known with certainty.
Oxidative damage and reperfusion
Reflow after blood vessel occlusion often causes an increase in O2 to levels that cannot be used by mitochondria under normal physiologic flow conditions. It has been shown that about 2% to 5% of the electron flow in isolated brain mitochondria produces O2− and H2O2 and are scavenged respectively by SODs and by glutathione peroxidase and catalase. Other antioxidants, including glutathione, ascorbic acid, and vitamin E, are also likely to be involved in the detoxification of free radicals. During reperfusion, perturbation of the antioxidative defense mechanisms is a result of the ROS overproduction, inactivation of detoxification systems, consumption of antioxidants, and failure to adequately replenish antioxidants in the ischemic brain tissue. Because of technical difficulties, the level of ROS after cerebral ischemia is generally assessed by indirect methods, including lipid peroxidation, protein oxidation, and DNA damage.
Based on the metal ion requirements and the anatomical distribution, three types of SODs exist in brain cells. CuZn-SOD is a cytosolic enzyme that requires both Cu and Zn as cofactors. It is a dimeric protein that is coded by the human CuZn-SOD transgene (SOD-1) on chromosome 21 in human cells. MnSOD is a mitochondrial enzyme with requirements for Mn2+. It is a tetrameric protein that is coded by the SOD-2 gene in chromosome 4 in human cells. Both CuZn-SOD and MnSOD from various sources have been fully characterized biochemically, and the cDNAs of both human enzymes have been successfully cloned. A Cu-containing SOD (SOD-3) has been identified in the extracellular space, and its gene has been successfully cloned. As specified for O2−, CuZn-SOD has been used extensively to reduce brain tissue injury induced by reperfusion. The extremely short half-life of CuZn-SOD (6 minutes) in circulating blood and its failure to pass the blood—brain barrier make it difficult to use this enzyme therapy in stroke. However, a modified enzyme with an increased half-life, such as polyethylene glycol-conjugated SOD, has been successfully used to reduce infarct volume in rats that have been subjected to focal ischemia. Liposome-entrapped SOD has an increased half-life (4.2 hours), blood—brain barrier permeability, and cellular uptake, and has also proved to be an effective treatment in reducing the severity of focal ischemic damage. Yet, in some instances, modified SOD (i.e., polyethylene glycol-conjugated SOD) has been used with conflicting data. The fact that the results are mixed makes it imperative to use other experimental strategies so that the role of SOD can be fully established in cerebral ischemia.
Degradation of membrane phospholipid and reperfusion.
Energy failure and [Ca2+]i overload are major triggers for membrane phospholipid degradation in cerebral ischemia (Wang and Feuerstein, 1995), fitting well with data where severe cerebral ischemia produced an increased LP (lactate/pyruvate) ratio (mitochondrial dysfunction/energy perturbation), followed by a glutamate increase in extracellular fluid and eventually increased [glycerol]. The persisting LP-ratio increase after MCAO suggests an ongoing damage process, irrespective of the degree of reperfusion. This may explain the sustained increase in (interstitial) glycerol after severe cerebral ischemia. The morphologic progression of injury during reperfusion led to the hypothesis that accelerated structural damage during reperfusion is a consequence of excessive generation of ROS followed by lipid peroxidation, and suggests that selectively vulnerable neurons are especially prone to ROS-induced damage (specifically lipid peroxidation) during reperfusion because (1) they are deficient in glutathione peroxidase and (2) they are surrounded by iron-laden supporting cells that release this iron during reperfusion. The changes in the conformation of free fatty acids (FFA) resulting from lipid peroxidation would then alter the permeability and fluidity of cell membranes. In addition, and probably more importantly, membranes contain receptors, ion channels, and other proteins whose functions are likely to be compromised by alterations in the lipid membrane.
Free fatty acids and in particular free AA are released during cerebral ischemia as a consequence of the activity of both phospholipase C (activated by depolarization) and phospholipase A2 (activated by increased Ca2+) (Abe et al., 1987), and elevated concentrations of these FFA persist into the early reperfusion. The rate of lipolysis in the selectively vulnerable zones is significantly greater than in other brain areas (Krause et al., 1988). Reperfusion-initiated metabolism of free AA may be the major source of O2−. Cyclooxygenase catalyzes the addition of two molecules of O2 to a PUFA and produces prostaglandin PGG, which is rapidly peroxidized to PGH with concomitant release of O2−. Thus, FFA that have been peroxidized are better substrates for phospholipase. Moreover, some of the products of lipid peroxidation stimulate the activity of phospholipase, and the activities of the lipid repair enzymes lysophosphatidylcholine acyltransferase and fatty acyl CoA-synthase are substantially inhibited during reperfusion. Additionally, lipid hydroperoxides interfere with reacylatin (salvage pathway) of neuronal phospholipids.
Lipid peroxidation is a set of radical-mediated chemical chain reactions whereby the double bonds in the PUFA side chains are rearranged. In the presence of a transition metal (such as F3+), lipid peroxidation chain reactions can expand geometrically. The glia have abundant stores of oxidized F2+, mostly in ferritin and trans-ferrin. Although O2− is not itself a potent oxidizer, it does promote reduction of F2+ and F3+ release from ferritin, and iron delocalization into low-molecular-weight species is seen in the brain during reperfusion. N2O2, formed by the reaction of O2− with NO (produced by nNOS or iNOS, is implicated as the lipid peroxidation-initiated ROS during reperfusion (Tanaka et al., 1997). N2O2- mediated nitrosylation of tyrosine resides is also seen in reperfused selectively vulnerable neurons (Zaleska and Floyd, 1985).
Membrane lipids are extensively peroxidized by iron-dependent radical reactions during reperfusion (Sakamoto et al., 1991). PBN (a radical spin trap) formation adducts peak at 5 minutes of reperfusion after 10 or 20 minutes of common carotid occlusion in rats, and total PBN adducts increase with increasing ischemic time. Lipid peroxidation generates PUFA hydroperoxides and alcohols that are subsequently degraded into several aldehydic products, including malondialdehyde (Kruman et al., 1999) and 4-hydroxynonenal. Such lipid peroxidation products are seen by 15 to 30 minutes reperfusion, persist for up to 72 hours, and are associated with substantial loss of PUFAs, development of gaps in the plasmalemma, or failure of ion partitioning. These products and markers of progressive membrane damage are substantially inhibited by pharmacological iron chelators.
Neurons and terminal differentiation of reperfusion DNA repair pathways.
In very general terms, the removal of oxidative DNA lesions by the base-excision-repair pathway requires recognition and excision of lesion from the DNA double helix by endonucleases or glycosylates (Schulz et al., 1995), followed by hydrolysis mediated by AP endonucleases and clipping of the ribosephosphate moiety to generate a gap for repair synthesis using DNA polymerases. The repair process is completed by the ligase that connects new and old DNA strands. In the nucleotide-excision-repair pathway, DNA lesion-specific endonucleases excise the damaged and adjacent nucleotide, followed by repair synthesis of at least 20 nucleotides in one repair patch. However, base-excision repair generally excises the damaged base, and incorporates one nucleotide in the presence of DNA polymerase-β, but no more than 10 nucleotides in the absence of functional DNA PM-β. The repair synthesis is mediated by DNA-dependent DNA polymerase-α, -β, -δ, -ε, and -γ, with DNA polymerase-β considered the most abundant DNA polymerase in brain tissue. These DNA polymerases act on the 3′-OH end of single-stranded breaks to incorporate the correct deoxynu-cleoside triphosphates directed by the sequence in the template. In mitochondria, DNA polymerase-γ is responsible for DNA replication and repair synthesis. It is reported that DNA ligation could be dependent on the presence of poly(ADP-ribose)polymerase (PARP) because an inhibition of PARP alters the rate of repair (LaPlaca et al., 1999). Nevertheless, the mechanism of this action is unclear in brain tissue. Breaks in DNA strands after ischemic damage activate PARP and cause the depletion of nicotinamide adenindinucleotide (NAD, a substrate of PARP) and ATP. Base-excision repair is disrupted with PARP-bound DNA. However, the increase of sister-chromatid exchange in PARP-deficient cells suggests that binding of PARP to DNA is required for orderly ligation of DNA. Activation of PARP enhances necrosis but reduces apoptosis after ischemic damage (Nouspikel and Hanawalt, 2000). Thus, the participation of PARP in gene repair in the brain may be a double-edged sword. Gene-repair pathways are probably present and functional in the brain, because several proteins that are involved in all types of repair are induced by brain injury and cerebral ischemia (Liu et al., 1996). Evidence of DNA repair has been presented after cerebral ischemia (Schulz et al., 1995). Ischemia-induced oxidative DNA lesions in the transcribed strand of the c-fos gene are repaired initially more slowly than the nontranscribed strand, but almost all are repaired within 2 to 3 hours (Liu et al., 1996). The rate of oxidative DNA lesion repair in nuclear and mitochondrial DNA might be similar (Liu et al., 1996). When the burden of repair in brain tissue is increased by fivefold after 30 minutes of cerebral ischemia, the frequency of somatic cell mutations resulting from repair errors is increased from the “background” frequency of 1.5 mutations per 105 genes to 7.7 mutations per 105 genes in a IACI reporter gene in the transgenic Big Blue mouse strain (Feig and Loeb, 1993). Therefore, the frequency of mutations in somatic cells in vivo correlates positively with oxidative DNA lesion levels in the brain. Mutations are randomly distributed in this reporter gene, but approximately 80% occur at G or C bases, with C to T transitions and frame shift mutations. Errors during DNA synthesis in vitro in the presence of purified DNA polymerase-β consist mainly of one-base-deletions or insertions, which result in frame shifts (Gillardon et al., 1996). The causal role of DNA polymerase in the formation of frame shifts, however, remains to be established in injured brain tissue.
A major role of DNA repair is to ensure transcription of mRNA from intact genes. Gene transcription might be pivotal in maintaining neuronal viability after reperfusion. Gene activation is induced by oxidative stress (Hengerer et al., 1990), and the genes undergoing transcription contain oxidative DNA lesions (Liu et al., 1996). It is believed that a gene with strand breaks or cross-links is unable to serve as a template during transcription and that the cell must repair these lesions before transcription can occur. However, several investigators have reported an elevation of gene transcripts during the first few hours of reperfusion when strand breaks are detected (Liu et al., 1996). Although DNA strand breaks and cross-links inhibit transcription, modified bases change the coding properties so that most polymerases insert random nucleotides opposite the base, and that adenine nucleotide is most likely to be inserted opposite the 8-hydroxy-2-deoxy-guanosine lesion during RNA transcription (Gillardon et al., 1996). This bypass insertion could be another mechanism that generates faulty transcripts with mismatched sequences. What is the probability that a full-length mRNA be transcribed from its nuclear gene with oxidative DNA lesions? One example is the expression of AP-1, a transcription factor, which is composed of proteins encoded by c-fos and/or other immediate early genes and transactivates late effector genes that are either detrimental (Hengerer et al., 1990) or salutary to cell viability (Yang et al., 1994). Under physiologic conditions, little mRNA encoding FOS is detected in the brain. Levels increase after cerebral ischemia, probably as a result of transcription of its nuclear gene. One hypothesis is that if the nuclear genes contain oxidative DNA lesions, the newly transcribed mRNA could be faulty. This proposal is supported by the finding that it takes over 1 hour for the increase in FOS/AP-1 activity to become apparent after injury-induced increase in c-fos mRNA (Lipton, 1999). It is thought that this delay is caused by an inhibition as a result of translation blockage or a reduced global protein synthesis. This blockage coincides with the duration of detecting excessive oxidative DNA lesions in c-fos, that is, the blockage expires when the majority of oxidative DNA lesions in nuclear genes are repaired (Heiss et al., 1997).
Hyperperfusion and reperfusion
Hyperperfusion after reperfusion is a well-recognized phenomenon. It is thought to arise as a result of disturbed cerebral autoregulation. Heiss et al. (1997) published an important study on hyperperfusion observed after temporary MCAO of varying duration (30, 60, or 120 minutes) in the cat. In the 30-minutes group, all animals survived the observation period, and reperfusion was characterized by a transient reactive hyperperfusion (relative to preocclusion values) with fast CBF normalization: no ischemia or only small cerebral ischemia in the deep nuclei were found after histologic analysis. After 60 and 120 minutes of MCAO, the degree of hyper-perfusion was related to the severity of prior focal ischemia (reaching up to 300% of preocclusion values), and cortical/subcortical infarcts of varying sizes developed; approximately 50% of animals survived this duration of cerebral ischemia. The animals that eventually died of brain tissue swelling exhibited higher CBF during the hyperperfusion period than those that survived, although this difference was not statistically significant. Based on these observations, it appears that moderate hyperperfusion (reactive hyperemia) in the reperfusion phase was well tolerated when CBF increase did not exceed approximately 125% of the preischemic control values, whereas more severe periods of hyperperfusion tended to be associated with larger infarcts, brain swelling, and early death. However, because in this study the degree of hyperperfusion was clearly related to the severity of prior focal ischemia, it cannot be concluded that the more severe hyperperfusion per se was responsible for the worse outcome. Thus, it is likely that severe brain tissue swelling was a consequence of reperfusion in a severely damaged vascular bed, not just the degree of hyperperfusion. Consequently, when postischemic hyperperfusion is not severe and is of a short duration, late-stage metabolic defects, as well as the final infarction volume, are smaller (Schroeter et al., 1994). In addition, the forced reperfusion by reopening of the MCA cannot salvage already irreversibly damaged tissue but may cause additional damage by inducing brain tissue edema. This latter effect may be aggravated by severe and prolonged hyperperfusion in paralyzed vessels.
Graf et al. (1997) have further studied different patterns of postischemic hyperperfusion after transient MCAO (60 minutes) in the cat and could identify three distinct patterns of hyperperfusion based on positron emission tomography studies. Type 1 showed immediate hyperperfusion lasting for the whole observation period (24 hours). In type 2, the period of hyperperfusion was transient and often followed by hypoperfusion in regions that previously were hyperperfused. In type 3, hyperperfused regions grew progressively with time. These findings suggest that the occurrence of postischemic hyperperfusion did not contribute to worse outcome (Fig. 3).
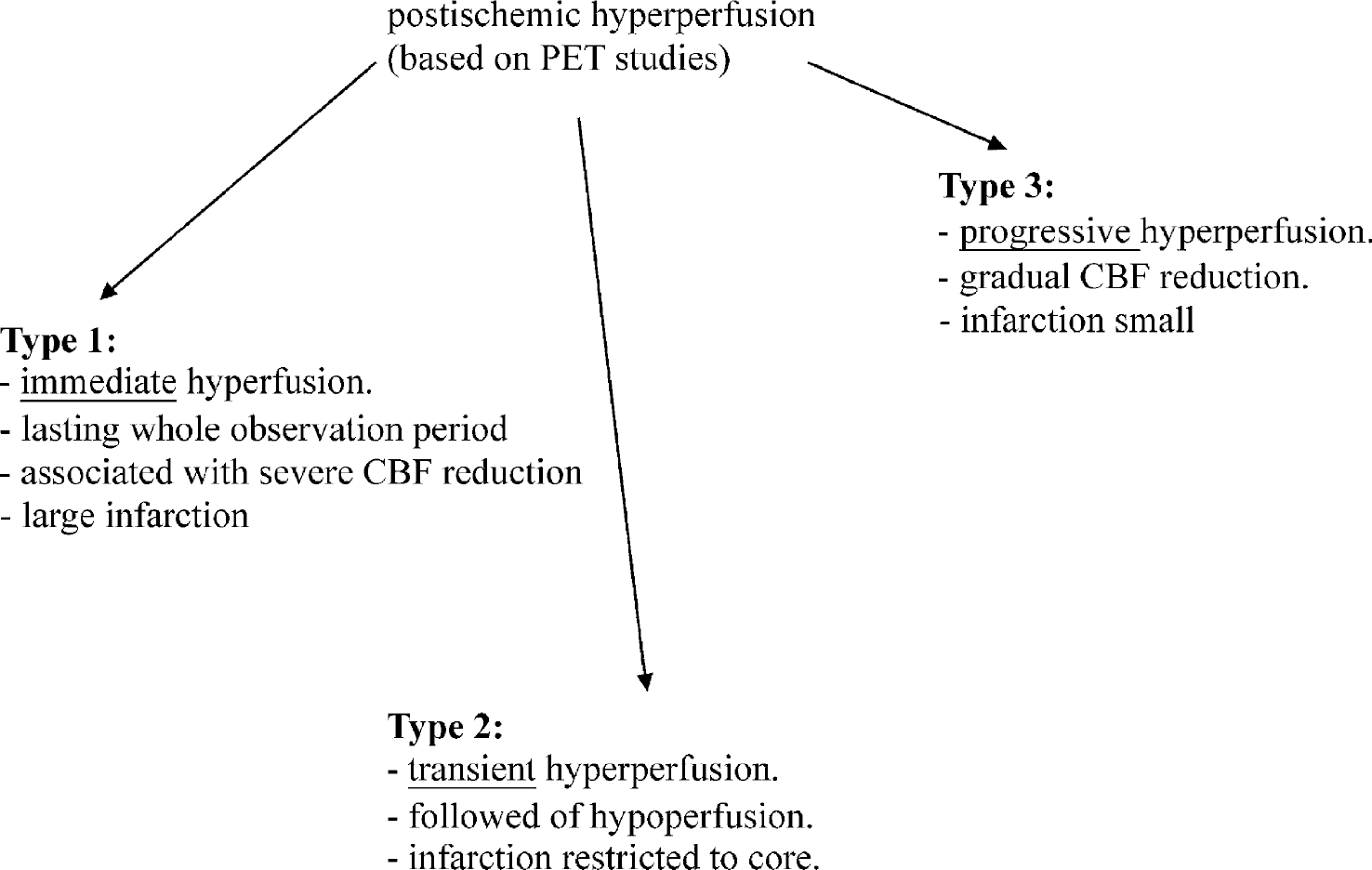
Hyperfusion and reperfusion. PET, positron emission tomography.
Cytochrome c release and reperfusion
Reperfusion induces mitochondrial respiratory dysfunction and can trigger apoptosis via mitochondrial permeability, which causes the liberation of apoptogenic factors from mitochondria. The release of cytochrome c and other mitochondrial proteins from the intermembrane space to the cytosol is commonly a critical step in the death of cells by apoptosis that can result from disruption of the outer mitochondrial membrane due to swelling associated with induction of the permeability transition pore. However, alternative mechanisms that are independent of the permeability transition pore and of mitochondrial swelling are apparently often involved (Sasaki et al., 2000). Bax and some other member of the bcl-family of proteins have been implicated in this release process. However, at 4 hours of reperfusion after 90 minutes of MCAO, cytochrome c was seen in the cytoplasm of some neurons, particularly in the striatum (Babu et al., 2000). By 24 hours of reperfusion, many cells throughout the MCA territory were affected. However, even at this time the released cytochrome c represented only a small proportion of that remaining in the mitochondrial fraction. In some of the affected cells, the staining was associated with DNA fragmentation suggesting apoptosis. However, there was not a strong association between the appearance of cytoplasmatic cytochrome c and the distribution of apoptotic cells, as seen in this and other studies.
Subsequent investigations have generally showed similar findings in permanent and transient focal ischemia in rats (Fujimura et al., 1998) and mice (Fujimura et al., 1999). Cytochrome c release was seen at earlier times in mice, probably reflecting a more rapid progression to infarction in this species. Interestingly, transgenic mice lacking one copy of the gene for the mitochondrial MnSOD were more susceptible to focal ischemia damage and exhibited greater cytochrome c release during early cerebral ischemia (Fujimura et al., 2000). Conversely, less release was seen in mice overexpressing the cytoplasmatic form of SOD (Fujimura et al., 2000). This modification is neuroprotective, further suggesting an association between the extent of damage and cytoplasmatic cytochrome c appearance. The delayed cytoplasmatic cytochrome c appearance after transient focal ischemia in cats suggests that this does not result from induction of the permeability transition pore, which, as discussed previously, is most likely to develop during the earliest stages of reperfusion. Furthermore, cytochrome c release has been reported during permanent arterial occlusion, which probably does not lead to transition pore opening.
The issue of whether the observed increased levels of cytoplasmatic cytochrome c are contributors to cell death or simply consequences of irreversible damage is not able to be resolved from the investigations reported to date. The number of cells exhibiting cytoplasmatic cytochrome c increased substantially in most studies after extended periods of ischemia or reperfusion, at times when the majority of cells are apparently already irreversibly damaged. Furthermore, this change developed in cells within the core tissue after permanent cerebral ischemia, where mechanisms other than cytoplasmic c release are likely to underlie most cell death. These findings suggest that cytoplasmic c release into the cytoplasm is probably not important for the loss of viability in many of the cells in which increased levels have been observed. It also remains possible that cytoplasmic c release triggers much of the apoptotic machinery and contributes to the death of other cells even though the conditions do not allow the development of the full apoptotic morphology. Consistent with this possibility, the activation of caspases and DNA fragmentation have been found to occur later than cytoplasmic c appearance in the cytoplasm during permanent MCAO (Fujimura et al., 1999). Losses of mitochondrial cytoplasmic c have further been shown to increase the production of ROS by mitochondria, which could also contribute to damage in some cells. Additional investigations are clearly needed to further evaluate the role of cytoplasmic c release in ischemic damage.
MECHAMISM OF CELL DEATH DURING REPERFUSION
Apoptosis
Apoptosis is a process of self-destructive cell death that involves activation of a mechanism encoded in the genomes of all higher eukaryotes. Appreciation of the importance of apoptosis in mammalian systems has only developed during the last 5 to 7 years, and some of the details of the process remain poorly understood. Nevertheless, it has become clear that the process of apoptosis is intimately involved in reperfusion and is an energy dependent process. It results from activation of cascades of detrimental biochemical events that include perturbation of Ca2+ homeostasis leading to increased excitotoxicity, malfunction of endoplasmic reticulum and mitochondria, elevation of oxidative stress causing DNA damage, alteration in proapoptotic gene expression, and activation of the effector cysteine proteases (caspases) and endonucleases leading to the final degradation of the genome. Apoptosis is initiated by oxidant stress and cytoplasmic c release (Cai and Jones, 1998). Downstream signaling and enzymatic mechanisms, including activation of caspases, are all regulated by oxidant mechanisms (Dong et al., 2000). Apoptosis depends on the availability of cellular ATP, so reperfusion could potentiate apoptosis by restoring cellular energy. Antioxidants inhibit DNA fragmentation and PARP cleavage, suggesting that ROS are responsible for these manifestations. Reperfusion itself enhances expression of CD95L (APO-1/Fas), a transmembrane protein that induces apoptosis by ligand binding and subsequent activation of caspases (Vogt et al., 1998).
Mitochondria are critical to the initiation of apoptosis in reoxygenated cells. Increased ROS production, cytoplasmic c release from mitochondria, and cell death during reperfusion are linked. Mitochondria from apoptotic cells produce increased quantities of O2− because of a switch from normal four-electron reduction of O2 to one- electron reduction after cytoplasmic c release (Dong et al., 2000). Overexpression of the antiapoptotic protein bcl-2 prevents increased O2− production associated with apoptosis. Cytoplasmic c loss from the mitochondrial matrix leads to activation of caspases, which participate in the apoptotic program. Capsase-3, −8, and −9 are activated by ischemia, possibly through Ca2+-induced activation of other proteases. Serine protease inhibitors suppress cytoplasmic c-mediated capsase-9 activation and apoptosis during reperfusion (Schaller et al., 2003).
In animal models of focal ischemia, the contribution of apoptotic death to damage has been most directly shown from the use of TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick-end labeling) staining, which detects DNA fragmentation, coupled with the identification of relevant morphologic changes including a regular pattern of chromatin condensation and apoptotic bodies. However, TUNEL will also stain cells that have undergone necrosis, and therefore does not represent a specific method to detect apoptotic cell death. Apoptotic cells defined in this way have been found scattered throughout the ischemic territory after both permanent and transient MCAO (Li et al., 1995). These cells are more apparent in the perifocal tissue and particularly in the inner border regions immediately adjacent to the ischemic core. Almost all of these apoptotic cells have been identified as neurons (Li et al., 1995). Cells exhibiting these changes have been detected within a few hours of reperfusion after focal ischemia of 1 to 2 hours but peak in number after reperfusion periods of 24 to 48 hours. A similarly delayed appearance of apoptosis is also produced with permanent occlusion (Li et al., 1995), although apparently fewer cells are affected under these conditions (Cai and Jones, 1998). Even at extended periods of reperfusion, cells exhibiting the features of apoptosis are in a substantial minority in all affected regions. A study in which the type of cell loss was defined based on morphological changes found ratios between apoptotic and necrotic cells of 1:9 in core regions and 1:6 in the inner border of the previously ischemic core (Cai and Jones, 1998).
Necrosis
Inhibition of apoptosis, because of lack of energy due to hypoxia-induced ATP depletion, may predispose neurones to necrosis and both mechanisms of cell death typically overlap (Schaller et al., 2003). DNA laddering into 180-bp fragments may occur in necrosis, although nucleosomal laddering is more characteristic of apoptosis. Multiple assays, including morphologic examination at the ultrastructural level, are required to define the actual characteristics (apoptotic, necrotic, or overlap) of cell death due to reperfusion. Cells are capable of undergoing necrosis directly after reperfusion, and necrosis is also related to increased ROS production.
POSSIBLE FUTURE THERAPEUTIC MANAGEMENT AND REPERFUSION
The previously mentioned pathophysiologic concept argues against the hypothesis of single-drug intervention for therapy for reperfusion that has characterized the research approaches of basic science laboratories, the pharmaceutical industry, and clinical trials. Antioxidants and NO synthase inhibitors can reduce the rate of membrane damage, but there is no reason to believe that they induce survival, signal transduction, inhibit the activity of cal-pain or caspase, or induce rapid recovery of translational competence. Similarly, caspase inhibitors may block the progression of apoptotic mechanisms (Fig. 4), but again there is no reason to expect that they will inhibit either lipid peroxidation or calpain activity or induce rapid recovery of translational competence. Peptide growth factors can induce early recovery of translational competence, induce survival signal-transduction, and could affect transcription for physiologic inhibitors of calpain and radical damage; however, because signal transduction, transcription, and translation take time, it is probably foolish to let calpain remain active and lipid peroxidation progress unimpeded while awaiting full expression of the molecular response to growth factor signaling. Indeed, radical stress can itself induce a 90% reduction in insulin-induced phosphorylation of ser-473 on Akt.
Antiapoptotic signaling. PCNA, proliferating cell nuclear antigen.
These considerations argue for a future possibility of a multidrug therapeutic approach that will probably have to include (1) peptide growth factors for early induction of survival signal transduction and recovery of translational competence, (2) inhibition of calpain, (3) inhibition of radical damage, and (4) caspase inhibition. Rigorous studies to establish such an approach will require evaluation of neurologic and histopathologic outcome as well as of molecular endpoints associated with each intervention component to establish the necessary role of each drug. Such a multifactorial therapeutic approach to the molecular injury mechanisms now appears essential for the development of substantially improved reperfusion.
CONCLUSIONS
Adequate oxygenation is critical to cellular viability. The paradox of reperfusion injury can be understood in terms of counteradaptive changes occurring in cerebral ischemia that predispose to cellular dysfunction, apoptosis, and necrosis during reperfusion. These changes include enhanced reduction of the mitochondrial respiratory chain, conversion of XD to XO, downregulation of antioxidant systems, increased ROS formation, and activation of deleterious signaling pathways. Specific molecular mechanisms and pathways involving ROS and NO synthase that lead to necrosis or apoptosis during reperfusion have been identified. After reperfusion, glucose and oxygen delivery are no longer limited but impairments in energy metabolism apparently persist in some tissue that will become infarcted. Ongoing decreases in mitochondrial functional capacity provide one likely contributor to the altered metabolism, especially under conditions leading to extensive brain damage.
Cell death in the brain after arterial occlusion clearly does not involve a single mechanism. In the core tissue during cerebral ischemia, the major limitations on the availability of oxygen and glucose place a more severe restriction on mitochondrial function. The resultant inability of cells to maintain the production of ATP and the associated redistribution of ions across plasma membranes does not immediately produce irreversible damage to cells. Indeed, based on animal studies, many cells seem to tolerate this state for periods up to 1 hour or even longer. Nonetheless, these derangements are incompatible with long-term cell survival and are probably the major cause of the death of cells in the ischemic core during permanent artery occlusion and reperfusion.