
Abstract
Previously, the authors cloned and characterized murine brain-specific angiogenesis inhibitor 1 (mBAI1). In this study, the authors cloned mBAI2 and analyzed its functional characteristics. Northern and Western blot analyses demonstrated a unique developmental expression pattern of mBAI2 in the brain. The expression level of mBAI2 appeared to increase as the development of the brain progressed. Reverse transcription-polymerase chain reaction (RT-PCR) analyses demonstrated the existence of alternative splice variants of mBAI2, which were defective in parts of type I repeat of thrombospondin or the third cytoplasmic loop of the seven-span transmembrane domain that were considered essential to the functions of mBAI2. The expressions of spliced variants in the brain were differently regulated compared with wild-type mBAI2 during development and ischemic conditions. In situ hybridization analyses of the brain showed the same localization of BAI2 as BAI1, such as in most neurons of cerebral cortex. In the in vivo focal cerebral ischemia model and the in vitro hypoxic cell culture model with cobalt, BAI2 expression decreased after hypoxia and preceded the increased expression of vascular endothelial growth factor (VEGF). RT-PCR analysis of antisense BAI2 cDNA-transfected SHSY5Y cells showed an increased VEGF expression as well as a decreased BAI2 expression. Immunohistochemical study of focal ischemic cortex showed that the regional localization of decreased BAI2 was related to the formation of new vessels. These results suggest that the brain-specific developmental expression pattern of angiostatic BAI2 is correlated with the decreased neovascularization in the adult brain, and that angiostatic BAI2 participates in the ischemia-induced brain angiogenesis in concert with angiogenic VEGF.
Keywords
Recently, we found that the whole encoding region of PAHX-AP1, a Refsum disease gene product-related protein, interacted with the cytoplasmic region of mBAI1 through yeast two-hybrid screening, and isolated the murine homologue of human BAI1 (hBAI1) (Koh et al., 2001). The hBAI1 was isolated as a novel brain-specific gene by identifying genomic DNA fragments containing functional p53 binding sites (Nishimori et al., 1997; Tokino et al., 1994). A seven-span transmembrane region (STR) and two functional elements, a RGD motif and thrombospondin type 1 repeats (TSRs), were well conserved between hBAI1 and mBAI1 (Koh et al., 2001). The TSR can inhibit experimental angiogenesis induced by bFGF in the rat cornea (Tolsma et al., 1993), and have been observed in several proteins involved in the guidance of nerve growth cones and axonal growth, such as UNC-5 and F-spondin (Klar et al., 1992; Leung-Hagesteijn et al., 1992).
Two novel human genes homologous to hBAI1 have been identified and designated as hBAI2 and hBAI3. Analysis of their predicted protein showed that TSR and STR were well conserved among the three BAI genes. Like hBAI1, these two genes were specifically expressed in the brain, suggesting that the three hBAIs are closely related. However, the extracellular and cytoplasmic domains are relatively different among the three proteins (Shiratsuchi et al., 1997).
In this study, we cloned murine homologue of hBAI2 (mBAI2, 4.7 kb) by screening the mouse brain cDNA library using an hBAI2 cDNA fragment as a probe. The mBAI2 expression was ubiquitous in the embryonic tissues, but it was targeted to the brain in adulthood. As with mBAI1, the peak level of mBAI2 in the brain was observed 10 days after birth. This expression pattern of angiostatic BAI1 and BAI2 may be one of the causes resulting in decreased new vessel formation in the adult brain. In situ hybridization analyses of the brain showed the same localization of BAI2 as BAI1. Reverse transcription-polymerase chain reaction (RT-PCR) analyses demonstrated the existence of alternative splicing of mBAI2 in the N- and C-terminal region, which was not observed in mBAI1.
To date, the functions of neuron-specific BAI2 in the brain are unknown. Our previous study showed that expression of BAI1 decreased on the ischemic side in the rat focal cerebral ischemia injury model produced by the occlusion of the middle cerebral artery (Koh et al., 2001). We attempted to find out whether BAI2 is involved in angiogenesis after focal cerebral ischemia in rats, and investigated the hypothesis that hypoxia induces angiogenesis through regulated expression of angiostatic and angiogenic genes to counteract the neurologic problems associated with ischemia. The BAI2 expression decreased in the ischemic cerebral cortex earlier than that of BAI1. During postischemic angiogenesis, increased vascular endothelial growth factor (VEGF) expression appeared following the decreased BAI2 expression. The time sequence of these angiostatic and angiogenic gene expressions in ischemic cerebral cortex was supported by results of the in vitro hypoxia-mimic human neuronal cell culture model, in which cobalt was given. The findings that increased VEGF expression observed in the antisense BAI2 cDNA-transfected cells and decreased BAI2 expression related to neovascularization after focal ischemia indicated the potential negative influence of angiostatic BAI2 on angiogenic VEGF expression.
Our findings demonstrate that cerebral ischemia could trigger hypoxia-sensing and adaptive downstream molecular responses in central neurons through hypoxia-sensitive suppression of brain-specific angiostatic genes, such as BAI2 and BAI1, and through stimulation of angiogenic factors, such as VEGF.
MATERIALS AND METHODS
Isolation of murine BAI2 cDNA
A murine cDNA spanning nucleotides 4,242 through 5,003 was generated by RT-PCR using oligonucleotides based on the human sequence (Shiratsuchi et al., 1997). Two hundred nanograms of total mouse brain RNA was used, and the human sense and antisense primers used were 5' -AATATCCTGGTG-CCCATGGCAGCC-3' and 5'-AGCGAGCCCAGTGTCATA-GATTTG-3', respectively. The resulting 762-bp product was subcloned into the TA vector cloning system (Invitrogen, Carlsbad, CA, U.S.A.), and the identity of the cDNA was confirmed by sequence analysis. The Genbank BLAST homology-searching program was used to search for this cDNA sequence. The cDNA insert corresponded to the cytoplasmic region of mBAI2, and this murine cDNA fragment was then used to screen the mouse brain Λ ZAP II cDNA library to finally get the full-length cDNA of mBAI2. Also, this unique cDNA fragment of mBAI2 was used as a probe for Northern blot analyses, to make GST-fusion protein for production of anti-mBAI2 antibody, and as an antisense probe for in situ hybridization. The nucleotide sequence of mouse BAI2 has been submitted to GenBank.
RNA isolation and Northern blot analyses
To isolate total RNA, mouse (BALB/c) or rat (Sprague Dawley) tissues were homogenized with a polytron homogenizer in 4.0 mol/L guanidine thiocyanate, 1% β-mercaptoethanol, and RNA purified by centrifugation through 5.7 mol/L CsCl as described elsewhere (Kim et al., 1994). RNA samples were quantitated by spectrophotometry at 260 nm. For Northern blot analysis, total RNA (10 μg) was denatured with glyoxal, separated by size on 1.0% agarose gels, and transferred to Gene-screen (Dupont). Probes (mBAI2, nucleotide residues 4,242–5,003; mBAI1, nucleotide residues 4, 170–4, 958; human VEGF, nucleotide residues 171–639; mouse HIF-1α, aa residues 720–826) were radiolabeled by a nick translation method, and hybridization and signal visualizations were performed as described elsewhere (Kim et al., 1994). In all experiments, the integrity of the RNA samples was established by Northern blot analysis with a mouse β-actin probe or rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) probe.
Reverse transcription-polymerase chain reaction analysis
Total RNAs were prepared from normal or ischemic brain tissues or cultured cells. Reverse transcription was performed at 42°C for 60 minutes (200 ng total RNA; 100 pmol random primers; reverse transcriptase; Invitrogen). The RT-PCR exponential phase was determined on 30 to 35 cycles to allow quantitative comparisons among the cDNAs developed from identical reactions. All reactions involved an initial denaturation at 94°C for 5 minutes followed by 30 or 35 cycles at 94°C for 1 minute, exposure to an appropriate annealing temperature for 1 minute, and at 72°C for 2 minutes, on a Mastercycler Personal PCR system (Eppendorf, Germany). After making the first strand cDNA by reverse transcription, control PCR was performed on 25 to 27 cycles to allow quantitative comparisons among the cDNAs developed from identical reactions with primers for the housekeeping gene, such as β-actin or GAPDH. After confirming the same quantity of GAPDH or β-actin bands among them by analyzing the samples on agarose gels, main PCR reactions were processed using specific primers for desired genes. The annealing temperature was 60°C for mBAI2, GAPDH, and β-actin, and 62 °C for hBAI1, hBAI2, VEGF, and HIF-1α. The amplification products were analyzed on agarose gels and visualized by ultraviolet epifluorescence following ethidium bromide staining. The PCR primers were as follows: S1, 5'-GATGATATCGCCGCGCTCGTC-3' and AS1, 5'-AGCCAGGTCCAGACGCAGGAT-3' for human, rat, and mouse β-actin; S1, 5'-TATGACAACTCCCTCAAGAT-3' and AS1, 5-AGATCCACAACGGATACATT-3' for human, rat, and mouse GAPDH; S1, 5'-GAGCAGCAATGACCTGTTC-3' and AS1, 5'-CCATGTCATCAACATCACGTAC-3' for TSR of mBAI2; S1, 5'-GCTCATCGGGATTATCGTC-3' and AS1, 5'-GGCAATAGAGTGCTTGAAGAG-3' for the third loop of STR in mBAI2; S1, 5'-ATGACCGACTTCGAGAAGGACG-3' and AS1, 5'-TCTGCGGCATC-TGGTCAATGTG-3' for hBAI1; S1, 5'-GTGTCCAGCCTTCCATGAGATG-3' and AS1, 5'-TTTCCGCATCCACCATGAAGC-3' for hBAI2; S1, 5'-TGCCATCC-AATCGAGACCCTG-3' and AS1, 5'-ATC-TGGTTCCCGAAACGCTGAG-3' for human VEGF; S1, 5'-AAGCCCTGAAAGCGCAAGTCC-3' and AS1, 5'-TGCAT-GATCGTCTGGCTGCTG-3' for human HIF-1α, respectively.
Production of the anti-mBAI2 antibody and Western blot analysis
The GST-mBAI2 fusion construct was prepared by amplifying the nucleotide residues 4,242 to 5,003 (this region is unique for mBAI2) of murine BAI2 into the unique BamH I and EcoR I sites of pGEX-2T, and was purified as previously described (Koh et al., 2001). Rabbit polyclonal antiserum recognizing mBAI2 was prepared using the GST-mBAI2 fusion protein. The serum recognizing mBAI2 was filtered through a passage to the column with GST-mBAI2 fusion protein, and that column was eluted with a low-pH buffer to obtain the anti-GST-mBAI2 antibody. It was filtered again through a passage to the column with only GST protein to remove the anti-GST antibody component as best as possible. Cell lysates were prepared from mouse tissues or ischemic cerebral cortex using a lysis buffer containing 1% Triton X-100, and resolved by SDS-PAGE. Resolved proteins were transferred to a nitrocellulose membrane and blotted with anti-BAI2 serum and anti-rabbit Ig-HRP (Amersham) as previously described (Lee et al., 2000). The blot was reprobed with antiactin antibody (Sigma) to control for loading.
In situ hybridization
Sprague-Dawley rats (200 to 230 g) were anesthetized with an intraperitoneal injection of sodium pentobarbital (50 mg/kg), and brains were fixed by in vivo perfusion of the abdominal aorta with 4% paraformaldehyde in a phosphate-buffered saline (PBS) for 10 minutes. Brains were excised and immersed in the same fixative for 3 hours at 4°C. The blocks were washed in PBS, dehydrated in a graded series of ethanol washes, and embedded in paraffin. Tissue sections were cut at 6 μm and mounted on gelatin-coated glass slides. Sense and antisense probes specific for the BAI2 were generated from the recombinant molecules (nucleotide residues 3,829–4,065) using T3 and T7 RNA polymerases in the presence of digoxigenin-11-UTP (Roche Molecular Biochemicals, Indianapolis, IN, U.S.A.). In situ hybridization was performed as described previously (Koh et al., 2001). Briefly, the tissue sections were deproteinated and acetylated. Prehybridization was conducted at 48°C for 4 hours in a humidified chamber. The slides were then hybridized with 20-ng/μL digoxigenin-11-UTP-labeled riboprobe in a hybridization buffer at 48°C for 14 to 16 hours. Hybridizations with the sense probes were performed parallel to the antisense probes on alternate sections. Unbound probe was removed by sequential washes of SSC with or without 20-μg/mL ribonuclease. RNA-RNA hybrids were immunodetected with a 1:500 dilution of antidigoxigenin alkaline phosphatase conjugate (Roche), followed by incubation with nitro blue tetrazolium salt and 5-bromo-4-chloro-3-indolyl phosphate. After mounting in a crystal mount medium, the sections were photographed on a light photomicroscope (Vanox-S, Olympus, Tokyo, Japan).
The rat focal ischemia model: occlusion of the middle cerebral artery
Anesthesia was induced with 4% halothane in an anesthetic chamber and maintained with 1% halothane in 100% O2 using a rodent mask (Stoelting). Under anesthesia, the rat was placed in the supine position and a midline cervical incision was made. The right common carotid artery (CCA) was exposed and the external carotid artery (ECA) was transected 2 mm distal from the carotid bifurcation after being ligated by 4–0 silk suture. The internal carotid artery (ICA) was then isolated. The CCA and ICA were occluded with microvascular clips. A 3-cm length of 4–0 monofilament suture with a slightly enlarged tip was introduced into a hole in the ECA and the microvascular clip in the ICA was removed. The suture was then gently advanced about 18 mm into the ICA and circle of Willis to cross the opening of the middle cerebral artery and the operative wound was closed. Rectal temperature was monitored and maintained at 37°C ± 0.5°C with a homeothermic blanket control unit (Harvard Apparatus, U.K.) during the surgical procedure and during recovery from the anesthesia. Rats were killed after the ischemic period (30 minutes or 1, 2, 4, 8, or 24 hours) and brains were exposed. The brain slices were stained with 2% triphenyl tetrazolium chloride to visualize and measure the infarct volumes in each group (Koh et al., 2001). The ischemic portion of the cerebral cortex due to the occlusion of the middle cerebral artery and the contralateral portion of the normal cerebral cortex were removed for total RNA preparation.
Cell culture and cobalt chloride treatment
The dose-dependent hypoxia-mimic effect of cobalt chloride was confirmed by incubating SHSY5Y neuroblastoma cells with increasing doses of cobalt chloride (100, 200, and 400 μmol/L) as described previously (Sandner et al., 1997), and by observing the cobalt dosage-dependent increase in VEGF expression through Northern blot analysis (data not shown). A 200-μmol/L concentration of cobalt chloride was chosen for this experiment. The SHSY5Y neuroblastoma cell was grown in DMEM supplemented with 10% FBS, and cobalt chloride treatment was performed by incubating SHSY5Y cells with cobalt chloride (200 μmol/L). Fresh culture medium was added at the beginning of incubation. After the cobalt chloride treatment period (30 minutes or 1, 2, 4, 8, or 24 hours), cells were harvested and RNA extractions were performed immediately.
Construction of antisense BAI2 plasmid and transfection
The pREP4–anti-BAI2 plasmid was made by inserting the whole encoding region of the BAI2 inversely into the pREP4 vector (Invitrogen), which has an RSV promoter. For stable transfection of the SHSY5Y neuroblastoma cells, 5 × 10 cells in a 75-cm2 flask were transfected by the calcium phosphate method with 10 μg pREP4–anti-BAI2 plasmid. Colonies surviving in the hygromycin-containing medium were isolated. RT-PCR and Western blot analyses were performed to choose the clone that expressed low levels of BAI2.
Immunohistochemistry
Immunohistochemistry was performed using an immunoperoxidase procedure (VECTA ABC Kit, Vector Laboratories). The brain tissue sections were deparaffinized, rehydrated, rinsed, and treated with 3% H2O2 in 60% methanol for 15 minutes to quench endogenous peroxidase activity. After washing, the sections were blocked in PBS containing 5% normal goat or horse serum for 1 hour. The sections were incubated for 12 to 14 hours with the antibodies for the BAI2, CD34 (DAKO) diluted in PBS with 0.3% bovine serum albumin. For a negative control, the sections were incubated in PBS containing only 5% normal goat or horse serum. For a positive control for the CD34, human benign prostate hypertrophy specimen was used. The sections were then rinsed, and were incubated sequentially for 30 minutes each with the biotinylated secondary antibody and the ABC reagents, followed by a 5-minute incubation with the DAB. Sections were counterstained with Mayer hematoxylin and photographed on a light microscope.
Cloning of the murine BAI2 cDNA
A set of oligonucleotide primers capable of amplifying the unique cytoplasmic region of the human BAI2 transcript was used to amplify the corresponding region of the murine BAI2 mRNA. The resulting 762-bp amplification product was subcloned and sequenced to confirm its identity (nucleotide residues 4,242 to 5,003 of mBAI2). This fragment was then used to screen a mouse brain Λ ZAP II cDNA library and several positive clones were obtained, and database surveys identified a high degree of the deduced amino acid (aa) sequence identity between one of the positive clones (#60) and hBAI2. This murine cDNA fragment was then used to rescreen the mouse brain cDNA library, and several overlapping clones (#76, #77, #79, and #81) were isolated (data not shown). Clone 76 had start codon and clone 81 had a stop codon. All of the overlapping clones spanned a total of 4,694 bp.
Sequence analysis of the cDNA identified an open reading frame that could direct the synthesis of a protein of 1,560 aas, of which the relative molecular mass was 170 kd. The termination codon of the open reading frame was located at nucleotides 4,682 to 4,684. Database surveys identified a high degree of the deduced aa sequence identity (96%, Fig. 1) over the full length of the molecule between our cloned gene product and hBAI2. Human BAI2 protein has another 12 aas (1,572 aa), especially in the cytoplasmic tail region (1,499–1,515 aa) compared with mouse BAI2. Based on this striking degree of homology, we have tentatively identified our cloned gene product as the murine BAI2. The mBAI2 and mBAI1 proteins have sequences that are 40% identical. mBAI2 has four TSRs, while mBAI1 contains five. mBAI2 was devoid of the first TSR of mBAI1, though its functional significance was unknown. Interestingly, clone 77 was devoid of the first and second TSR of mBAI2. Like mBAI1, mBAI2 contained STR, but clone 79 was deleted in 33 aas between the fifth and sixth STR. This region corresponded to the third cytoplasmic loop of STR, and is important for the interaction of G protein in the receptors coupled to G proteins (Stefan and Blumer, 1994). However, this third loop is much shorter in mBAI1 than in mBAI2, and the size of this loop of mBAI1 is the same as that of the spliced variant of mBAI2. These alternative splicings were confirmed by RT-PCR (Fig. 4). The deduced aa sequences of the mBAI2, hBAI2, and mBAI1 genes are shown in Fig. 1. TSR in the extended extracellular domain and STR were located at the same positions and were highly conserved between them. However, the cytoplasmic region (1,311–1,416 aa) of mBAI2 was almost divergent between the mBAI2 and mBAI1 genes. This finding indicates that BAI-interacting proteins, which bind to this cytoplasmic region of BAI1 and BAI2, might be different between the mBAI2 and mBAI1 genes.

Comparison of deduced aa sequences of the mouse BAI2 (mBAI2) with human BAI2 (hBAI2) and mouse BAI1 (mBAI1). The hBAI2 and mBAI1 are shown below the mouse sequence; the identical aas among them are indicated in black, and gaps introduced for maximal alignment are marked with dashes. mBAI2 is a 1,560-aa protein with a deduced molecular mass of 170 kd. The aas in the thrombospondin type I repeats (TSR) and seven-span transmembrane regions (STR) are well conserved among them.
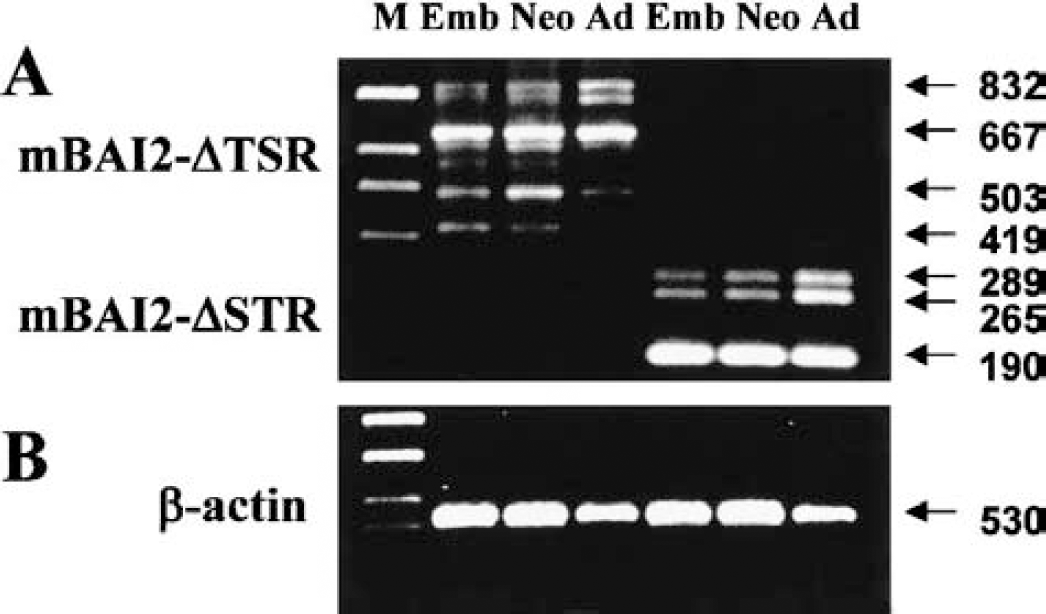
RT-PCR analyses of developmental expression of alternatively spliced variants of mBAI2 in the brain.
Brain-specific expression pattern of mBAI2
A Northern blot tissue survey was performed using a cDNA probe spanning nucleotides 4,242 through 5,003 to ascertain the developmental pattern of mBAI2 expression. A ≈60 transcript was observed in most tissues from embryonic-day-18 mice. A particularly high level was present in the brain and a moderate level was found in the skin, kidney, skeletal muscle, and thymus (Fig. 2A, upper panel). By neonatal day 1, the pattern of mBAI2 expression had dramatically changed. Expression in the brain was still increased, whereas expressions in other tissues were acutely downregulated (Fig. 2B, upper panel). Most adult (3 months old) tissues expressed little or none of the 6.0-kb transcript, although a very high level was detected in the brain (Fig. 2C, upper panel). The blots were rehybridized with an β-actin probe to confirm the fidelity of the samples (Figs. 2A–2C, lower panel). These results indicate that the mRNAs encoding mBAI2 are the most abundant in brain regardless of developmental stage, and are downregulated in other tissues. It is of interest to note that mBAI2 and mBAI1 genes showed the same brain-specific expression pattern.
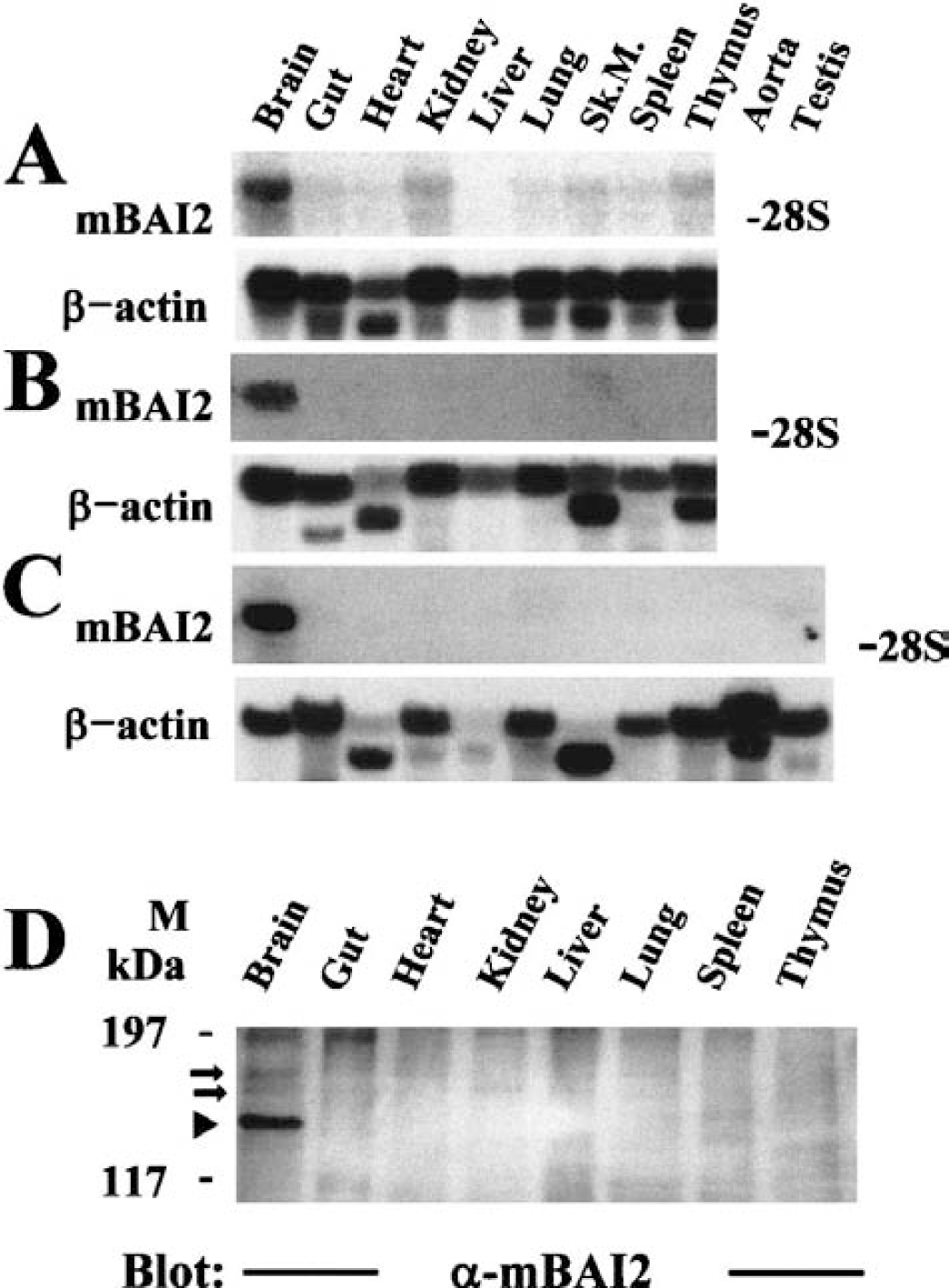
Northern and Western blot analyses of mBAI2 expression in various mouse tissues. Total RNA isolated from various tissues of embryonal (
To monitor the brain-specific expression pattern of mBAI2, Western blot analysis of tissue distribution was performed using a polyclonal anti-mBAI2 antibody raised against aas 1,277 to 1,529 of mBAI2 fused to GST. This region is localized in the cytoplasmic portion of mBAI2 and is unique to mBAI2. As shown in Fig. 2D, only the brain expressed the BAI2 protein that appeared as a =175-kd band, but there were other lower bands (=168 and =155 kd) in the brain recognized by a polyclonal anti-mBAI2 antibody. These bands may result from alternatively spliced variants of mBAI2, which were devoid of the first and/or second TSR (=168-kd band) and of the third loop of STR (=155-kd band). However, these spliced transcripts were not observed in the Northern blot assays because the differences of molecular size between the wild-type BAI2 and variants were relatively small, and the Northern message of mBAI2 was a little thick.
Developmental-specific expression pattern of mBAI2 in the brain
To further characterize the developmental expression pattern of mBAI2 in the brain, RNAs prepared from brains of embryonic-day-18, early postnatal period (first and third day after birth), before and after the eyelid opening (neonatal day 10, 15, 22, and 29), 8-week, and 3-month-old rats were hybridized with cDNA probes from mBAI2, mBAI1, and β-actin genes. The expression level of mBAI2 increased after birth and reached its highest level before eyelid opening (10 days old, Fig. 3A). However, the mBAI2 expression was slightly decreased after eyelid opening, and this level was maintained through adult life. Thus, the expression of mBAI2 showed the same pattern as mBAI1 (Fig. 3B). High levels of β-actin in the embryonic and early neonatal brains reflect the high cellular activities in these periods (Fig. 3C).
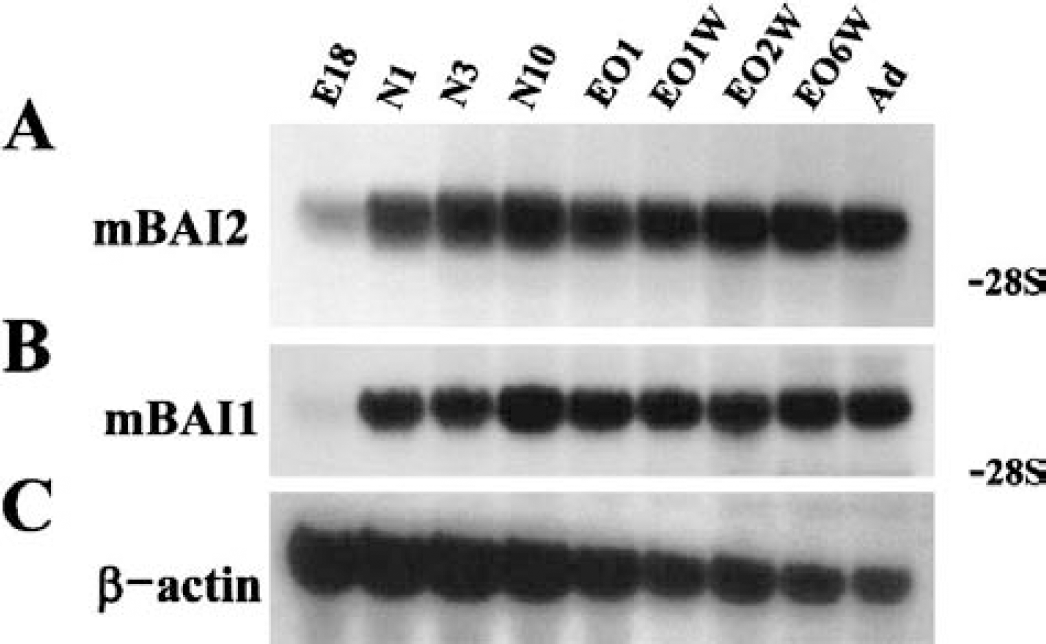
Northern blot analyses of the developmental pattern of mBAI2 in the rat brain. Total brain RNA from various stages of development was hybridized with an mBAI2 cDNA probe
Reverse transcription-polymerase chain reaction analyses of the developmental expression of alternatively spliced variants of mBAI2 in the brain
During screening of mBAI2, we obtained several in-frame alternatively spliced cDNA clones containing deleted TSRs or the third loop of STR. To determine whether the sequence heterogeneity observed in the cDNA clones could also be detected in steady-state mRNA populations, and whether wild-type BAI2 and these spliced variants were changed during brain development, we performed RT-PCR analyses in mouse brain using specific primers for TSRs or STR. Figure 4 and Table 1 show several alternative splicings of regions involving TSRs and STR. RT-PCR analyses of adult brain RNA using primers flanking the TSRs produced a 832-bp amplification product corresponding to the nondeleted sequence observed in cDNA clone 76, as well as a prominent 503-bp product corresponding to the deleted sequence observed in cDNA clone 77 (Fig. 4A, Table 1). There were other prominent 667-bp products corresponding to the first-TSR- or second-TSR-deleted sequence that were not observed in the cDNA clone (Fig. 4A, Table 1). Also, RT-PCR analyses of adult brain RNA using primers flanking the third loop of STR produced a 289-bp amplification product corresponding to the nondeleted sequence observed in cDNA clone 81, as well as a prominent 190-bp product corresponding to the deleted sequence observed in cDNA clone 79 (Fig. 4A, Table 1). There was another amplification product that did not have eight aas in this third loop of STR (265-bp product) that was not observed in cDNA clone and mBAI1. Also, through RT-PCR analyses using primers flanking the whole STR, we found another spliced product that did not have 66 aas of STR resulting in deletion of the third loop and sixth transmembrane region (Fig. 4A, Table 1). The identity of these RT-PCR products was confirmed by sequence analysis, and the deduced aa sequences encoded by these products are shown in Table 1. The location and comparison of deleted aas in these spliced variants of mBAI2 are depicted in Figure 5.
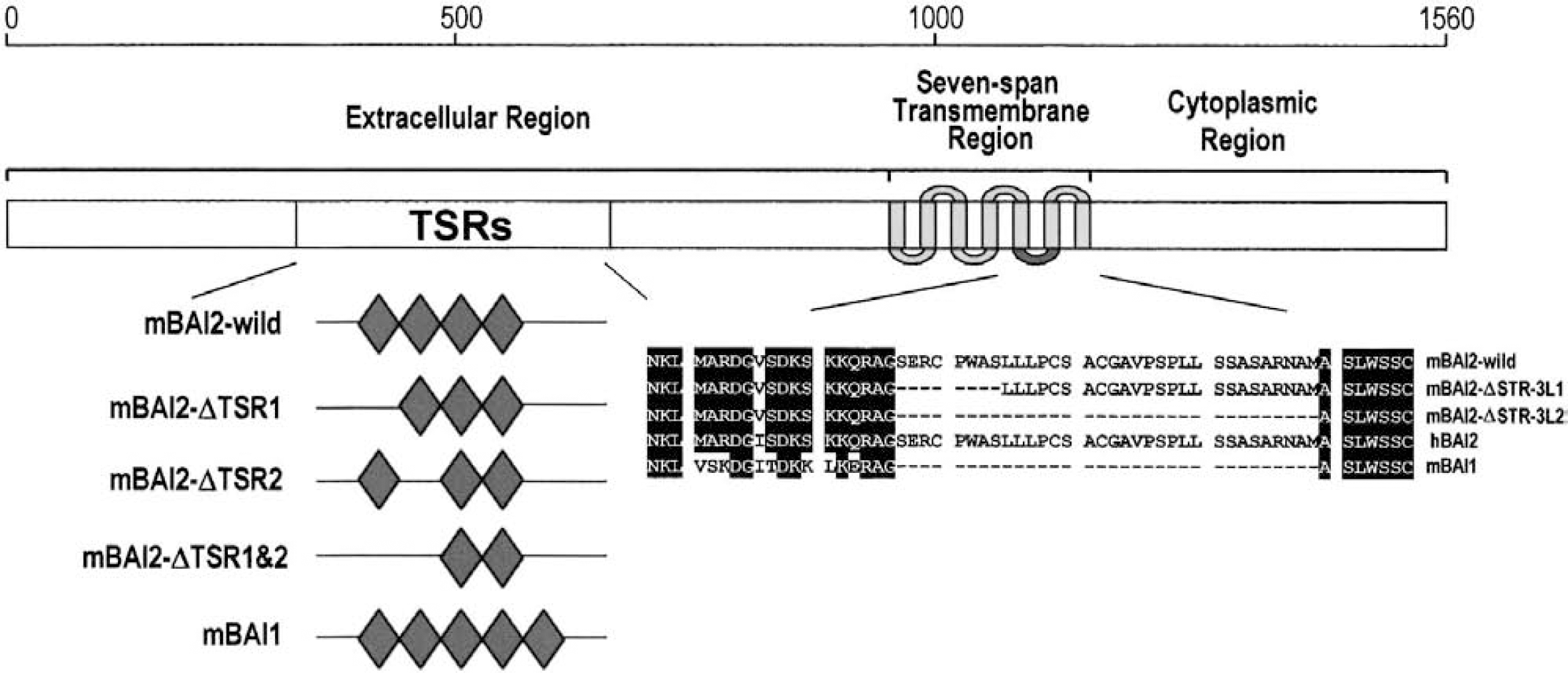
Schematic representation of the molecular structure of wild-type mBAI2 and alternatively spliced variants, which had TSR- or STR-deleted sequences, and comparison of TSR or aa sequences in the corresponding sites of hBAI2 and mBAI1. mBAI2-wild indicates mBAI2 that has a nondeleted sequence in the TSR or STR. mBAI2-ΔTSR1 indicates mBAI2 that has a first-TSR-deleted sequence. mBAI2-ΔTSR2 indicates mBAI2 that has second-TSR-deleted sequence. mBAI2-ΔTSR1&2 indicates mBAI2 that has first-TSR– and second-TSR-deleted sequences. mBAI2-ΔSTR-3L-1 indicates mBAI2 that did not have eight aas in the third loop of the seven-span transmembrane region. mBAI2-ΔSTR-3L-2 indicates mBAI2 that did not have the third loop of the seven-span transmembrane region.
Comparison of deleted amino acids among wild-type and spliced variants of mBAI2
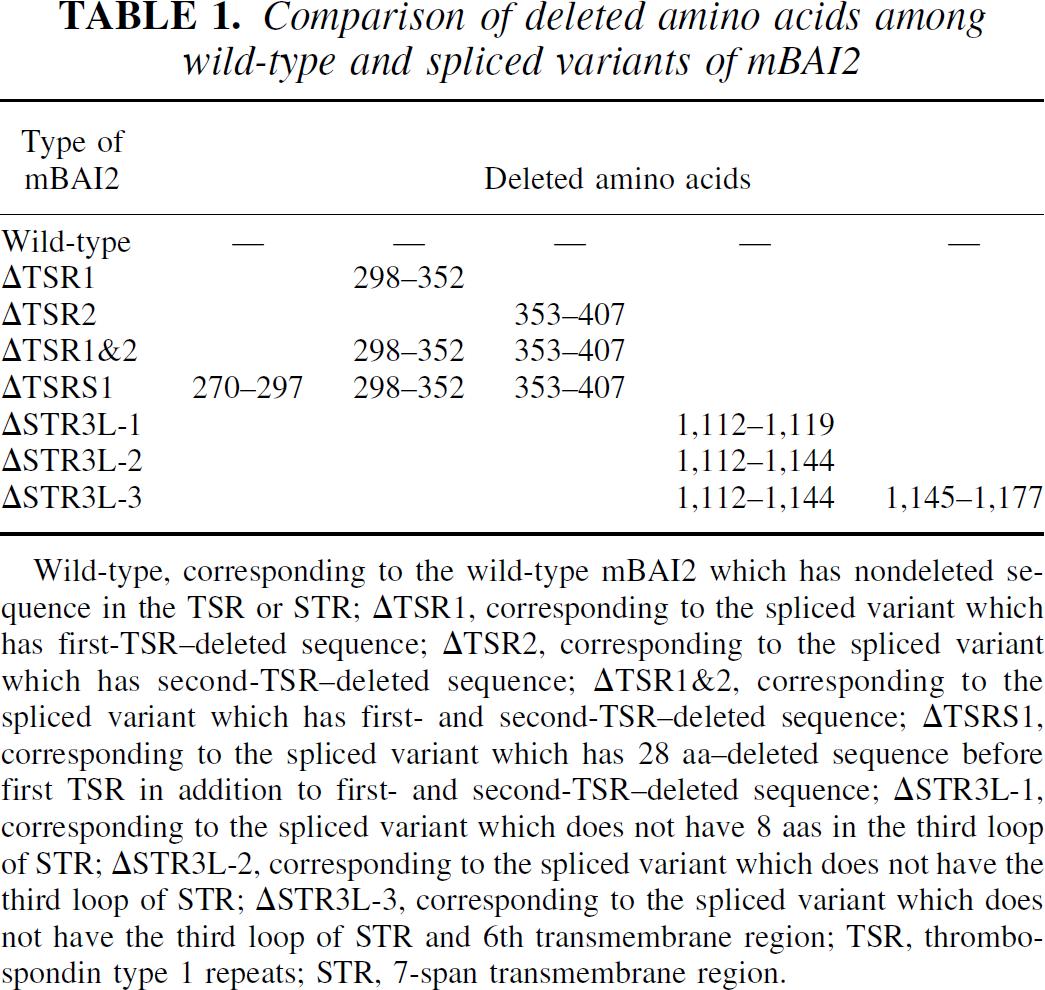
Wild-type, corresponding to the wild-type mBAI2 which has nondeleted sequence in the TSR or STR; ΔTSR1, corresponding to the spliced variant which has first-TSR-deleted sequence; ΔTSR2, corresponding to the spliced variant which has second-TSR-deleted sequence; ΔTSR1&2, corresponding to the spliced variant which has first- and second-TSR-deleted sequence; ΔTSRS1, corresponding to the spliced variant which has 28 aa-deleted sequence before first TSR in addition to first- and second-TSR-deleted sequence; ΔSTR3L-1, corresponding to the spliced variant which does not have 8 aas in the third loop of STR; ΔSTR3L-2, corresponding to the spliced variant which does not have the third loop of STR; ΔSTR3L-3, corresponding to the spliced variant which does not have the third loop of STR and 6th transmembrane region; TSR, thrombospondin type 1 repeats; STR, 7-span transmembrane region.
The wild-type BAI2, which has intact TSR or STR, developmentally increased from embryonic to adult brain. However, spliced variants, which did not have first or second TSR or the third cytoplasmic loop, showed no change in expression during brain development. The spliced variant, which did not have first and second TSR, increased in the neonatal brain compared to the embryonic or adult brain. However, the spliced variant that did not have 28 aas before the first TSR in addition to the deletion of the first and second TSR sequence was downregulated in the adult brain compared to embryonic or neonatal brain. The spliced variant, which did not have part (eight aas) of the third cytoplasmic loop, increased more in the adult brain than in the embryonic or neonatal brain. Slightly high levels of β-actin band in the embryonic and neonatal brains (Fig. 4B) reflect the high cellular activities in these periods (Fig. 3C). These results suggest that alternative splicing of the mBAI2 product can produce several proteins lacking parts of TSR or the third loop of STR, and that expression of these variants in the brain is under developmental regulation.
Localization of BAI2 mRNA in the brain
In situ hybridization with an antisense riboprobe spanning mBAI2-specific nucleotides 3,829 through 4,065 was used to determine the expression pattern of the BAI2 in the rat brain. BAI2 was expressed throughout most neurons of the whole cerebral cortex, but a high level was present in layer II to III, as was BAI1 (Figs. 6 and 7A). It was also present in high levels in the pyramidal neurons of all fields of the hippocampus, and in the granule cell and polymorphic layers of dentate gyrus (Figs. 6 and 7B), medial and lateral septum (Fig. 7C), and medial and lateral habenular nucleus (Fig. 7D). In the cerebellum, the BAI2 signal was most abundant in the Purkinje cell layer (Fig. 7E) and in lateral and interposed deep cerebellar nuclei (Fig. 7F), but diffuse weak and very weak signals were observed in the granular and molecular layers, respectively (Fig. 7E). Most neurons of the red nucleus showed intense hybridization signals (Fig. 7G).
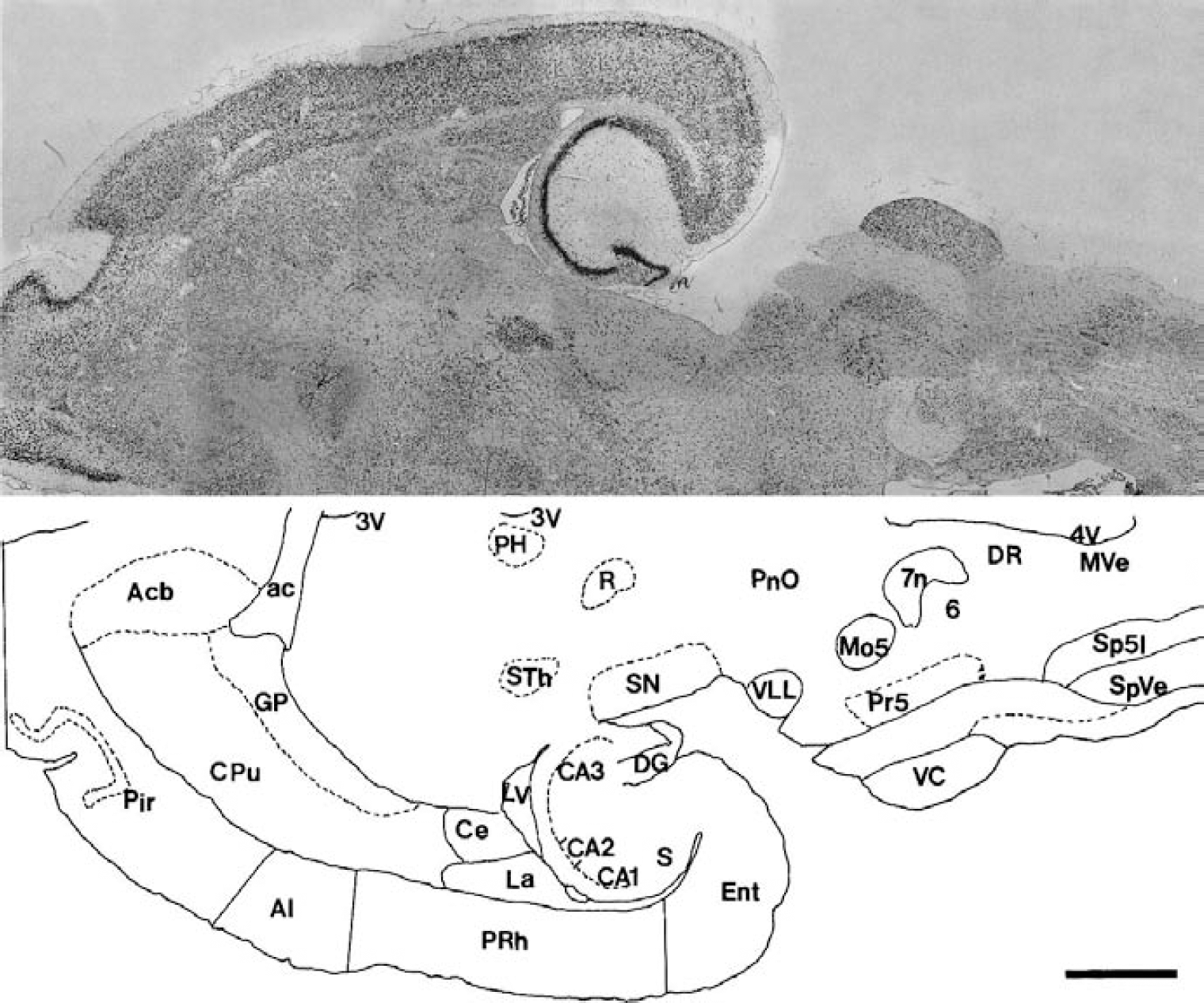
In situ hybridization localization of BAI2 mRNA (upper panel) and the camera lucida drawing (lower panel) of labeled structures in the adult rat brain stem. Digoxigenin-labeled antisense BAI2 probe was hybridized under high-stringency conditions to the horizontal section of brain. 3V, third ventricle; 4V, fourth ventricle; 6, abducent nucleus; 7n, facial nerve; ac, anterior commissure; Acb, accumbens nucleus; AI, agranular insular cortex; CA1, CA2, CA3; CA1, CA2, CA3 field of the Ammon horn; Ce, central amygdaloid nucleus; CPu, caudate putamen; DG, dentate gyrus; DR, dorsal raphe nucleus; Ent, entorhinal cortex; GP, globus pallidus; La, lateral amygdaloid nucleus; LV, lateral ventricle; Mo5, motor trigeminal nucleus; MVe, medial vestibular nucleus; PH, posterior hypothalamic area; Pir, piriform cortex; PnO, oral pontine reticular nucleus; Pr5, principal sensory trigeminal nucleus; PRh, perirhinal cortex; R, red nucleus; S, subiculum; SN, substantia nigra; Sp5I, interpolar part of the spinal trigeminal nucleus; SpVe, spinal vestibular nucleus; STh, subthalamic nucleus; VC, ventral cochlear nucleus; VLL, ventral nucleus of lateral lemniscus. Scale bar = 3 mm.
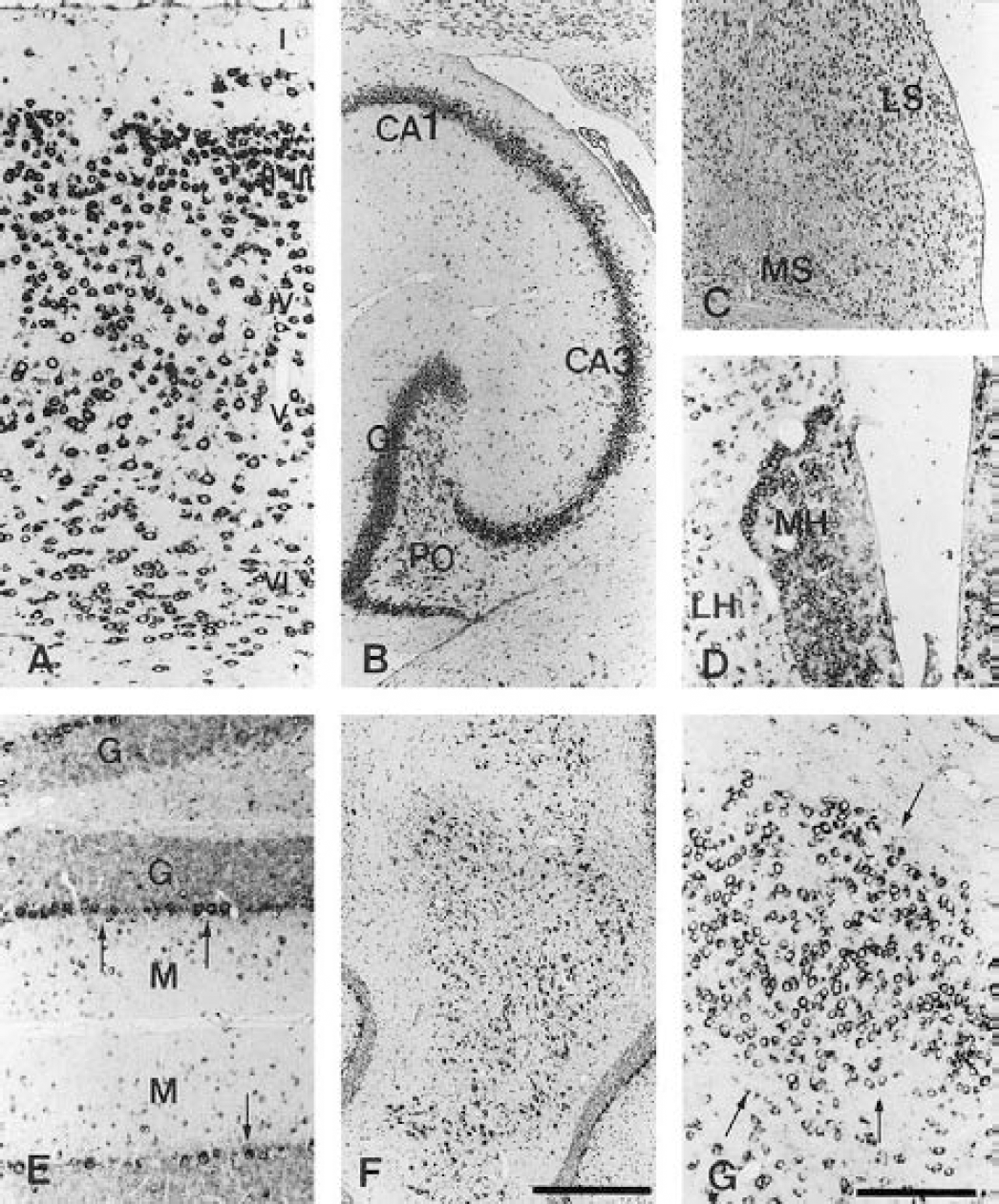
In situ hybridization localization of BAI2 mRNA in several regions of adult rat brain.
BAI2 was also expressed in several nuclei of the brain stem (Fig. 6). It was highly expressed in the trigeminal motor and sensory (Figs. 8A–8C), cochlear (Fig. 8D), hypoglossal (Fig. 8E), and dorsal raphe nuclei (Fig. 8F). It was also expressed in the dorsal medullary reticular formation, pontine reticular nucleus, and vestibular nucleus. In the retina, BAI2 was highly expressed in the ganglion cell layer, as well as in the inner and outer nuclear layers (data not shown). These observations indicate that BAI2 is a neuron-specific protein like BAI1, and that the localization of BAI2 expression in the brain coincided with that of BAI1.
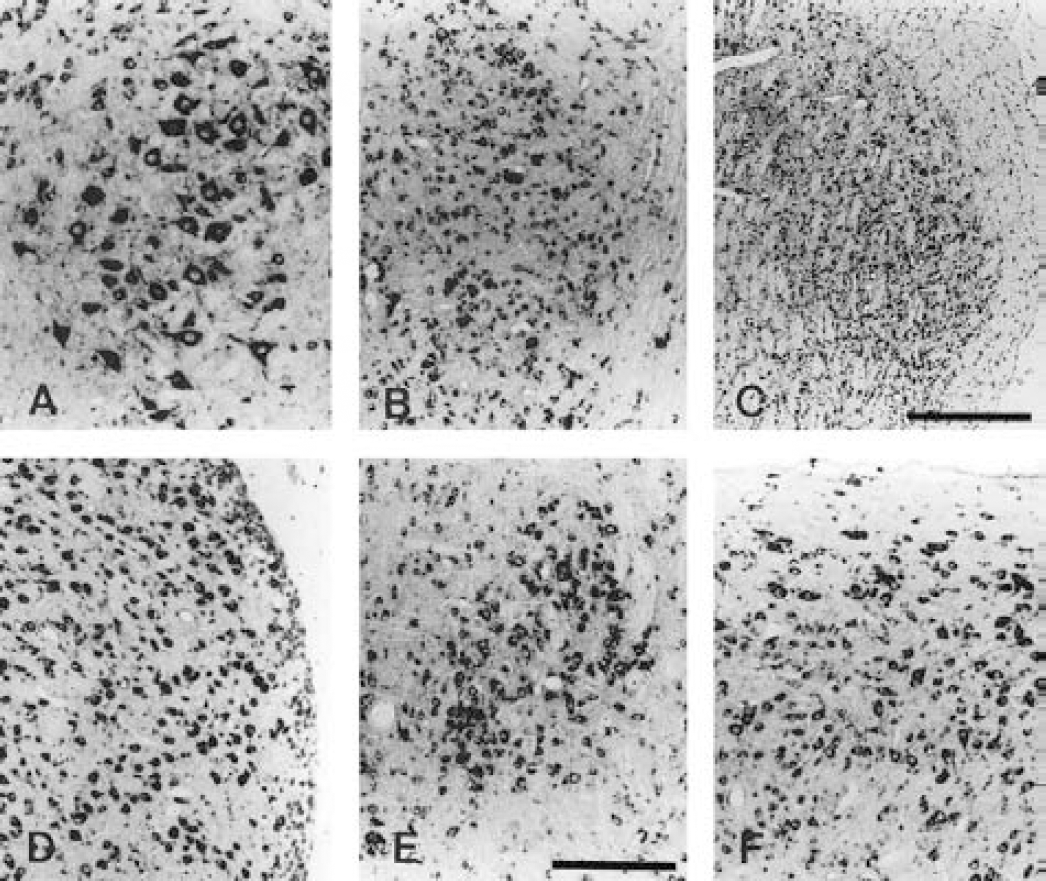
Expression of BAI2 mRNA in several nuclei of adult rat brain stem.
Changes in BAI2 in the focal ischemia model
Analysis of BAI2 and BAI1 showed four and five TSRs in the extracellular region, respectively. It has been reported that peptides corresponding to the TSRs or GST-TSR fusion proteins inhibit experimental angiogenesis in the rat cornea induced by bFGF (Nishimori et al., 1997; Tolsma et al., 1993). To investigate the role of BAI2 in ischemic cerebral neovascularization, we examined whether BAI2 expression was changed in the rat focal cerebral ischemia injury model produced by the occlusion of the middle cerebral artery. The expression of BAI2 initially decreased in the ischemic side after 1 hour, and the decreased level was maintained until 2 hours; expression was recovered to the control level at 4 hours, after which a slightly decreased level was maintained until 24 hours (Fig. 9A). BAI2 also slightly decreased in the contralateral side after 8 hours. However, expression of BAI1 decreased only in the ischemic side after 24 hours (Fig. 9B). This finding indicates that BAI2 and BAI1 are involved in the early and late stages of neovascularization of cerebral cortex after ischemia, respectively. This difference suggests that the functional significance of BAI2 and BAI1 may differ.
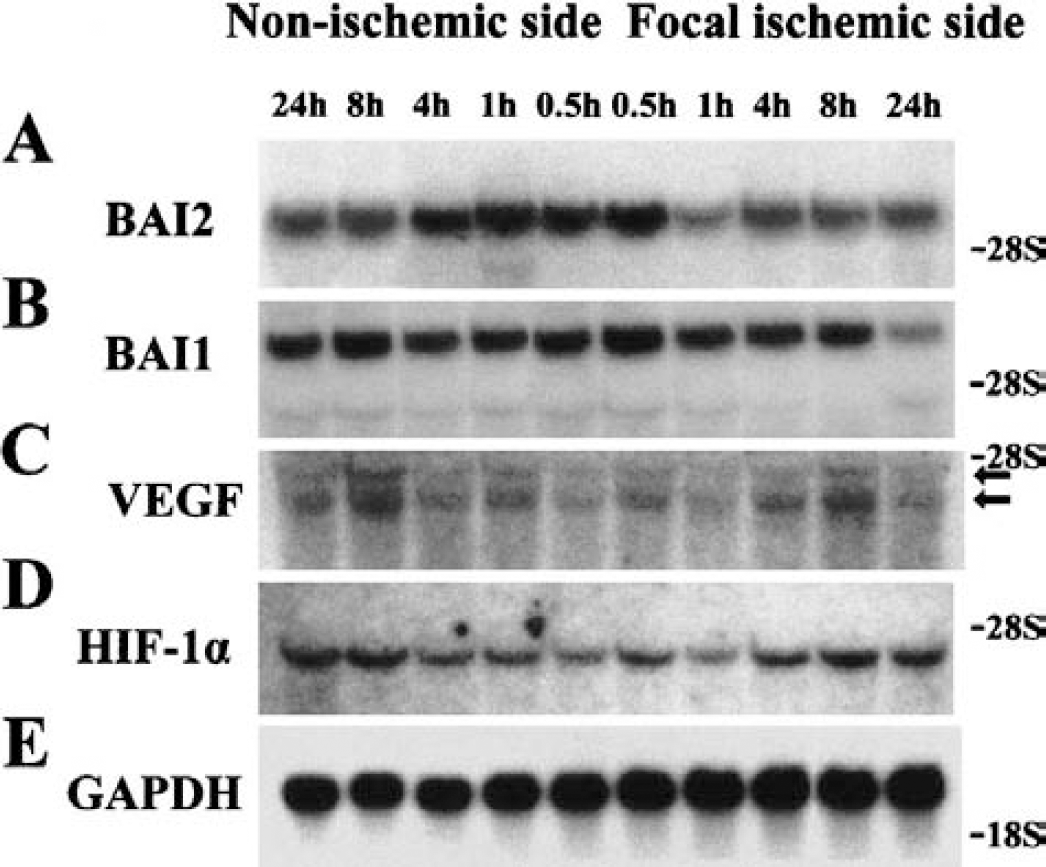
Northern blot analyses of BAI2, BAI1, and VEGFin the ischemic cerebral tissues. The changes of BAI2, BAI1, and VEGF expressions were examined in a focal cerebral ischemia model by occlusion of the middle cerebral artery in rats. The data show one out of three representative experiments.
Western blot analysis of BAI2 in the ischemic cerebral tissues was done to validate that the observation made at the mRNA level is also present at the protein level. The expression of BAI2 decreased in the ischemic side after 1 hour compared with sham-operated cerebral cortex, the decreased level was maintained at 2 hours, and expression slowly recovered after 8 hours (Fig. 10A).
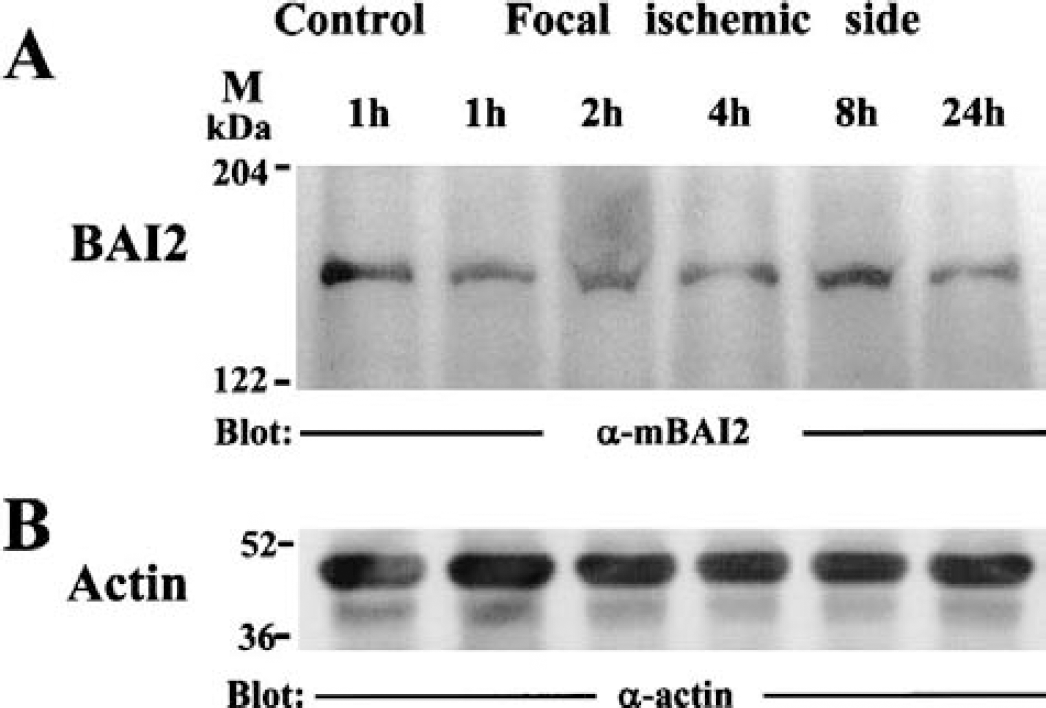
Western blot analysis of BAI2 in the ischemic cerebral tissues. The change of BAI2 expression was examined in the portion of ischemic cerebral cortex.
Vascular endothelial growth factor is an angiogenic and neurotrophic peptide that has been implicated in hypoxia-mediated angiogenesis under physiologic and pathologic conditions, and its expression is transcriptionally induced in hypoxic tissues through the action of hypoxia-inducible factor-1α (HIF-1α; Detmar, 2000). To determine if this signaling pathway is also activated in the ischemic brain, we examined the expressions of VEGF and HIF-1α in this in vivo ischemia model. Northern analysis showed that VEGF reached its peak level in the ischemic and nonischemic contralateral cerebral cortex after 8 hours, but returned to basal levels at 24 hours (Fig. 9C). However, HIF-1α expression increased in the ischemic and nonischemic cerebral cortex after 4 hours (Fig. 9D), and the time sequence of its increased expression after ischemia was situated between those of BAI2 and BAI1. The blots were rehybridized with a GAPDH probe to confirm the relative amounts and fidelity of the samples (Fig. 9E).
Changes in alternatively spliced variants of BAI2 mRNA in the focal ischemia model
RT-PCR analyses of alternatively spliced variants of mBAI2 suggested that there are several mBAI2 proteins lacking parts of TSR or the third loop of STR that may be important for mBAI2 functions (Table 1, Fig. 5). RT-PCR analysis was used to examine whether these spliced BAI2 products were changed in the in vivo focal ischemia model compared with the wild-type BAI2. The wild-type BAI2 expression decreased on the ischemic side after initiation of ischemia and was undetectable throughout the time sequence of focal ischemia (Figs. 11A and 11B). The spliced variants devoid of first or second TSR also decreased after 4 hours. However, another variant devoid of first and second TSR was nearly undetectable because we used PCR primers from mouse origin and limited amplification cycles for the quantitative comparisons of bands (Fig. 11A). Also, the spliced variants lacking the third loop of STR decreased after 4 hours (Fig. 11B), but the overall changes of the spliced variants expressions were smaller than those of wild-type BAI2 after the ischemic period. The RT-PCR result of β-actin again confirmed the relative amounts and fidelity of the total RNA samples (Fig. 11C).
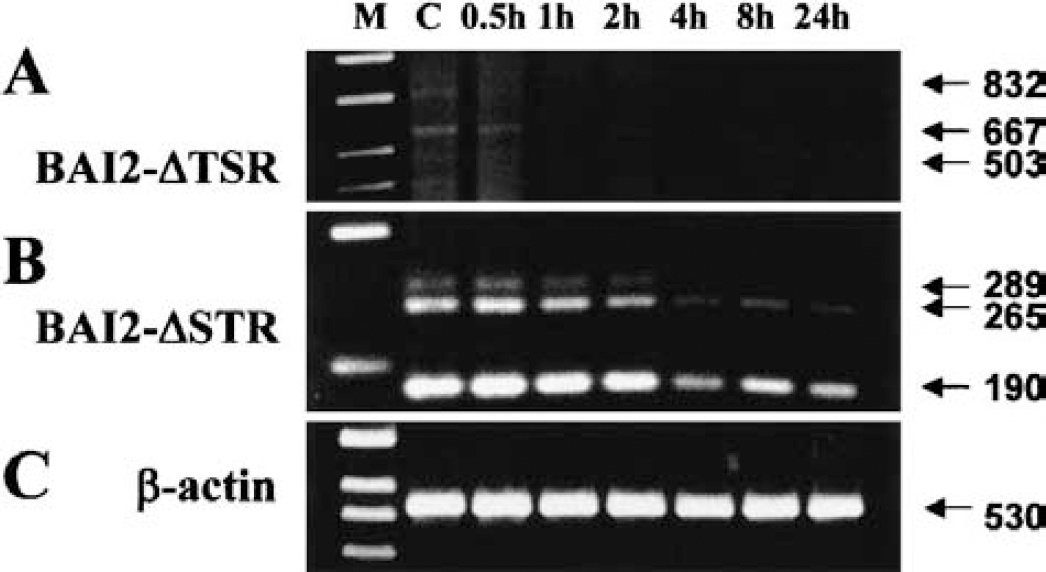
RT-PCR analyses of alternatively spliced variants of BAI2 mRNA in the focal cerebral ischemia model.
Changes in BAI2 and BAI1 mRNA in the hypoxic cell culture model with cobalt chloride
Cobalt mimics hypoxia and was used as a tool to study the role of oxygen sensing and signaling cascades in the regulation of hypoxia-inducible gene expression in the absence of hypoxia (Schuster et al., 1989). This metal can produce oxidative stress and may trigger signaling pathways resulting in the activation of the HIF-1 transcription factor and upregulation of hypoxia-related genes (Minchenko et al., 1994; Sandner et al., 1997). To support the in vivo results of the time sequence of angiostatic and angiogenic gene expressions in ischemic cerebral cortex, we used an in vitro hypoxia-mimic human SHSY5Y neuroblastoma cell culture model in which cobalt was given, and examined the time sequence of BAI2, BAI1, and VEGF expression following cobalt administration through RT-PCR analysis. The BAI2 expression acutely dropped at 0.5 hours, returned to the basal level at 2 hours, and dropped again at 8 hours (Fig. 12A), whereas BAI1 expression was sharply reduced at 2 hours but recovered to the control level at 8 hours (Fig. 12B). The VEGF expression increased from 4 hours, peaked 8 hours following cobalt administration, and returned to the basal level at 24 hours (Fig. 12C). HIF-1α increased from 2 hours and reached its peak level 8 hours following cobalt administration, but returned to the basal level at 24 hours (Fig. 12D). The RT-PCR result of β-actin confirmed the relative amounts and fidelity of the total RNA samples (Fig. 12E). The time-sequential changes of BAI2 and BAI1 expression in the in vitro hypoxic cell culture model did not exactly match those of the in vivo focal cerebral ischemia model. This difference might be caused by differences in the hypoxia-inducing method and the presence of the feedback system, which was absent in the in vitro culture model. However, earlier involvement of BAI2 in the angiogenesis after ischemia, and the appearance of increased VEGF and HIF-1α following decreased or absent BAI2, were matched between the in vivo and in vitro models. Thus, in vitro results support the in vivo focal ischemia finding that decreased BAI2 precedes the increased VEGF expression, and that the functional contribution of BAI2 and BAI1 in ischemia-induced angiogenesis might differ.
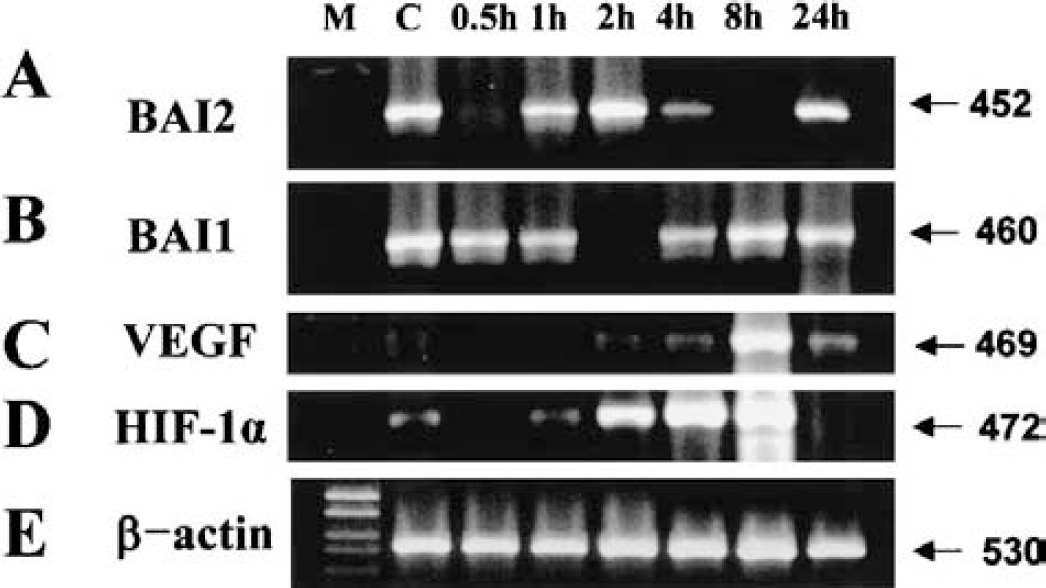
RT-PCR analyses of BAI2, BAI1, and VEGF mRNA in the hypoxic SHSY5Y neuroblastoma cell culture model with cobalt chloride.
Changes in BAI2 and VEGF mRNA in the antisense BAI2 plasmid-transfected SHSY5Y cells
To observe the causal link between decreased BAI2 expression and increased VEGF expression following cerebral ischemia, we performed RT-PCR analyses of the BAI2 and VEGF expressions in SHSY5Y neuroblastoma cells stably transfected with antisense BAI2 construct. Results showed that the expression of BAI2 decreased as a result of stable antisense BAI2 construct transfection, but VEGF expression increased at the same time (Fig 13A). This finding indicates that angiostatic BAI2 might suppress the VEGF expression in the normal physiologic condition.
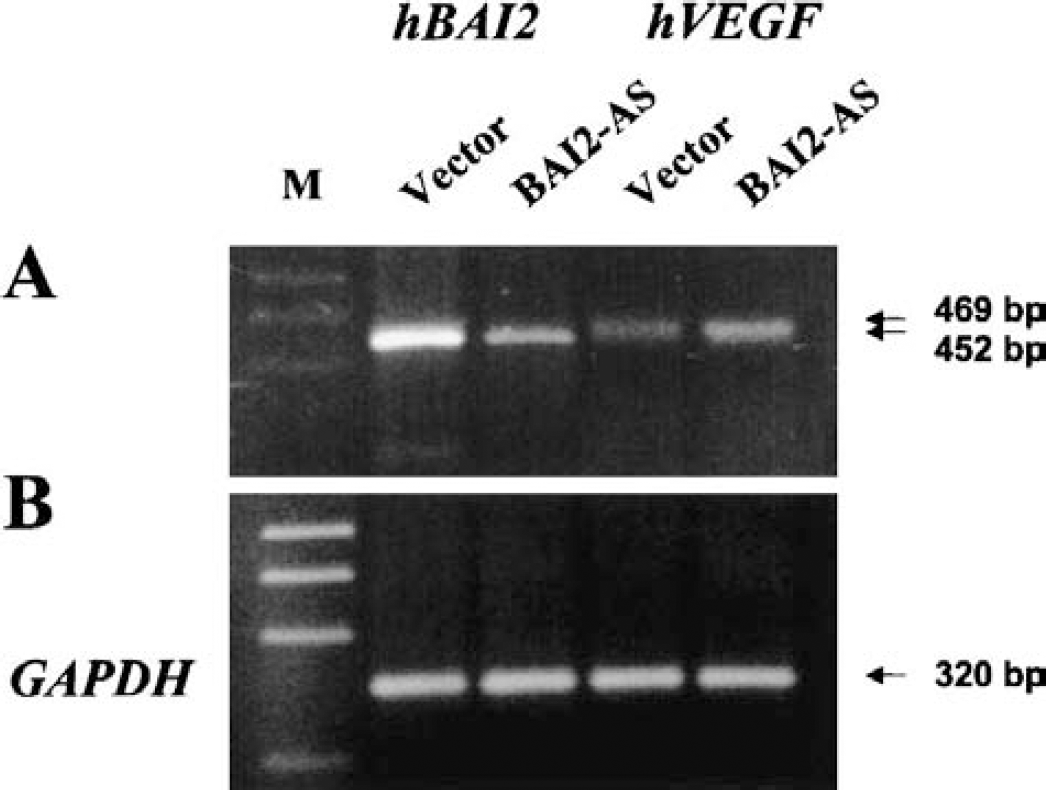
RT-PCR analyses of BAI2 and VEGF mRNA in the stably antisense BAI2 plasmid-transfected SHSY5Y cells.
Immunohistochemical analyses of BAI2 and CD4 localization in ischemic brain tissue
The changes in BAI2 and CD34 (a marker for new vessel) expressions were examined in the ipsilateral ischemic and contralateral normal cerebral cortex by occlusion of the middle cerebral artery in rats. Expression of BAI2 decreased in the ischemic cerebral cortex (Fig. 14B) and hippocampus (Fig. 14D) after 1 hour compared with the contralateral cerebral cortex (Fig. 14A) and hippocampus (Fig. 14C), respectively. Expressions of CD34 were induced in the ischemic cerebral cortex (Fig. 14F) after 1 hour compared with the contralateral normal cerebral cortex (Fig. 14G). These results indicate that decreased BAI2 expression was related to neovascularization in the cerebral cortex after focal ischemia.
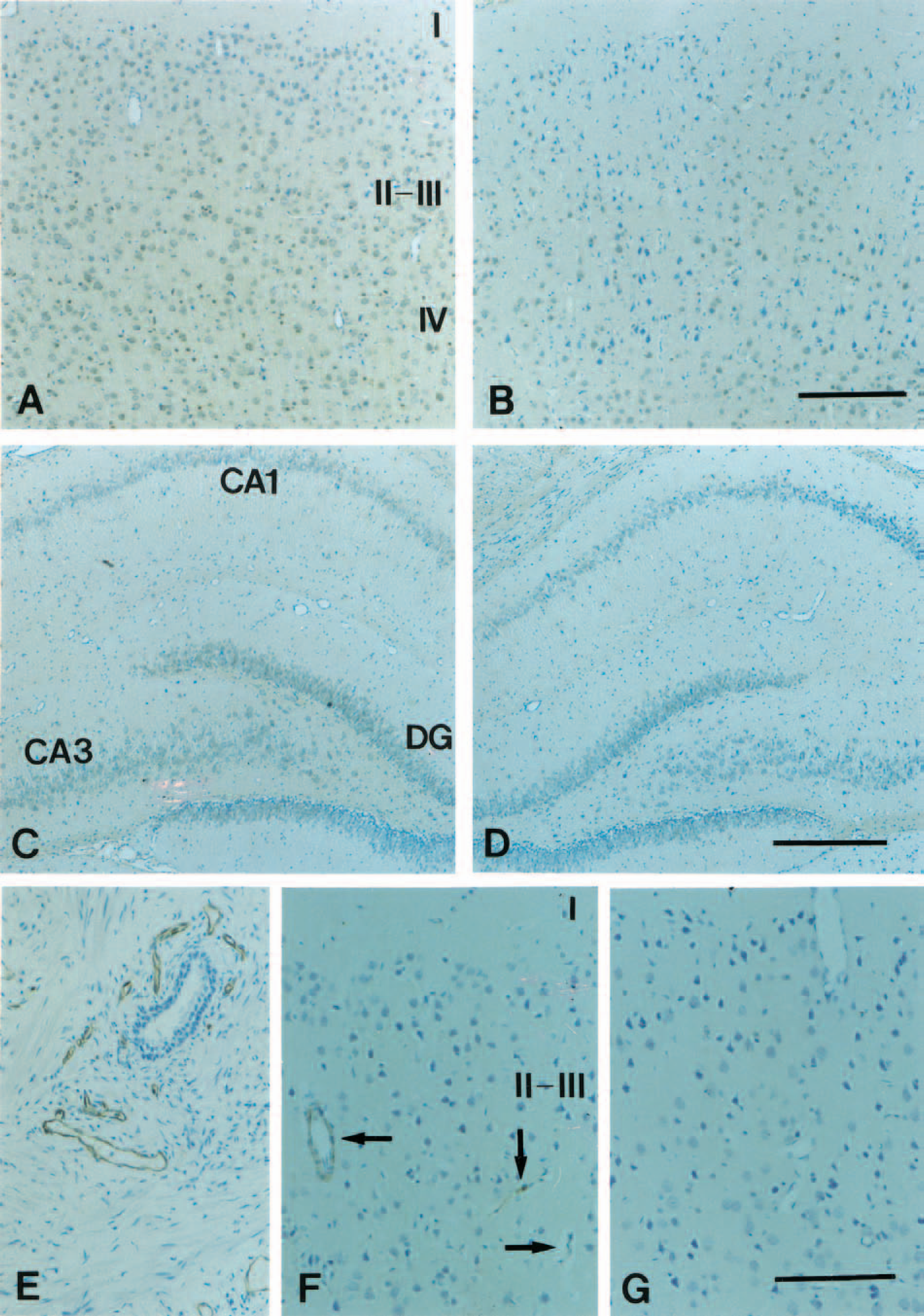
Immunohistochemical analyses of BAI2 and CD34 localizations in the ischemic rat brain tissues. The changes of BAI2 and CD34 (a marker for new vessel) expressions were examined in the ipsilateral ischemic and contralateral normal brain by occlusion of the middle cerebral artery in rats. Expression of BAI2 decreased in the cerebral cortex
DISCUSSION
In this study, we isolated the murine homologue of hBAI2. We revealed a unique expression of mBAI2 in the mouse adult brain, and highly conserved aa sequences between humans and mice (96%). In situ hybridization analyses showed that BAI2 is colocalized with BAI1 in the rat brain, such as in the most neurons of the cerebral cortex, olfactory bulb, dentate gyrus, hippocampus, caudate putamen, medial septum, Purkinje cell layer, lateral cerebellar nucleus, and trigeminal, abducent, facial, cochlear, hypoglossal nuclei, and retina. However, there are no reported functions of BAI2 in these subregions of brain. The predicted BAI2 protein includes extended extracellular and cytoplasmic domains, STR, and TSR. hBAI2 and mBAI2 have four TSRs, whereas hBAI1 and mBAI1 have five TSRs in their extracellular region. In contrast, the other angiostatic genes, thrombospondin-1 (TSP1) and thrombospondin-2 (TSP2), contain three TSRs. We found two kinds of alternatively spliced variants of mBAI2 through RT-PCR analysis. One type did not have the first and/or second loop of TSR in its extracellular region, whereas the other did not have the third cytoplasmic loop of STR.
Previous studies showed that the peptide sequence cysteine-serine-valine-threonine-cysteine-glycine (CSVTCG) within the TSR of TSP1 interacts with the surface-receptor glycoprotein CD36 (Asch et al., 1993) and mediates the in vitro and in vivo inhibitory effects of TSP1 on endothelial cells (ECs) (Dawson et al., 1997; Jimenez et al., 2000). The first TSR of BAI2 may be important to the antiangiogenic activity because it contains a motif (CSLTCG) similar to the binding site for CD36. Properdin has six TSRs, plays an important role in stabilizing the C3b-Bb convertase complex, and is crucial for full complement activity (Fearon and Austen, 1975). However, properdin lacking the fourth TSR, which contains a motif (CPVTCG) similar to the binding site for CD36 and sulfated glycoconjugates, is unable to stabilize the alternative pathway C3 convertase (Higgins et al., 1995). The TSR contains two subdomains that may independently influence the process of neovascularization, and synthetic peptides derived from the TSR have potent antiangiogenic activity in vivo and in assays of EC function (Shafiee et al., 2000; Tolsma et al., 1993). Moreover, TSP2, the other TSR-containing protein of the TSP family, has antiangiogenic activity (Volpert et al., 1995). However, TSP3, which lacks TSRs, had no inhibitory activity on human dermal microvascular EC proliferation, confirming that TSRs elicit the antiangiogenic activity of TSP1 (Qabar et al., 2000). Thus, these results suggest that the spliced variant of mBAI2, which did not have first TSR, might not have the antiangiogenic action of wild-type mBAI2. The third cytoplasmic loop is important to the interaction of G protein in the serpentine receptors coupled to G proteins, which have STR (Stefan and Blumer, 1994). However, this third loop is much shorter in mBAI1 than in mBAI2, and the size of this loop of mBAI1 is the same as that of the spliced variant of mBAI2. Thus, the spliced variant of mBAI2, which did not have this third loop, may not perform the certain essential function of the wild-type mBAI2. The finding also indicates that the functions of mBAI2 mediated by this cytoplasmic loop through interaction with G proteins may be different or absent in mBAI1. It is speculated that BAI2 may have more important functions in brain angiogenesis than BAI1, and the search for G proteins or interacting proteins in this cytoplasmic loop using yeast two-hybrid assay is under investigation.
The expression levels of wild-type and variant BAI2 during the development of the brain were different. Wild-type BAI2, which has intact TSR or STR, developmentally increased in the brain, while spliced variants lacking TSR or a third cytoplasmic loop showed little or no change in expression during brain development. Moreover, RT-PCR results showed only a slight decrease in the expression of the spliced variants after the focal cerebral ischemic period, while expression of wild-type BAI2 was acutely reduced after ischemia. However, alternative splicing patterns in the N- and C-terminal region were not observed in the mBAI1 because we did not clone such spliced mBAI1 variants. However, according to RT-PCR analysis using the primers spanning these regions that were well conserved between mBA12 and mBAI1, these alternative patterns were observed in mBAI2 variants. These data indicate that the functions of spliced variant of mBAI2 might differ from that of wild-type mBAI2 protein, and the functional characterization of these spliced variants are under investigation.
Brain angiogenesis is a tightly controlled process that is regulated by neuroectodermal-derived growth factors that bind to tyrosine kinase receptors expressed on ECs. In the rat brain, angiogenesis is complete around postnatal day 20, but ECs can proliferate in the adult brain under pathologic conditions such as ischemia and tumor. Current evidence suggests that physiologic angiogenesis in the brain is regulated by mechanisms similar to those in pathologic angiogenesis (Plate, 1999). In the murine retina model, TSP1 was increased from postnatal day 13 to 15, corresponding to the time of development of retinal neovascularization (Suzuma et al., 1999). However, we observed that mBAI2 and mBAI1 were acutely upregulated after birth in the brain, and the peak expression level of these angiostatic genes occurred 10 days after birth. This finding indicates that this developmental regulation of brain-specific angiostatic genes might contribute to the completion of physiologic brain angiogenesis. Also, developmentally increased expressions of BAI2 and BAI1 in the adult brain suggest that they might have other neuronal functions, such as neuronal differentiation.
The ischemic brain might stimulate angiogenesis to compensate for impaired circulation, though the mechanisms controlling hypoxia-induced angiogenesis in the mammalian brain are not fully understood. It was reported that the hypoxia-induced upregulation of VEGF expression is mediated both by activation of VEGF gene transcription through HIF-1α, and by enhanced VEGF mRNA stability through specific interactions between a defined mRNA stability sequence in the 3' untranslated region and distinct mRNA-binding proteins (Detmar, 2000; Marti et al., 2000). In this study, VEGF expressions increased in the ischemic and nonischemic contralateral cerebral cortex after augmented HIF-1α expression, and its time sequence of contribution to the angiogenesis after ischemia is situated between that of BAI2 and BAI1. Also, results of in vitro hypoxic cell culture model supported the in vivo focal ischemia result that the increased VEGF expression appeared following decreased BAI2. The present findings that the decreased BAI2 precedes the increased VEGF expression, and other reports that VEGF plays a major role in ischemia-induced tissue neovascularization (Detmar, 2000), indicate that the decreased BAI2-mediated VEGF induction in ischemia-induced angiogenesis may be a feedback mechanism. Thus, it is hypothesized that a reciprocal relationship exists between BAI2 and VEGF such that they are constituents of a switch that regulates in concert many components of the angiogenesis after focal cerebral ischemia, and like HIF-1α, BAI2 may be one of the components upregulating VEGF expression. Our results of increased VEGF expression observed in the antisense BAI2 cDNA-transfected cells and decreased BAI2 expression correlated with neovascularization after focal ischemia indicate that angiostatic BAI2 suppress the VEGF expression in the normal condition, and support the hypothesis of a reciprocal relationship between BAI2 and VEGF. Thus, BAI2 may act as one of the important physiologic regulators in focal cerebral ischemia-induced angiogenesis. Furthermore, the roles of BAI2 in ischemia-induced cerebral angiogenesis are well conserved among mice, rats, and humans.
Marti et al. (2000) reported that VEGF expression was strongly upregulated in the ischemic border between 6 and 24 hours after permanent middle cerebral artery occlusion. Neuropilin-1 was significantly upregulated as early as 2 hours and persisted for at least 28 days after focal cerebral ischemia, primarily in ischemic neurons (Zhang et al., 2001). In present study, we observed that the expression of BAI1 was decreased only in the ischemic side after 24 hours, but BAI2 participated in the earlier phase of in vivo and in vitro ischemia-induced angiogenesis than BAI1. In a focal cerebral ischemia model in rats, a delayed increase of angiopoietin-1 was observed for up to 2 weeks after ischemia, and a biphasic expression of angiopoietin-2 was noted, peaking at 24 hours and 2 weeks after ischemia. The expression of Tie-2 and Tie-1 also increased starting 24 hours after reperfusion and remained elevated for up to 2 weeks after ischemia (Lin et al., 2000). These and our results strongly suggest that the angiogenic VEGF and neuropilin-1 system and the angiostatic BAI2 system, all of which were induced by hypoxia, contribute to the early phase of ischemia-induced angiogenesis, whereas the angiostatic BAI1 system and the angiogenic angiopoietin/Tie system seem to work in the later phase. The spatial and temporal correlations between these systems represent a physiologic response of the brain to counterbalance ischemia in order to protect neuronal tissue. Differential expression of these angiostatic and angiogenic genes suggests the involvement of complex regulatory mechanisms that remain to be characterized.