
Abstract
Astrocytes perform a variety of functions in the adult central nervous system (CNS) that contribute to the survival of neurons. Thus, it is likely that the activities of astrocytes affect the extent of brain damage after ischemic stroke. The authors tested this hypothesis by using a mouse ischemia model to compare the infarct volume produced in wild-type mice with that produced in mice lacking glial fibrillary acidic protein (GFAP), an astrocyte specific intermediate filament component. Astrocytes lacking GFAP have been shown to have defects in process formation, induction of the blood-brain barrier, and volume regulation; therefore, they might be compromised in their ability to protect the CNS after injury. The authors reported here that 48 hours after combined permanent middle cerebral artery occlusion (MCAO) and 15 minutes transient carotid artery occlusion (CAO) GFAP-null mice had a significantly (P < 0.001) larger cortical infarct volume (16.7 ± 2.2 mm3) than their wild-type littermates (10.1 ± 3.9 mm3). Laser-Doppler flowmetry revealed that the GFAP-null mice had a more extensive and profound decrease in cortical cerebral blood flow within 2 minutes after MCAO with CAO. These results indicated a high susceptibility to cerebral ischemia in GFAP-null mice and suggested an important role for astrocytes and GFAP in the progress of ischemic brain damage after focal cerebral ischemia with partial reperfusion.
Astrocytes perform a variety of neuroprotective functions in the adult brain. Perhaps paramount among these is protection against excitotoxic death by avidly removing extracellular glutamate from the synaptic cleft (Rothstein, 1996: Tanaka et al., 1997). Astrocytes also buffer the extracellular K+ concentration, process nutrients, regulate extracellular volume, and release a battery of neurotrophic factors (Eddleston and Mucke, 1993; Brenner et al., 1994a; Brenner 1994b). In addition, astrocytic endfeet closely contact capillary endothelial cells and vascular smooth muscles, thus they are well positioned to regulate capillary microcirculation (Sporbert et al., 1999). These, and perhaps other neuroprotective activities, may be critical in limiting the extent of damage after ischemic injury. Establishing a protective role for astrocytes in this process would be an important first step toward focusing attention on these cells as a target for therapy. The authors considered that the recent production of mice lacking glial fibrillary acidic protein (GFAP) (Gomi et al., 1995; Pekny et al., 1995; Liedtke et al., 1996; McCall et al, 1996), an astrocyte specific intermediate filament component, might have permitted testing of this hypothesis. Although the gross anatomy of the central nervous system (CNS) of GFAP-null mice is normal for up to one year of age (Gomi et al., 1995; Pekny et al., 1995; Shibuki et al., 1996), there are suggestions that their processes are shorter and thinner (Liedtke et al., 1996; McCall et al, 1996) and that their blood-brain barrier is defective (Liedtke et al., 1996; Pekny et al., 1998). GFAP mice are also abnormal in long-term depression (Shibuki et al., 1996) and long-term potentiation (McCall et al, 1996). These effects suggest an alteration in synaptic glutamate kinetics. Cell culture studies have found that astrocytes lacking both GFAP and vimentin are defective in taurine release (Ding et al., 1998), a mechanism considered critical for volume regulation. Although astrocytes deficient in GFAP alone released taurine normally, this could be an artefact of the up-regulation of vimentin that occurs in cultured astrocytes (Ding et al., 1998). In vivo, GFAP-null astrocytes may also be defective in volume regulation. Given the likelihood that GFAP-null mice could be compromised in astrocyte function, the authors compared the extent of ischemic brain damage in GFAP-null mice with that in wild-type littermates after permanent middle cerebral artery occlusion (MCAO) alone and in combination with transient carotid artery occlusion (CAO).
MATERIALS AND METHODS
GFAP-null mice
GFAP-null mice (B6,129-GfoptmlMes; McCall et al, 1996) were obtained from either A. Messing (University of Wisconsin, Madison, WI, U.S.A.) or Jackson Laboratories, Bar Harbor, Maine, U.S.A. Mice of mixed B6,129 genetic background heterozygous for the GFAP-null allele were crossed to generate the homozygous GFAP-null mice and wild type-littermates used for these experiments. Mature mice weighing 18.4 to 26.0 g from several different litters were included in each experimental group. Animals were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals [DHEW (DHHS) publication no. (NIH) 85-23, rev. 1985, Office of Science and Health Rep, DRR/NIH, Bethesda, MD 20205, U.S.A.]. Procedures involving animals were approved by the National Institutes of Health Animal Care and Use Committee.
Permanent middle cerebral artery occlusion with transient carotid artery occlusion in mice
Animals were anesthetized with 2.5% halothane for induction and 1.5% halothane for maintenance in 30% O2 and 70% N2O through a face mask. The middle cerebral artery was exposed as described by Welsh et al. (1987) and Nawashiro et al. (1997a, b, c). The left temporoparietal region of the head was shaved and a 5-mm incision was made between the orbit and ear. Under an operating microscope, an incision was made by dividing the temporal muscle, and the left lateral aspect of the skull was exposed by reflecting the temporal muscle and surrounding soft tissue. Using a high speed microdrill, a small burr hole (1 mm) was made through the outer surface of the semi-translucent skull just over the visibly identified middle cerebral artery between the inferior cerebral vein and olfactory tract. Saline was applied to the area throughout the procedure to prevent heat injury. The inner layer of the skull was removed with fine forceps, and left MCAO was performed by electrocoagulation using a small vessel cauterizer directly through the dura without damaging the brain surface. After MCAO, a 10-mm cervical ventral midline skin incision was made. The left common carotid artery and external carotid artery were exposed and isolated. A micro aneurysm clip, 85 gr pressure (Roboz Surgical Instrument, Rockville, MD, U.S.A.), was applied to the common carotid and the external carotid artery either for 15 or 30 minutes in wild-type mice, and for 15 or 30 minutes in GFAP-null mice. The time interval between MCAO and initiation of transient common CAO was 5 to 7 minutes. Reperfusion of the carotid artery was visually confirmed with the operating microscope. Cervical sham operations without application of clips were performed for the groups of mice that received permanent MCAO only. The duration of anesthesia in all animals was less than 60 minutes and all animals recovered active spontaneous movements within 30 minutes of its cessation. Throughout this period the rectal temperature was measured and maintained at 37°C ± 0.2°C with a heating blanket.
Measurement of local CBF by laser-Doppler flowmetry
To determine the real-time change of local cerebral blood flow (1CBF), cortical 1CBF was measured on the left frontal, parietal, and temporal cortices through the translucent skull by means of laser-Doppler flowmetry (Laserflo BPM403; VASAMEDICS, St. Paul, MN, U.S.A.). The cortical area which showed hypoperfusion, defined as less than 30% of the preischemic value of 1CBF, was mapped and recorded after MCAO, before and after reversible CAO. The spatial resolution of this technique was approximately 1 mm. Anatomical landmarks of the skull for brain surface mapping were sagittal suture, coronal suture, lambdoid suture, and superior temporal line. The area of hypoperfusion was quantified by a computer-assisted image analyzing system (NIH image 1.57). During anesthesia, the right femoral artery was catheterized using a P10 tube for continuous monitoring of arterial blood pressure.
Measurement of the size of cortical infarcts using 2,3,5-triphenyltetrazolium chloride
Forty-eight hours after MCAO the animals were injected with a lethal dose of thiopental (150 mg per kilogram of body mass, IP), the brains were removed and cut into 1-mm-thick coronal sections, and the sections were immersed in 2% triphenyltetrazolium chloride saline solution at 37°C for 30 minutes. The infarcted areas of the brain slice were measured by a computer-assisted image analyzing system (NIH image 1.57). The infarct volume was calculated by taking the sum of the infarct area of different brain slices multiplied by the thickness of the slices. The contribution of the edema volume to the infarct volume was corrected by subtracting the ipsilateral uninvolved hemisphere volume from the total contralateral hemisphere volume and dividing by the total contralateral hemisphere volume (Swanson et al., 1990; Lin et al., 1993). Infarct volumes in each group were analyzed by analysis of variance followed by the Bonferroni-Dunn test to ascertain significance between groups. Statistical significance was P < 0.05.
Comparison of cerebrovascular anatomy between GFAP-null mice and wild-type littermates
To investigate the difference in cerebrovascular anatomy, the circle of Willis was visualized using carbon black in GFAP-null mice (n = 10) and wild-type littermates (n = 6). After receiving deep anesthesia, animals were given an intracardiac injection of carbon black and were then killed.
RESULTS
After surgery, all animals were able to eat and drink without any support and showed no apparent motor weakness. Infarct volumes were measured 48 hours after surgery using triphenyltetrazolium chloride staining and corrected for the contribution of edema as described in the Methods section. The results (Fig. 1) showed that after permanent MCAO only there was no significant difference in infarct volume between wild-type and GFAP-null mice, but after permanent MCAO in addition to 15 minutes of CAO, the GFAP-null mice had a substantially greater infarct volume than the wild-type littermates (P < 0.001). Increasing the duration of CAO to 30 minutes for the wild-type mice produced an infarct volume similar to that for the GFAP-null animals with 15 minutes CAO. Furthermore, increasing the duration of CAO to 30 minutes for the GFAP-null mice produced an infarct volume similar to that for the GFAP-null animals with 15 minutes CAO.
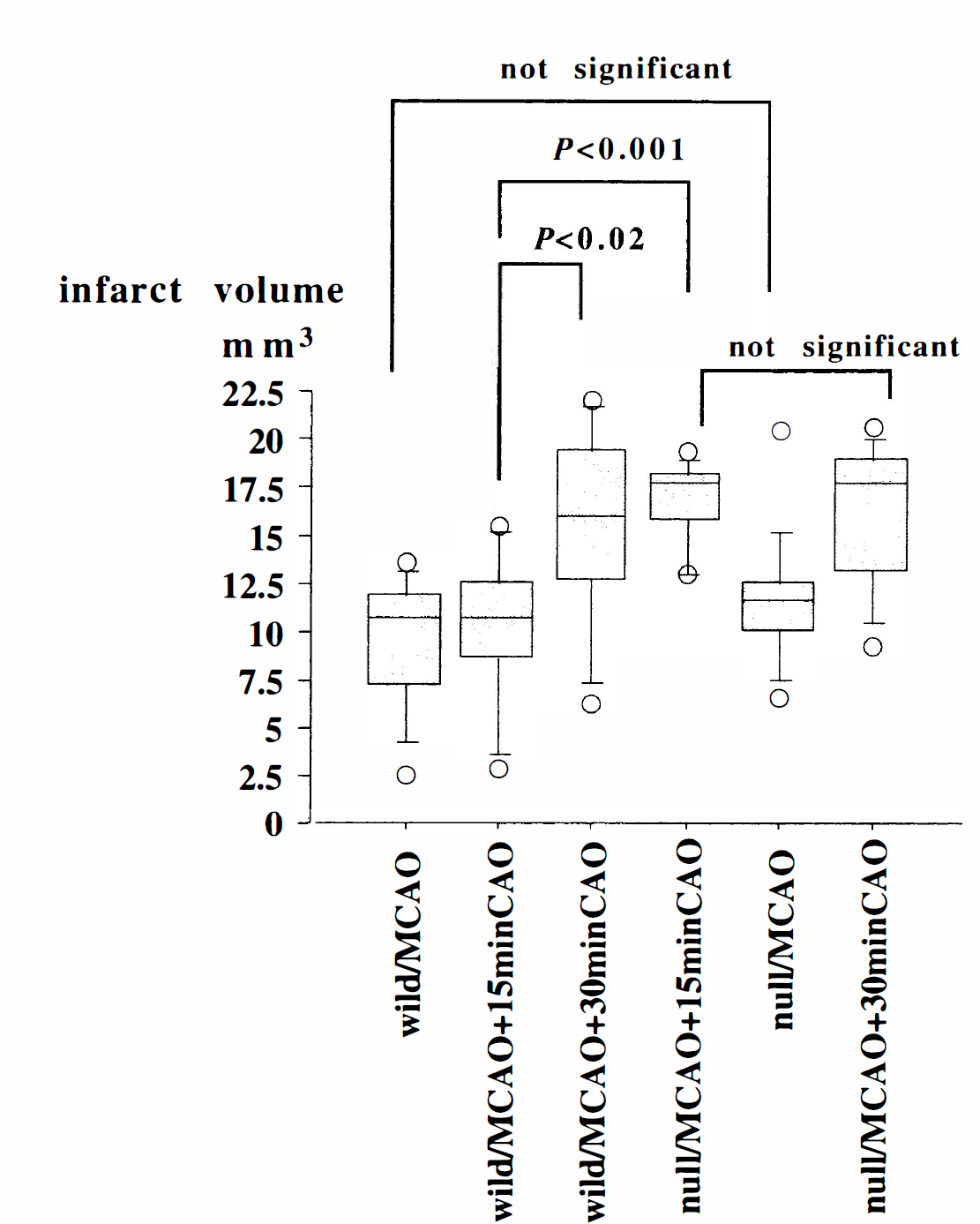
Infarct volumes for wild-type and GFAP-null mice after permanent MCAO with or without CAO. Average infarct volumes with dispersion (10, 25, 75, 90 percentile of variables) in each group are shown. The open circles in the graph indicate the lowest and highest individual values in each group. Significance was determined by analysis of variance. Wild, wild-type mice; null, GFAP-null mice. Means ± SD. Values of animals used were as follows: wild/MCAO = 9.6 ± 3.4 mm3 (n = 12); wild/MCAO + 15 minutes CAO = 10.1 ± 3.9 mm3 (n = 13); wild/MCAO + 30 minutes CAO = 15.4 ± 5.3 mm3 (n = 7); null/MCAO + 15 minutes CAO = 16.7 ± 2.2 mm3 (n = 10); null/MCAO = 11.4 ± 3.4 mm3 (n = 13); null/MCAO + 30 minutes CAO = 16.2 ± 3.8 mm3 (n = 9).
Local cerebral blood flow was measured by laser-Doppler flowmetry to determine if the differences in infarct volumes could be attributed to effects on 1CBF. Permanent MCAO alone, which produced no significant difference in infarct size between the wild-type and GFAP-null mice, also produced no significant difference in blood flow. For both the wild-type (n = 5) and GFAP-null mice (n = 5), the 1CBF decreased to approximately 10% of the preischemic value in the infarct core in the temporal cortex after permanent MCAO, and the regions of reduced 1CBF were similar (data not shown). In marked contrast, within 2 minutes after CAO was begun in combination with MCAO, GFAP-null mice (n = 5) showed a more widespread and profound decrease of 1CBF in the frontal and parietal cortices than did the wild-type mice (n = 5). This difference was sustained for over 10 minutes after reperfusion of the carotid artery. Representative maps of the hypoperfused areas yielded values of 25.4 ±5.0 mm2 in GFAP-null mice and 19.0 ±3.6 mm2 for wild-type mice (mean ± SD; P < 0.05). Systemic mean arterial blood pressure was not significantly different between wild-type and GFAP-null mice before MCAO (83 ±13 mm Hg and 79 ± 9 mm Hg, respectively) and 15 minutes after MCAO with CAO (90 ± 17 mm Hg and 87 ± 11 mm Hg, respectively).
The intracardiac injection of carbon black revealed patent anterior communicating artery in all GFAP-null and wild-type littermates. Bilateral patent posterior communicating arteries were discernible in 8 of 10 GFAP-null mice, whereas 4 out of 6 wild-type littermates showed patent right and left posterior communicating arteries. There were no significant differences in cerebrovascular anatomy between GFAP-null and wild-type littermates.
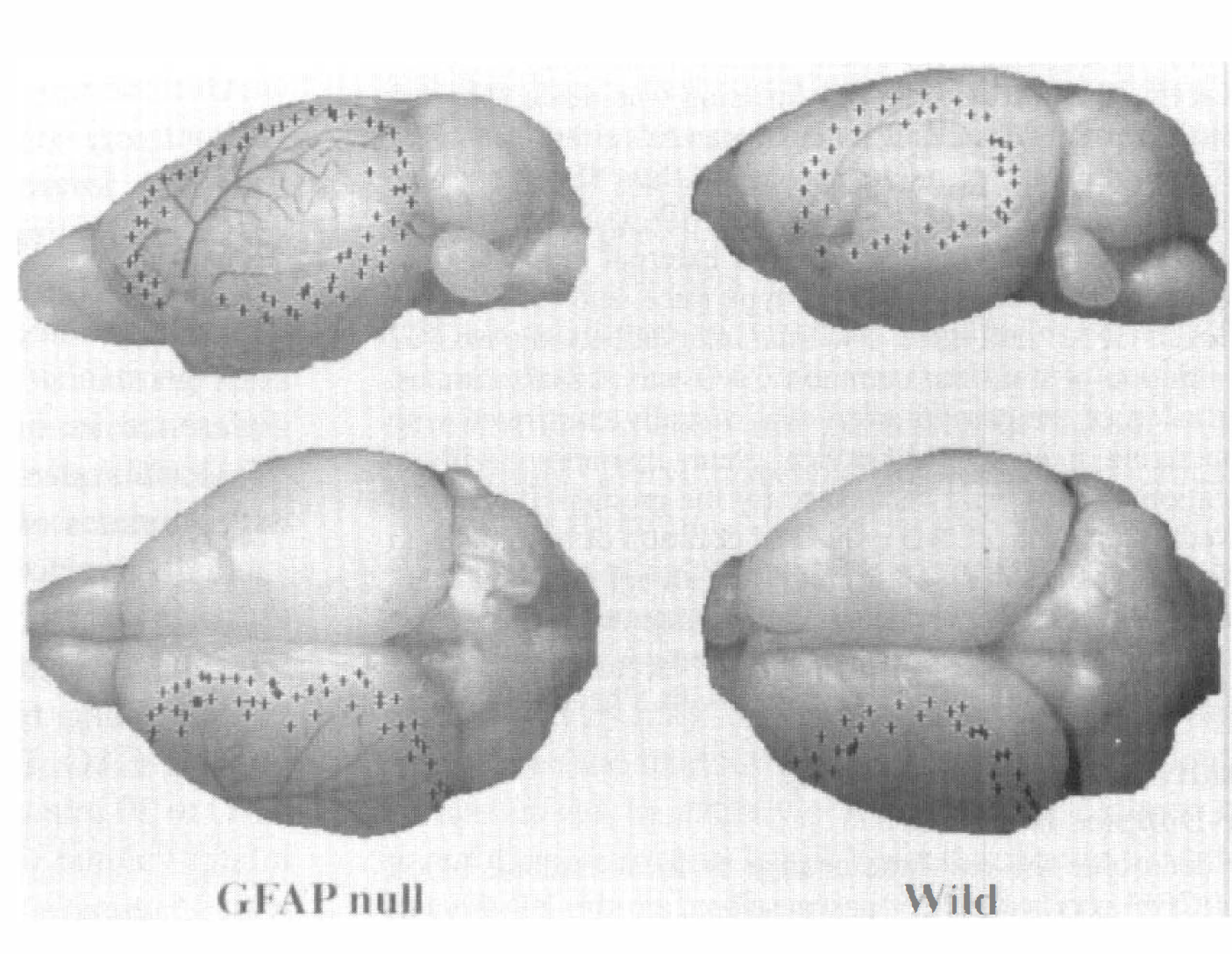
Hypoperfused area detected by laser-Doppler flowmetry in GFAP-null and wild-type mice after MCAO with 15 minutes CAO. Plus signs (+) on the brain surface indicate the margins of the region in which local cerebral blood flow was reduced to approximately 30% of preischemic values.
DISCUSSION
In this study we observed that GFAP-null mice were more sensitive to MCAO with CAO than were wild-type mice. This result suggested that astrocytes normally provided a protective function after ischemia, and that this function was compromised in GFAP-null mice. An alternative explanation was that the absence of GFAP results in aberrant development of some other CNS component, and it is the malfunctioning of this other component that causes greater sensitivity to ischemia. However, GFAP-null mice appear normal in development, reproduction, and lifespan; except for the very old mice, their central nervous system is normal with respect to gross morphology, including the vascular anatomy (Gomi et al., 1995; Pekny et al., 1995; Shibuki et al., 1996). Indeed, we found no significant difference in infarct volume between GFAP-null and wild-type mice after MCAO alone. Furthermore, we found no significant differences in cerebrovascular anatomy of the circle of Willis that could supply collateral circulation. Thus, an impaired physiologic response of GFAP-null astrocytes to ischemia seemed the most likely explanation for our observations.
As noted above, there were multiple candidate functions attributed to astrocytes that could affect neuronal survival after ischemia, including regulation of extracellular glutamate and K+, contributing to the blood-brain barrier, and control of brain volume. For example, rapid swelling of perivascular astrocytes was one potential mechanism for the shutdown of collateral flow noted in GFAP-null animals. Astrocytes have been shown to undergo swelling within 30 minutes of MCAO (Garcia et al., 1994; Pantoni et al., 1996), and the absence of GFAP may increase the susceptibility of these cells to cytotoxic edema (Ding et al., 1998).
A particularly attractive candidate for contributing factors that emerged from this study was an effect on cerebral blood flow. After MCAO with 15 minutes CAO, the significant difference in infarct size between GFAP-null and wild-type mice was paralleled by a greater drop in 1CBF in the GFAP-null mice within the penumbral region. This difference was observed within 2 minutes of the CAO, and lasted for at least 10 minutes after reperfusion. Significantly, after MCAO alone, which produced no difference in infarct volume between GFAP-null and wild-type mice, no difference in 1CBF was detected. Thus, the differential effect of the GFAP-null genotype on infarct volume correlated with its differential effect on collateral blood flow. This correlation, and the rapidity of the onset of the 1CBF reduction, suggested that an effect of astrocytes on regulating 1CBF could be the critical element to neuronal survival in the penumbral region after ischemia. Measurements of blood flow after MCAO in rats have previously led to the suggestion that a failure of the microcirculation in the penumbral region contributes to the extent of neuronal damage (Dawson et al., 1997).
Multiple mechanisms have been proposed for how astrocytes might regulate CBF including regulation of the Na-K-Cl cotransport activity in brain microvessel endothelial cells (BMEC) through phosphorylation and increased expression of the cotransport protein (Sun and O'Donnell, 1996), phosphorylation of a vasodilatorstimulated phosphoprotein in BMEC through nitric oxide/cGMP production (Sporbert et al., 1999), and modulation of BMEC expression of tPA, PAI-1, and thrombomodulin (Tran et al., 1998; Tran et al., 1996). With particular relevance to ischemia, Alkayed et al. (1997) suggest that the cerebral vasodilation induced by glutamate may be mediated by glutamate stimulation of formation and release of epoxyeicostrienoic acids by astrocytes. Excessive release of glutamate from neurons and astrocytes is thought to occur after ischemia and is thought to contribute to neuronal death in the penumbral region by excitotoxicity (Benveniste, 1991). Vasodilation mediated by glutamate could thus be a compensatory mechanism. The lower penumbral 1CBF in GFAP-null mice compared with the wild-type mice after combined MCAO with CAO could be caused by an inability of the GFAP-null mice to increase epoxyeicosatrienoic acid production in response to glutamate. A defect in response to glutamate has already been indicated by the alterations in long-term potentiation (McCall et al, 1996) and long-term depression (Shibuki et al., 1996) observed in these mice, and the greater susceptibility of GFAP-null mice to cervical cord hemorrhage after injury (Nawashiro et al., 1998) has suggested a defect in the interaction of astrocytic endfeet with endothelial cells.
Footnotes
Acknowledgments
The authors thank Dr. Albee Messing for supplying GFAP-null mice and wild-type mice at National Institutes of Health, and Dr. Takashi Sakagawa and Dr. Shigeru Okuyama for supplying GFAP-null and wild type-mice in Japan.