
Abstract
Tolerance to cerebral ischemia is achieved by preconditioning sublethal stresses, such as ischemia or hypoxia, paradigms in which the decrease of O2 availability may constitute an early signal inducing tolerance. In accordance with this concept, this study shows that hypoxia induces tolerance against focal permanent ischemia in adult mice. Normobaric hypoxia (8% O2 of 1-hour, 3-hour, or 6-hour duration), performed 24 hours before ischemia, reduces infarct volume by approximately 30% when compared with controls. To elucidate the mechanisms underlying this neuroprotection, the authors investigated the effects of preconditioning on cerebral expression of hypoxia-inducible factor-1α (HIF-1α) and its target genes, erythropoietin and vascular endothelial growth factor (VEGF). Hypoxia, whatever its duration (1 hour, 3 hours, 6 hours), rapidly increases the nuclear content of HIF-1α as well as the mRNA levels of erythropoietin and VEGF. Furthermore, erythropoietin and VEGF are upregulated at the protein level 24 hours after 6 hours of hypoxia. The authors' findings show that (1) hypoxia elicits a delayed, short-lasting (<72 hours) tolerance to focal permanent ischemia in the adult mouse brain; (2) HIF-1 target genes could contribute to the establishment of tolerance; and (3) this model might be a useful paradigm to further study the mechanisms of ischemic tolerance, to identify new therapeutic targets for stroke.
Ischemic tolerance is an adaptive process by which a tissue is rendered more resistant to a subsequent ischemia. Tolerance can be achieved by several preconditioning sublethal stresses performed before ischemia. This phenomenon, first described in the heart (Murry et al., 1986), has attracted considerable attention in brain research over the last ten years. The first description of ischemic tolerance in the brain was reported by Kitagawa et al. (1990) in a gerbil model of global ischemia. Since this report, this phenomenon has been widely described in many other animal models, including global and focal cerebral ischemia (Kitagawa et al., 1990; Kirino et al., 1991; Liu et al., 1992; Nishi et al., 1993; Glazier et al., 1994; Gidday et al., 1994; Matsushima and Hakim, 1995; Toyoda et al., 1997; Stagliano et al., 1999). Tolerance to cerebral ischemia can be achieved in vivo by other preconditioning stimuli ranging from sublethal hypothermia (Nishio et al., 2000), hyperthermia (Chopp et al., 1989; Ikeda et al., 1999), hyperbaric oxygenation (Prass et al., 2000), metabolic inhibitors (Wiegand et al., 1999), spreading depression (Kobayashi et al., 1995; Matsushima et al., 1996) as well as cytokines (Nawashiro et al., 1997; Yanamoto et al., 2000a; Bernaudin et al., 1999). It is noteworthy that most of these models have been described in the rat and there are only few studies of ischemic tolerance involving mice (Nawashiro et al., 1997; Prass et al., 2000; Miller et al., 2001). Ischemic preconditioning has been also described in vitro on neuronal cultures by mimicking the ischemic insult with sublethal oxygen and glucose deprivation (Bruer et al., 1997; Khaspekov et al., 1998) or with metabolic inhibitors (Weih et al., 1999).
In these in vitro models of preconditioning as well as during in vivo ischemic preconditioning, the reduction of oxygen availability may constitute one of the early cellular signals leading to tolerance. In vivo hypoxia has been reported to induce tolerance against global hypoxic–ischemic brain injury in the neonatal rat (Gidday et al., 1994; Ota et al., 1998; Vannucci et al., 1998; Bergeron et al., 2000). More recently, Miller et al. (2001) showed that hypoxic preconditioning also induces tolerance against focal transient cerebral ischemia in adult mice.
Because preconditioning often provides profound protection against ischemic brain injury, understanding its mechanisms may have important significance in the development of therapeutic strategies in stroke. Ischemic preconditioning seems to require de novo protein synthesis (Barone et al., 1998; Wiegand et al., 1999). A cascade of events, including N-methyl-
Results obtained on the neonatal rat brain suggest that hypoxia-inducible factor-1 (HIF-1) could be partly responsible for the induction of tolerance to ischemia by hypoxic preconditioning, because exposure of neonatal rats to 8% oxygen for 3 hours induces the expression of HIF-1 α and its target genes (Bergeron et al., 2000; Jones and Bergeron, 2001). In addition, desferrioxamine and CoCl2, two agents that are well known to promote HIF-1 activation (Ehleben et al., 1997), were also able to induce tolerance against hypoxia–ischemia in the neonate rat brain (Bergeron et al., 2000). HIF-1 is an important transcription factor that regulates gene expression in the brain and other tissues in response to decreases in oxygen availability. Moreover, HIF-1 and numerous of its target genes are increased after cerebral ischemia (Hayashi et al., 1997; Bergeron et al., 1999; Jin et al., 2000a; Marti et al., 2000). HIF-1 targets genes such as erythropoietin and vascular endothelial growth factor (VEGF) have been shown to protect the brain against ischemia (Hayashi et al., 1998; Bernaudin et al., 1999; Sakanaka et al., 1998; Sadamoto et al., 1998; Alafaci et al., 2000; Calapai et al., 2000; Jin et al., 2000b; Siren et al., 2001). This suggests that HIF-1 might be involved in the establishment of ischemic tolerance in the brain.
Although preexposure to hypoxia has been reported to induce tolerance to subsequent global ischemia in the newborn rat brain (Gidday et al., 1994; Ota et al., 1998; Vannucci et al., 1998; Bergeron et al., 2000) and, very recently, to focal transient cerebral ischemia in the adult mouse brain (Miller et al., 2001), it is not known whether hypoxia protects the adult brain against focal permanent cerebral ischemia. In the present work, we established a novel in vivo model of tolerance against focal permanent cerebral ischemia in the adult mouse by exposing animals to normobaric hypoxia and determined whether HIF-1 α and some of its target genes, erythropoietin and VEGF, were induced by this model of preconditioning. This model is useful to further study the mechanisms of ischemic tolerance in the adult brain and, thereby, to identify potential novel therapeutic targets in the field of stroke.
MATERIALS AND METHODS
All animal experiments were approved by the local ethics committee and governed by the pertinent national legislation. Male adult Swiss mice (30 ± 4 g, CERJ, Le Genest Saint Isle, France), having free access to food and water, were used for all experiments.
Hypoxic preconditioning treatment
Normobaric hypoxia (8% O2 during 1 hour, 3 hours, or 6 hours) was achieved by replacement of oxygen by nitrogen in a tight chamber (1.5 m3) in which oxygen fraction (Servomex, St. Denis, France), temperature, and hygrometry were continuously controlled. Exposure of mice to hypoxia led to an increase in their respiratory rate and a decrease in their locomotor activities. When withdrawn from the hypoxic chamber, the animals immediately recovered their respiration rate and behavior. No weight loss, as well as no evident histopathologic injury, was observed at any time after hypoxia.
Hypoxic preconditioning was performed on distinct animals before ischemia (Figs. 1A and 1B) or before analysis of HIF-1α, erythropoietin, and VEGF brain expression (Fig. 1C). In the latter case, the mice were killed by cervical dislocation and the brains were removed at various times (0 hours, 12 hours, 24 hours) after several durations (1 hour, 3 hours, 6 hours) of hypoxia. Killing of the animals was performed in the chamber when no reoxygenation was desired.
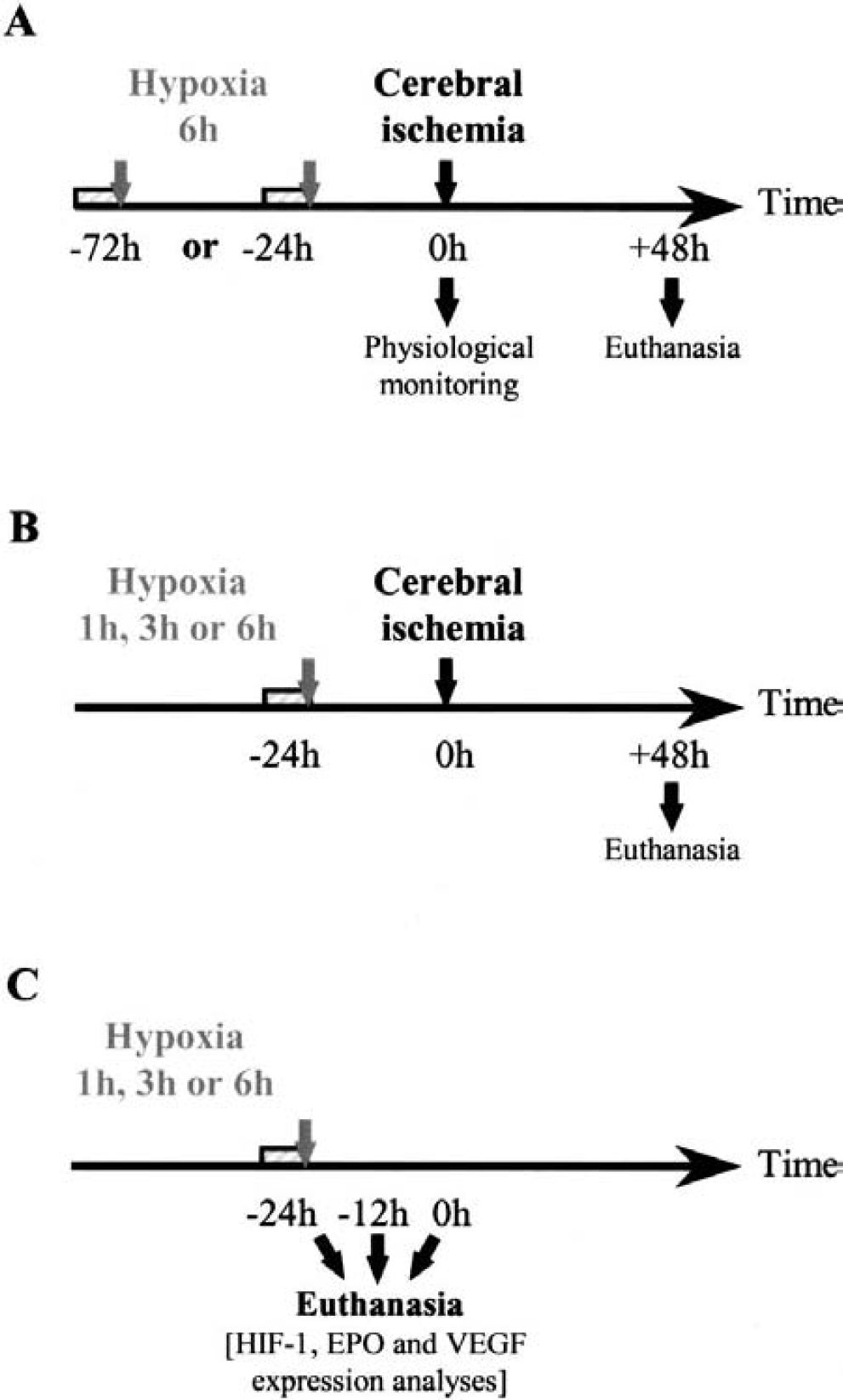
Schematic representations of the protocol used for studies on the hypoxic preconditioning effect on cerebral ischemia (
Focal cerebral ischemia
For each protocol, animals were divided into two groups: the preconditioning groups were subjected to hypoxia, whereas the control groups received only the ischemic insult. In an attempt to determine the time course of ischemic tolerance, hypoxia (6-hour duration) was performed either 24 hours or 72 hours before ischemia (Fig. 1A). Because the delay of 24 hours was the most efficient in terms of neuroprotection, it was chosen for the other experiments. The duration of hypoxia was then decreased to 3 hours and 1 hour to determine the minimal duration necessary to induce tolerance (Fig. 1B). Focal ischemia was induced by permanent occlusion of the left middle cerebral artery under halothane (1%–1.5% in N2O/O2: 2/1) anesthesia, as reported previously (Welsh et al., 1987; Bernaudin et al., 1999). Briefly, a skin incision was made between the orbit and ear. Under an operating microscope, an incision was made dividing the temporal muscle, and the left lateral aspect of the skull was exposed by reflecting the temporal muscle and surrounding soft tissue. The distal course of the middle cerebral artery was then visible through the translucent skull. A small craniotomy was performed with a cooled dental drill. The left middle cerebral artery was then coagulated by bipolar diathermy, the muscle and soft tissue were replaced, and the incision was sutured. Under halothane anesthesia, mice were killed 48 hours later and the brains were removed and stored at −80°C until use.
Physiological parameters
During surgery, rectal temperature was measured and maintained around normal values (37.1 ± 0.4°C) by a homeothermic blanket (Harvard, Les Vlis, France). To avoid the increase in surgery duration because of a more extensive physiologic monitoring, distinct animals from those receiving the ischemic insult were used to control cardiovascular (mean arterial pressure; Stoelting monitor; Phymer, Paris, France) and biochemical (blood gases, pH, hematocrit; Ciba Corning 238 analyzer) parameters. Measurements were performed in the same conditions of anesthesia as those used for the ischemia experiments (i.e., dose and duration), in mice subjected (n = 7) or not (n = 7) to preconditioning hypoxia (8% O2, 6 hours) 24 hours earlier.
Infarct volume measurement
Coronal brain sections (20 μm) were cut on a cryostat, stained with thionin and the infarcted areas of each section were measured with a Rag 200 image analyzer (Biocom) in a masked manner. Total infarct volumes were calculated after integration of infarcted areas with the distance between each section level analyzed (400 μm) with edema correction. Statistical significance was determined using analysis of variance followed by a Fisher protected least significant difference test. Data are presented as mean ± SD.
RNA isolation and semiquantitative reverse transcription–polymerase chain reaction analysis
Total RNA was prepared from whole hemispheres by phenol/chloroform extraction using the RNAxel extraction kit (Eurobio, Les Vlis, France) and total RNA (1 μg) was reverse-transcribed with Promega's RT (reverse transcription) system (Promega, Lyon, France). The resulting cDNAs (1 to 3 μL) were subjected to amplification in a total volume of 50 μL containing 10X buffer, 1.5 to 4 mmol/L of MgCl2 depending on the primers, 0.2 mmol/L each of dNTP, 2.5 U Taq polymerase (Promega), and a pair of specific primers (0.2 μmol/L each). Primers for erythropoietin, VEGF, and β-actin have been used as published, respectively (Ringheim et al., 1995; Marti and Risau, 1998; Bernaudin et al., 2000). The polymerase chain reaction (PCR) temperature profile consisted of 25 to 40 cycles at 95°C for 30 seconds, 55°C or 58°C for 30 seconds, and 72°C for 60 seconds, followed by a final extension period at 72°C. Forty microliters of the PCR products were separated by 1.5% agarose gel electrophoresis and stained with ethidium bromide. RT–PCR products were 449 bp for erythropoietin, 644 bp for VEGF164, 512 bp for VEGF120, and 539 bp for β-actin.
Preparation of total, cytosolic, and nuclear protein extracts
For total protein extracts, whole hemispheres tissue lysates were prepared as previously described (Hayashi et al., 1997) at different times after hypoxia. For cellular cytosolic and nuclear protein extracts, whole hemispheres were lysed after different times of hypoxia and reoxygenation in the following buffer: 20 mmol/L HEPES, 10 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L dithiothreitol, 0.2% NP40, 10% glycerol, 1 mmol/L PMSF, 0.1 mmol/L vanadate, and 1 μg/mL protease inhibitors in distilled water (Sigma, L'Isle d'Abeau Chesnes, France). After 5 minutes on ice, samples were centrifuged for 10 minutes at 13,000g. Supernatants (cytosolic extracts) were kept at −80°C. Pellets were resuspended in 50 μL of buffer containing: 350 mmol/L NaCl, 20% glycerol, 20 mmol/L HEPES, 10 mmol/L KCl, 1 mmol/L EDTA, 1 mmol/L PMSF, 0.1 mmol/L vanadate and 1 μg/mL protease inhibitors in distilled water. Suspension was mixed vigorously with tips and incubated on ice for 30 minutes. Then samples were centrifuged for 10 minutes at 13,000 g and supernatants (nuclear extracts) were kept at −80°C.
Western blot analysis
Thirty to 60 μg of protein was separated by sodium dodecylsulfate–polyacrylamide gel electrophoresis as described previously (Hayashi et al., 1997). Western blot analyses were performed using rabbit polyclonal antibodies for erythropoietin (1:500; R&D System Europe Ltd., Oxon, U.K.), VEGF (1:150; Santa Cruz Biotechnology, Heidelberg, Germany), and HIF-1α (1:100; gift from Dr. Jacques Pouysségur, France). Peroxidase-coupled anti-rabbit immunoglobulin G (1:80,000; Sigma) was used in secondary incubations followed by the detection of reactive bands by chemiluminescence (NEN, Paris, France).
RESULTS
Hypoxic preconditioning induces tolerance to focal cerebral ischemia in the adult mouse
Normobaric hypoxia (8% O2, 6 hours), performed 24 hours before focal permanent cerebral ischemia, reduced infarct volume when compared with the control group (−31%, P < 0.01) (Figs. 2A and 2C). When hypoxia was performed earlier, i.e., 72 hours before ischemia, the decrease in infarct volume (−12%) was not significant, suggesting that the tolerance tended to disappear (Fig. 2A). In addition, a shorter period of hypoxia (1 hour or 3 hours instead of 6 hours), performed 24 hours before ischemia, also reduced the infarct volume when compared with animals not exposed to hypoxia (−36%, P < 0.01 or −22%, P < 0.05, respectively) (Fig. 2B). There was no correlation between the duration of hypoxia (1 hour, 3 hours, 6 hours) and the infarct volume: the extent of neuroprotection obtained in mice subjected to either 1 hour, 3 hours, or 6 hours of hypoxia was not statistically different.
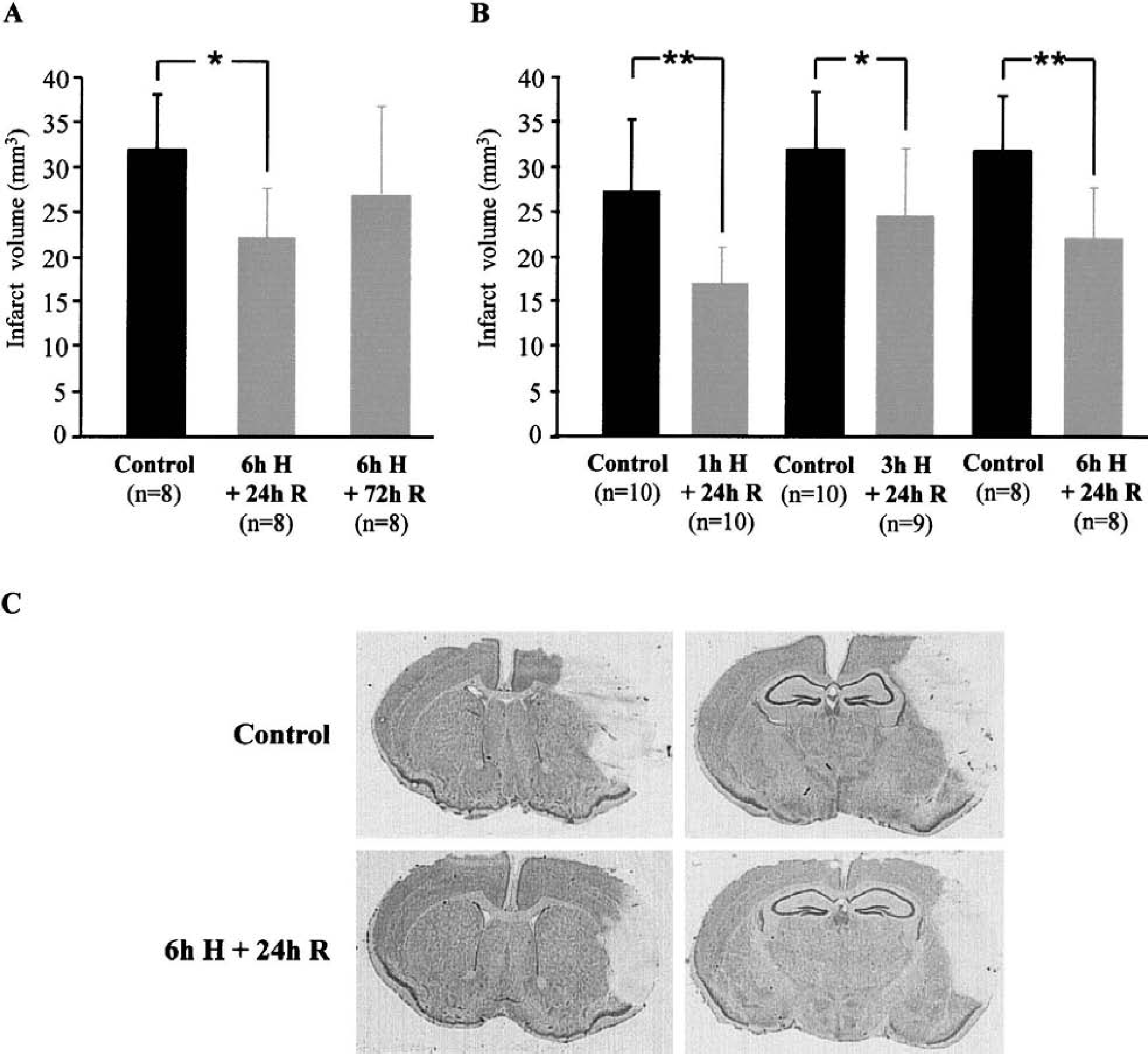
Effect of preconditioning normobaric hypoxia (8% O2) on infarct volumes obtained after focal permanent cerebral ischemia.
The neuroprotective effect of preconditioning cannot be explained by changes in systemic cardiovascular and biochemical parameters at the moment of ischemia because hypoxia, when performed 24 hours earlier, did not modify rectal temperature, blood gases, pH, mean arterial pressure, and hematocrit when compared with control animals (Table 1).
Effects of preconditioning normobaric hypoxia (8% O 2 , 6 hours) on physiologic and biochemical parameters
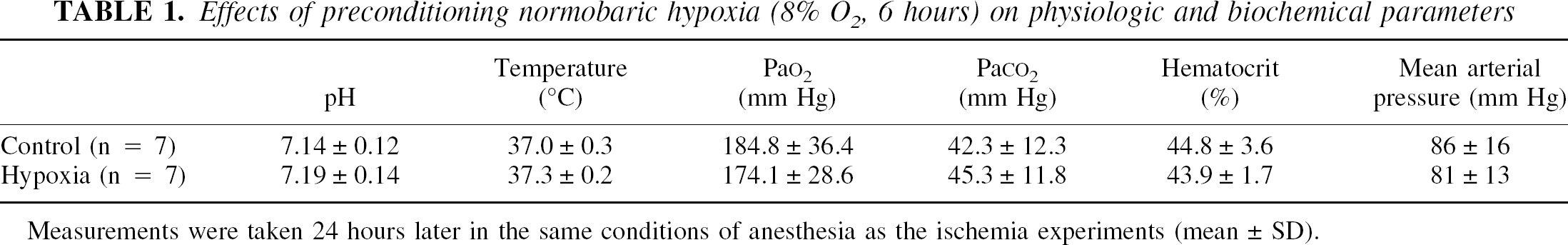
Measurements were taken 24 hours later in the same conditions of anesthesia as the ischemia experiments (mean ± SD).
Hypoxic preconditioning induces hypoxia-inducible factor-1α activation
Exposure of mice to hypoxia (8% O2; 1 hour, 3 hours, and 6 hours) induced an increase in HIF-1α expression in nuclear extracts of the whole hemisphere, when compared with control animals, suggesting the activation of the transcription factor. This increase in HIF-1α content was not detected in the cytoplasm, but only in the nucleus, suggesting either a rapid translocation to the nucleus or a stabilization of the protein already present in the nucleus (Fig. 3A). Increase in HIF-1α expression started as early as 1 hour after the beginning of hypoxia, was maximal at 3 hours and maintained after 6 hours of hypoxia. Although reoxygenation markedly reduced HIF-1α increase expression induced by hypoxia, nuclear HIF-1α expression was still evident 12 hours and 24 hours after exposition to hypoxia (Fig. 3B).
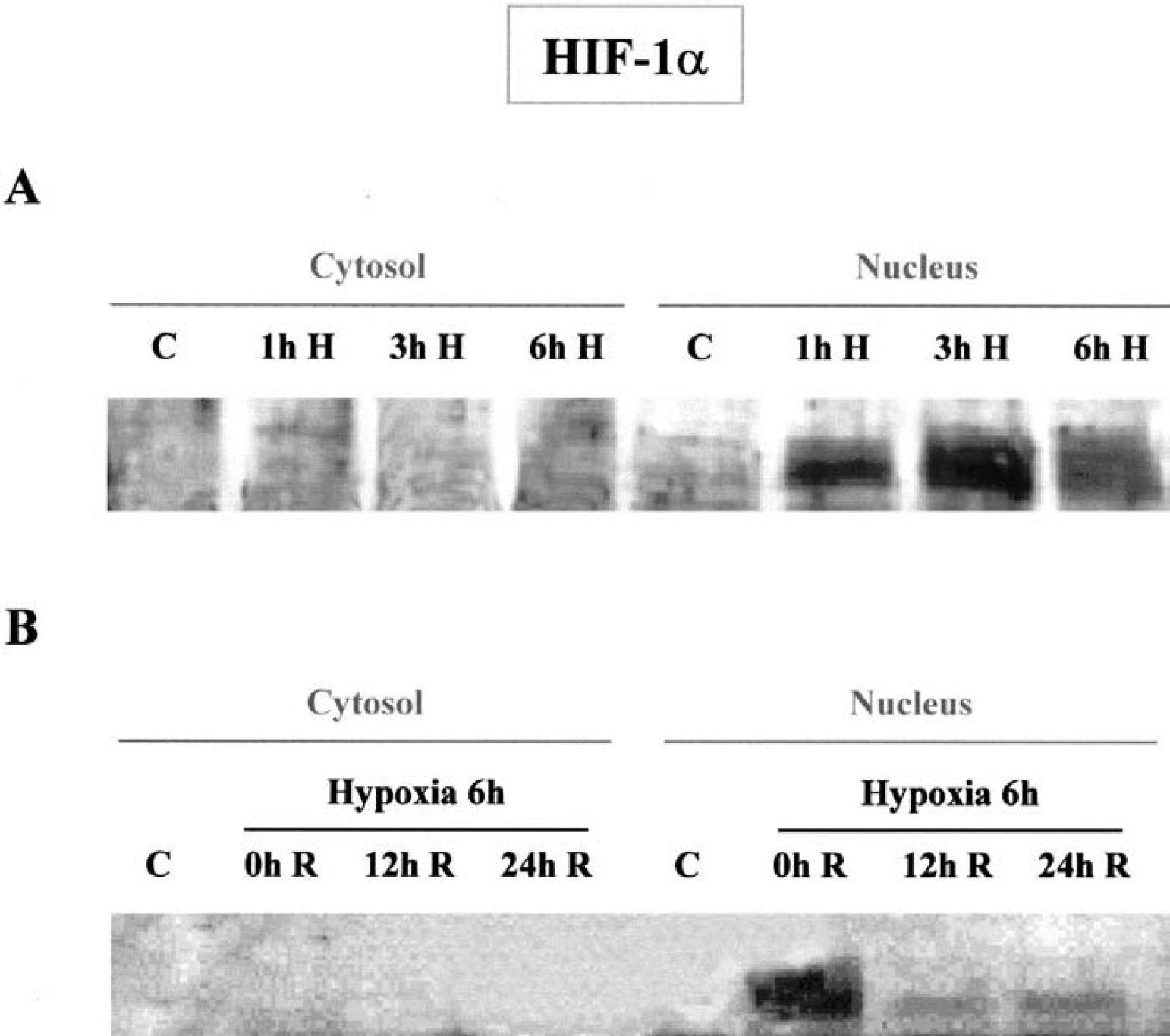
Western blot analysis of hypoxia-inducible factor-α (HIF-1α) subunit in cytosolic and nuclear extracts of the whole brain hemisphere of mice subjected, or not, to normobaric hypoxia (8% O2).
Regulation of VEGF and erythropoietin expression by hypoxic preconditioning
The results obtained by RT–PCR show that hypoxia (8% O2; 1 hour, 3 hours, and 6 hours) increased erythropoietin mRNA levels when compared with normoxic control (Fig. 4A). Interestingly, the increase observed after 6 hours of hypoxia disappeared after 12 hours and 24 hours of reoxygenation (Fig. 4B).
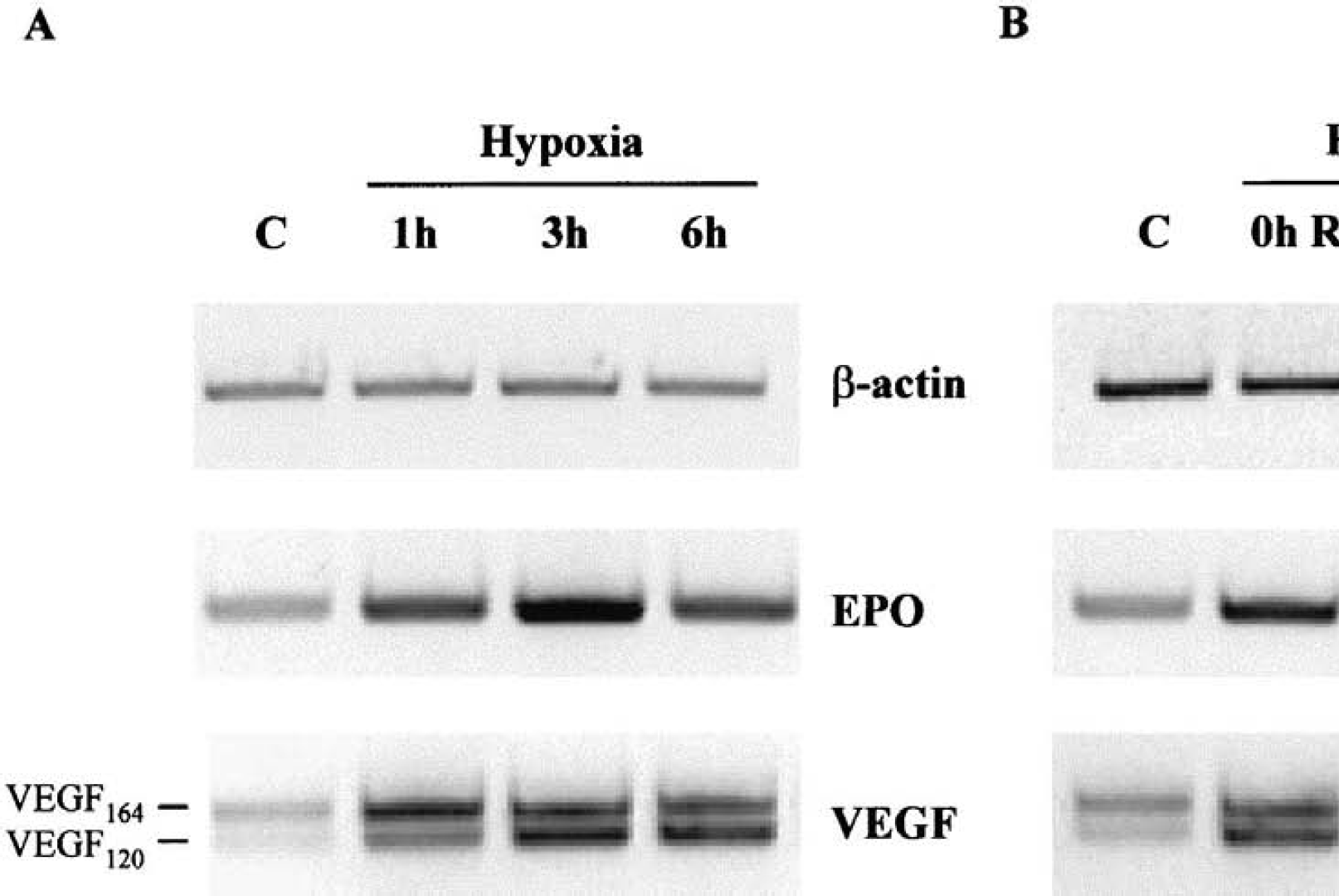
Reverse transcriptase–polymerase chain reaction (RT-PCR) analysis of erythropoietin (EPO), vascular endothelial growth factor (VEGF), and β-actin mRNA expression in the whole brain hemisphere of mice subjected, or not, to normobaric hypoxia (8% O2).
Two bands, corresponding to the two mRNA of VEGF164 and VEGF120 isoforms, were detected by RT–PCR. Both of them were also increased after 1 hour, 3 hours, and 6 hours of hypoxia (Fig. 4A) but no significant increase was observed after 6 hours hypoxia followed by 12 hours or 24 hours of reoxygenation (Fig. 4B).
Because confirmation of protein expression and time course of translation is of primary importance, we analyzed by Western blotting the expression of erythropoietin and VEGF at the protein level in the whole brain from total protein extract.
As shown in Fig. 5, 6 hours of hypoxia lead to an increase of brain erythropoietin expression after 24 hours of reoxygenation. Under reducing conditions, Western blot analysis showed a single band of VEGF protein with a molecular mass of 23 kd, which could correspond to the VEGF164 isoform. Expression of this VEGF isoform was increased as early as 6 hours of hypoxia and was maintained upregulated after 24 hours of reoxygenation (Fig. 5).
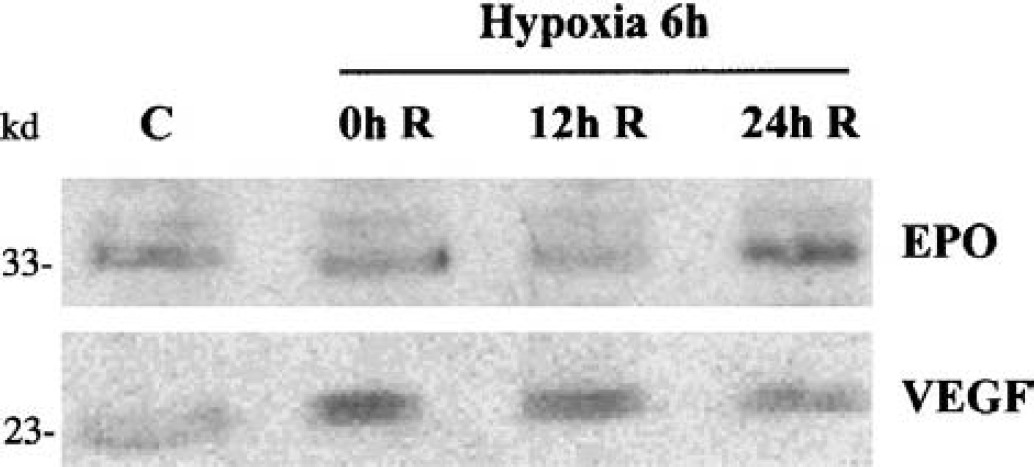
Western blot analysis of erythropoietin (EPO) and vascular endothelial growth factor (VEGF) protein expression in total extracts from whole brain hemisphere of mice subjected, or not, to 6 hours of normobaric hypoxia (8% O2) followed by 0 hours, 12 hours, or 24 hours of reoxygenation. The Western blots presented are representative of results obtained from three individual experiments. C, control; H, hypoxia; R, reoxygenation.
DISCUSSION
Induction of ischemic tolerance after hypoxic challenge has already been reported in vitro and in vivo. However, in the latter case, this model of tolerance had been described until now against global hypoxia–ischemia in the neonate. Very recently, one study reported the preconditioning effect of hypoxia in the adult mice, but only against transient focal cerebral ischemia (Miller et al., 2001). Our study shows therefore for the first time that hypoxic preconditioning protects the adult brain against a permanent model of focal ischemia. In addition to the model, this study reports two phenomena that had been described earlier but not in the hypoxic conditions shown to induce tolerance in the adult brain. Indeed, our work shows that (1) the degree and duration of hypoxia leading to tolerance to focal ischemia are also able to induce the expression of HIF-1α and its target genes erythropoietin and VEGF; and (2) the time course of erythropoietin and VEGF induction is compatible with the time course of tolerance induction (i.e., the presence of the two proteins when ischemia is performed).
Although it is described that hypoxia can induce adaptive mechanisms to protect the brain against ischemia in the neonatal brain (Gidday et al., 1994; Vannucci et al., 1998; Bergeron et al., 2000), the effect of hypoxic preconditioning on cerebral ischemia in the adult brain is still unclear. Recently, Miller et al. (2001) showed that hypoxic preconditioning (11% O2, 2 hours) performed 48 hours before the ischemic insult can also induce tolerance to focal transient, but not permanent cerebral ischemia in mice. In the present work, we established a novel model of tolerance against focal permanent cerebral ischemia in the adult mouse by exposing animals to normobaric hypoxia (8% O2, 6 hours), 24 hours before the insult. Indeed, exposure of mice to hypoxia induced a delayed (24 hours) tolerance to focal permanent cerebral ischemia. The discrepancy between our results and those of Miller et al. (2001) could be explained by the difference of the degree of hypoxia that we used (8% instead of 11% O2), suggesting that systemic hypoxia stimulus should be stronger to induce tolerance against the permanent occlusion of a major cerebral artery compared with the transient occlusion. In addition, the delay between hypoxia and ischemia determines the efficacy of preconditioning: our results suggest that tolerance achieved by exposition to 6-hour hypoxia is short lasting (<72 hours). One might suggest that the delay between preconditioning and ischemia should be shorter than 48 hours to show a tolerance against this model of permanent ischemia. Therefore, the discrepancy between our results and those of Miller et al. (2001) could be also explained by the difference of the delay between the hypoxic preconditioning and ischemia that we used (24 hours instead of 48 hours). Our results show that reduction of the hypoxic preconditioning duration to 1 hour or 3 hours is still enough to decrease the infarct volume compared with the control group. There was no correlation between the duration of hypoxia (1 hour, 3 hours, 6 hours) and the infarct volume. The duration of preconditioning hypoxia described in the literature is approximately 2 to 3 hours, which is longer than our shortest hypoxic episode. Although we did not determine the minimal duration of hypoxia that is necessary to activate this cascade of events, our results show that 1 hour of exposition hypoxia is enough to induce tolerance, and that this neuroprotection cannot be improved by increasing the duration of hypoxia.
As hypoxic preconditioning is a noninvasive and reproducible model for ischemic tolerance, this model could be very useful to further study the mechanisms of ischemic tolerance in the adult brain and, thereby, to identify potential new therapeutic targets in the field of cerebral ischemia.
HIF-1 is a heterodimer made of two protein subunits, HIF-1α and HIF-1β (Wang et al., 1995). HIF-1β is constitutively expressed, whereas HIF-1α expression is tightly regulated by cellular oxygen concentration (Huang et al., 1996). Thus, HIF-1α determines HIF-1 DNA-binding activity and transcriptional activity during hypoxia. We investigated the effect of hypoxic preconditioning on HIF-1α expression in the adult brain. Our results show that the temporal profile of ischemic tolerance parallels the pattern of HIF-1α activation. Indeed, we showed that hypoxic preconditioning of 1 hour, 3 hours, or 6 hours induced an increase of HIF-1α expression and translocation into the nucleus. These results are in accordance with the study of Bergeron et al. (1999), describing an increase in HIF-1α expression after a slightly more severe normobaric hypoxia (6% O2, 4.5 hours) in the adult rat brain. Moreover, Chavez et al. (2000) showed an increase of HIF-1α expression in the brain cortex of adult rats after 6 hours of hypobaric hypoxia (10% O2). As we showed that 1 hour, 3 hours, or 6 hours of hypoxic preconditioning can induce ischemic tolerance, our results suggest that this transcription factor might be implicated in the establishment of tolerance in this model.
To further investigate the role of HIF-1 and its target genes in the tolerance mechanisms, we thus analyzed the expression of two well-known HIF-1 target genes: VEGF and erythropoietin. Our data revealed an increase in erythropoietin and VEGF mRNA expression after the hypoxic preconditioning of 1-hour, 3-hour, or 6-hour duration. The reoxygenation after the 6-hour hypoxic treatment reduced this increase at the mRNA level, but an increase at the protein level of both erythropoietin and VEGF is occurring during this period and is still present when the ischemic insult is performed. Therefore, these results suggest that the erythropoietin and VEGF increase induced by preconditioning hypoxia could protect the brain against the subsequent focal permanent cerebral ischemia. These results are in accordance with our previous data showing that intracerebral erythropoietin pretreatment protects the brain against permanent focal cerebral ischemia (Bernaudin et al., 1999) and the several other recent in vivo and in vitro studies showing a protective effect of erythropoietin in brain ischemia (Sakanaka et al., 1998; Sadamoto et al., 1998; Alafaci et al., 2000; Calapai et al., 2000; Siren et al., 2001; Sinor and Greenberg, 2000). Erythropoietin appears to protect against ischemia through a direct effect on neurons (Morishita et al., 1997; Bernaudin et al., 1999; Sinor and Greenberg, 2000; Digicaylioglu and Lipton, 2001). This effect could be mediated by the inhibition of the glutamate exocytosis (Kawakami et al., in press). MAPK and PI(3)kinase/Akt pathways seem to be crucial for the neuroprotective effect of erythropoietin because specific inhibitors of these pathways largely abolished the erythropoietin-induced protection against hypoxia-induced cell death in primary neuronal cultures (Siren et al., 2001). In addition, recent data (Digicaylioglu and Lipton, 2001) showed that erythropoietin-mediated neuroprotection involves crosstalk between Jak2 and NF-κB signaling cascades. Similarly, in vitro and in vivo data reported a protective effect of VEGF against cerebral ischemia (Hayashi et al., 1998; Jin et al., 2000b). In addition to its angiogenic effect, VEGF directly protects the neurons from ischemia, and this effect also could be mediated by the phosphatidylinositol 3-kinase (PI3-K)/Akt and the mitogen-activated protein kinase kinase/extracellular-signal-regulated kinase pathways (Jin et al., 2000b; Matsuzaki et al., 2001).
Interestingly, two opposite stresses, i.e., hyperoxygenation (Prass et al., 2000) and hypoxia, lead to similar response of the brain, i.e., the activation of endogenous neuroprotective mechanisms. As pointed out earlier, many different stimuli are able to induce tolerance against cerebral ischemia and it is actually not clear whether these models activate distinct pathways or if all of them share a common signal. According to the latter hypothesis, the oxygen free radicals constitute potential common candidates: indeed, their production is induced by several preconditioning stimuli, including hyperbaric oxygenation (Noda et al., 1983), and free radicals are known to activate certain transcription factors (such as NF-κB) that may contribute to tolerance (Blondeau et al., 2001). Several studies have shown that hypoxia induces free-radical production, such as NO, and activation of NF-κB (Simakajornboon et al., 2001) in the brain. In particular, Rauca et al. (2000) reported the importance of free-radical production during hypoxia preconditioning (9% O2, 1 h). Moreover, reactive oxygen species, such as NO, have been shown to increase HIF-1α expression (Haddad and Land, 2001; Sandau et al., 2001), which would suggest that HIF-1α might also be implicated in other models of preconditioning associated with free-radical production. Even though we did not measure the production of oxygen free radicals, nor the NF-κB activation, during or after hypoxia, an increase of free-radical signaling by hypoxic preconditioning cannot be excluded. Such a hypothesis could reconcile our results with the model of tolerance induced by hyperbaric oxygenation. Conversely, it is also conceivable that these apparently two contradictory models of preconditioning, hypoxia and hyperoxia, lead to tolerance by completely distinct mechanisms.
Besides the cascade of events after HIF-1α activation, other mechanisms could explain the preconditioning effects of hypoxia. These hypotheses are based on studies in the literature reporting the protection afforded by hypoxia against stresses other than ischemia. For example, exposure to moderate hypoxia (10% O2) protects the rat brain against hippocampal damage induced by subsequent severe hypoxia (3% O2) (Gorgias et al., 1996). This tolerance is closely related to an increase in the cerebral endogenous antioxidant defense, a mechanism that might contribute to the neuroprotection observed against focal ischemia in our conditions. Because normobaric hypoxia (9% O2, 8 hours) has been reported to induce tolerance against kainate-induced seizures (Pohle and Rauca, 1994), one might suppose that the mechanisms of hypoxic preconditioning involve a reduction of the excitotoxic events during focal ischemia. Such a hypothesis has already been suggested for other models of ischemic tolerance (Douen et al., 2000). Although we did not investigate these hypotheses, we cannot exclude a potential implication of these signaling pathways in the development of ischemic tolerance.
Finally, it is worth noting that hypoxia constitutes a systemic stress, affecting organs other than the brain, which may contribute to the neuroprotection against cerebral ischemia. Indeed, several systemic responses to hypoxia are commonly observed: the hypoxic ventilatory response, which is mediated by peripheral chemoreceptors; an elevation in cardiac pulse rate and output; dilatation of blood vessels; and the liver and kidney responses to hypoxia. All of these systemic modifications, by altering the levels of circulating mediators, could play a role in the later development of ischemic tolerance within the brain. As presented in Table 1, our results show, however, that there were at the time of ischemia (24 hours after hypoxia) no alterations in the systemic cardiovascular and biochemical parameters that classically influence the brain outcome in this disease.
In conclusion, our present work shows for the first time that, in the adult brain, hypoxic preconditioning (8% O2, 1 hour, 3 hours, or 6 hours) protects against permanent focal cerebral ischemia and that this tolerance is associated with changes in the expression of HIF-1α and its target genes, erythropoietin and VEGF. Although these results do not show a direct cause-relation effect of HIF-1 target genes on tolerance induction, the increased expression of these genes at the time of tolerance appearance provides an indirect argument speaking for the possible implication of HIF-1α and its target genes erythropoietin and VEGF in this phenomenon. Therefore, this study constitutes a first step allowing further experiments, using either a pharmacologic or a genetic approach to inhibit the activity of erythropoietin and/or VEGF, which could further argue for the involvement of these agents in tolerance. The implication of other genes that are regulated by hypoxia, under, or not, the control of HIF-1, should be further investigated. In this context, the effects of preconditioning hypoxia on the expression of GLUT-1 or glycolytic enzymes or both might be studied. Indeed, the initial event occurring during cerebral ischemia is a decrease of cerebral blood flow, leading to a failure of glucose delivery to the brain. An increase in glucose transport capacities could contribute to a better preservation of glucose supply, and therefore neuronal integrity, during ischemia (Lawrence et al., 1996). Furthermore, hypoxia has been shown to stimulate GLUT-1 expression both in vitro (Loike et al., 1992; Bruckner et al., 1999) and in vivo (Harik et al., 1994). Because the effects of preconditioning on the metabolic state of the brain are far from elucidated, the implication of GLUT-1 in our model of ischemic tolerance might be of interest. Furthermore, although not directly related to ischemic tolerance, it has been reported that a protein neosynthesis is implicated in the preconditioning effects of hypoxia (9% O2) against kainate-induced seizures (Emerson et al., 1999). Likewise, the implication of a protein neosynthesis in this model of tolerance would be of interest.
Understanding the mechanisms induced by hypoxia that lead to brain protection against ischemia might be very useful to identify potential new drug treatments not only in the field of adult cerebral ischemia, but also in the field of oxygen starvation of newborns.
Footnotes
Acknowledgments:
The authors thank Dr. Frank R. Sharp (Department of Neurology, University of Cincinnati, Cincinnati, U.S.A.) for helpful comments on the article.