
Abstract
In a previous study, we reported that the sustained increase in CBF concomitant with seizures induced by kainate is mainly due to the potent vasodilator nitric oxide (NO). However, the production site of NO acting at cerebral vessels was undetermined. In the present study, we investigated whether NO responsible for the cerebral vasodilation is of either neuronal or endothelial origin. We used a putative selective inhibitor of neuronal NO synthase, 7-nitro indazole (7-NI). CBF was measured continuously in parietal cortex by means of laser Doppler flowmetry in awake rats. Systemic variables and electroencephalograms were monitored. Kainate (10 mg/kg i.p.) was given to rats previously treated with saline (n = 8) or 7-NI (25 mg/kg i.p., n = 8) or L-arginine (300 mg/kg i.p., n = 8) followed 30 min later by 7-NI (25 mg/kg i.p.). Under basal conditions, 7-NI decreased CBF by 27% without modifying the mean arterial blood pressure. Under kainate, 7-NI prevented significant increases in CBF throughout the seizures despite sustained paroxysmal electrical activity. L-arginine, the substrate in the production of NO, prevented any decrease in CBF under 7-NI in basal conditions and partially, but nonsignificantly, reversed the cerebrovascular influence of 7-NI during seizures. In a separate group of rats (n = 6), inhibition of cortical NO synthase activity by 7-NI was assayed at 73%. The present results show that neurons are the source of NO responsible for the cerebrovascular response to seizure activity after kainate systemic injection.
In cerebral cortex, nitric oxide (NO) is constitutively produced in vascular endothelial cells and in some scattered neurons (Bredt et al., 1990) by activation of type III and type I NO synthases, respectively (Nathan and Xie, 1994). NO is a highly diffusible and potent vasodilator, whose synthesis may be nonselectively inhibited by L-arginine analogues, which decrease CBF and increase arterial blood pressure (Iadecola et al., 1994). The hypothesis of a tonic vasodilatory effect exerted by NO on cerebral blood vessels has thus been proposed. As far as seizures are concerned, several studies using analogues of L-arginine to inhibit NO synthesis have shown that NO mediates the increase in CBF following systemic administration of pentylenetetrazole (Faraci et al., 1993) or focal application of bicuculline (Pereira de Vasconcelos et al., 1995). However, there is no general agreement on this point regarding epileptogenic compounds reducing GABAergic neurotransmission (Wang et al., 1994). In rats undergoing limbic seizures induced by kainate (KA), we have previously shown a sustained increase in CBF (Pinard et al., 1987) that is significantly reduced by Nω-nitro-L-arginine methylester (L-NAME) (Rigaud-Monnet et al., 1994), indicating that NO is of major importance in CBF control in that type of seizure. But the principal NO production site has remained undetermined, since L-arginine analogues such as L-NAME block both type I and type III NO synthases.
On the one hand, the endothelium of blood vessels is the production site of numerous vasoactive factors, including NO. This cell layer is of major importance in the control of CBF, notably mediating the vascular response to cholinergic stimulation. On the other hand, there is morphological evidence that supports the hypothesis that neuronally derived NO participates in the control of CBF. Although only 2% of cortical neurons have been shown to contain type I NO synthase (Bredt et al., 1990), the presence of the enzyme along the entire length of the neuronal processes, the close association of NO synthase–containing neurons with the basal lamina of intraparenchymal microvessels (Iadecola et al., 1993), and the widespread diffusion of NO, calculated by modeling to be capable of traversing ≤100 μm within brain tissue (Wood and Garthwaite, 1994), make a role for neuronally derived NO in CBF regulation very likely. In addition, NO synthase immunoreactivity has been found in adventitial nerve fibers of cerebral arteries (Nozaki et al., 1993), which also raises the possibility that NO mediates neurogenic vasodilation in vivo. The aim of the present study was to determine whether NO, implicated in the control of CBF in seizures induced by KA, originates from neurons. The pharmacological tool used to selectively block type I NO synthase was 7-nitro indazole (7-NI), a putative inhibitor of neuronal NO synthase recently characterized in mice (Moore et al., 1993) and rats (Babbedge et al., 1993; Mackenzie et al., 1994). 7-NI interacts with both the pteridine and the substrate sites of NO synthase (Klatt et al., 1994).
EXPERIMENTAL PROCEDURES
All experimental procedures were carried out in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Experiments were performed under permit no. 02934 from the French Ministery of Agriculture. The protocols received full review and approval by the CNRS Animal Care and Use Committee before the experiments were conducted.
Surgical preparation
Male Wistar rats weighing 280–320 g (Charles River) were used in this study. They had access to food and water ad libitum and were housed in individual cages. Anesthesia was induced with 4% halothane and maintained with 1.5% halothane in 70% N2O–30% O2. Catheters were inserted into the femoral artery and vein and channeled subcutaneously to exit at the neck. The skull was exposed, and a 4 × 4-mm area of the parietal bone was thinned to translucency with a saline-cooled drill. Two small screws were sealed with dental cement on the frontal bone, in order to fix a proprietary miniature stereotaxic apparatus (Bonvento et al., 1994) used for positioning the laser probe. Three silver electrodes were also placed in the bone for EEG recording.
Experimental protocol
One day later, the awake, spontaneously breathing rats were placed in hammocks where they could freely move their heads and limbs. The arterial catheter was connected to a pressure transducer (Statham P23Db) for continuous monitoring of mean arterial blood pressure (MABP) and regular measurement (every 30 min) of arterial blood gases and pH (Corning 178). The EEG measurements were recorded on a polygraph (ECM).
The miniature stereotaxic apparatus was fixed to each animal's head, allowing for laser probe positioning over the thinned bone, using a microscope to avoid the large pial vessels. CBF was monitored in the parietal cortex by laser Doppler flowmetry (Moor Instruments). The signals from the flowmeter were monitored online via an analog-to-digital converter in a microcomputer for analysis of continuous CBF measurements. CBF was measured in arbitrary units. A baseline CBF value of 280–350 arbitrary units was typical. The cerebrovascular reactivity to hypercapnia was systematically tested using 5% carbon dioxide in oxygen for 3 min.
After a 90-min period of rest, the rats were randomly given saline (n = 8), 7-NI (25 mg/kg i.p., n = 8), or L-arginine (L-arg) (300 mg/kg i.p., n = 8) followed 30 min later by 7-NI (25 mg/kg i.p.) and an intravenous infusion of L-arg (5 mg/kg/min). In all 7-NI-treated rats, 7-NI was repeated twice at 1-h intervals. Kainate (10 mg/kg i.p.) was given 30 min after the first injection of saline or 7-NI in all three treatment groups. All variables were monitored throughout the experiment: under basal conditions, under treatment, and for 120 min after KA injection. All CBF data were collected and transformed to percentage changes from control values recorded during the 10 min before the first injection.
Determination of NO synthase activity
NO synthase catalytic activity was measured in an additional series of animals (n = 6) by the technique described by Bredt and Snyder (1989), as modified by Clavier and colleagues (1994). Brain biopsy samples were obtained from parietal cortex of rats given either saline (n = 3) or 7-NI (25 mg/kg i.p.) (n = 3) 30 min previously. In all experiments and for each tissue studied, samples from saline-treated and 7-NI-treated rats were always processed in parallel. NO synthase activity was assessed in vitro by determining the conversion of [14C]L-arginine to [14C]L-citrulline, the formation of which is stoichiometric with NO synthesis. Brain tissue was sonicated in a Tris-EDTA (ethylenediamine-tetraacetic acid) buffer, at pH 7.4, for 30–40 s. Tissue homogenate was then centrifuged at 10,000 g/min for 15 min at 4°C, and the supernatant was sampled. In brief, 25 μl of supernatant was added to 100 μl of reaction mixture containing 1 μmol [14C]L-arginine, 1 mmol NADPH, and 1 mmol calcium chloride and incubated for 30 min at 37°C. The reaction was stopped by addition of 2 ml of stop buffer (30 mmol/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES]), 3 mmol EDTA (pH 5.2). [14C]L-citrulline was eluted over a chromatographic column using Dowex 50X8-100 resin, Na+ form, at pH 7.0. The radioactivity in the samples was determined in a Packard liquid scintillation counter. Total citrulline recovered was calculated from specific activity of the [14C]L-arginine, correcting for counting efficiency, and was expressed as picomoles per minute per milligram protein. To assess that citrulline production was due to NO synthase activity, parallel samples were processed in the presence of 100 μmol L-NAME, demonstrating complete (>99%) inhibition of L-arg conversion. Protein concentration was measured by the method of Bradford (1976).
Materials
L-arg, Dowex 50X8-100 resin, KA, and L-NAME were obtained from Sigma Chemical Co. L-[U-14C]arginine (317 mCi/mmol) was obtained from Amersham. 7-NI was obtained from Research Biochemical International. L-arg and KA were dissolved in normal saline, and the pH was adjusted to 7.4. 7-NI was suspended in peanut oil by sonication.
Statistical analysis
Concerning CBF results, an analysis of variance (ANOVA) followed by Dunnett's test was used for each group to compare basal values with subsequent values (i.e., under either treatment and during seizures). Intergroup comparisons between basal values, between values under either treatment, and between values during seizures were performed using Sheffe's or Tukey's test. Differences in NO synthase activity among groups were determined by a one-way ANOVA followed by an unpaired Student's t test. To compare the delay in the onset of spike activity and status epilepticus between drug-treated and saline-treated groups, an ANOVA, followed by Dunnett's test, was used. A value of p < 0.05 was considered significant. All data are presented as the mean ± SD.
RESULTS
NO synthase activity
Baseline cortical NO synthase activity was 1.51 ± 0.38 and 0.41 ± 0.18 pmol/mg protein/min, respectively, in control rats and in 7-NI-treated rats. Intraperitoneal treatment with 7-NI induced a significant reduction of NO synthase activity (73%) in the parietal cortex.
Systemic and cerebrovascular variables
Control conditions.
All physiological variables were within normal limits. Arterial blood pressure, blood gases, and pH were similar in the three groups of rats under basal conditions (Table 1). Neither 7-NI alone nor the combined action of L-arg plus 7-NI significantly modified systemic variables. No abnormality was seen in the EEG. No marked change in behavioral activity was observed. Administration of L-arg produced no significant change in CBF compared either with basal values or with values obtained after saline administration in the control group. Administration of 7-NI produced a 27% significant reduction of CBF in rats untreated with L-arg. In contrast, when administration of 7-NI followed treatment with L-arg (L-arg + 7-NI), CBF did not significantly change.
Mean values (±SD) of arterial partial pressures of carbon dioxide (PaCO2) and oxygen (PaO2), pH, and mean arterial blood pressure (MABP), before (t = 0) and at various times after KA injection, in rats previously treated with saline (control), or 7-nitro indazole (7-NI), or L-arginine + 7-nitro indazole (L-arg + 7-NI) a
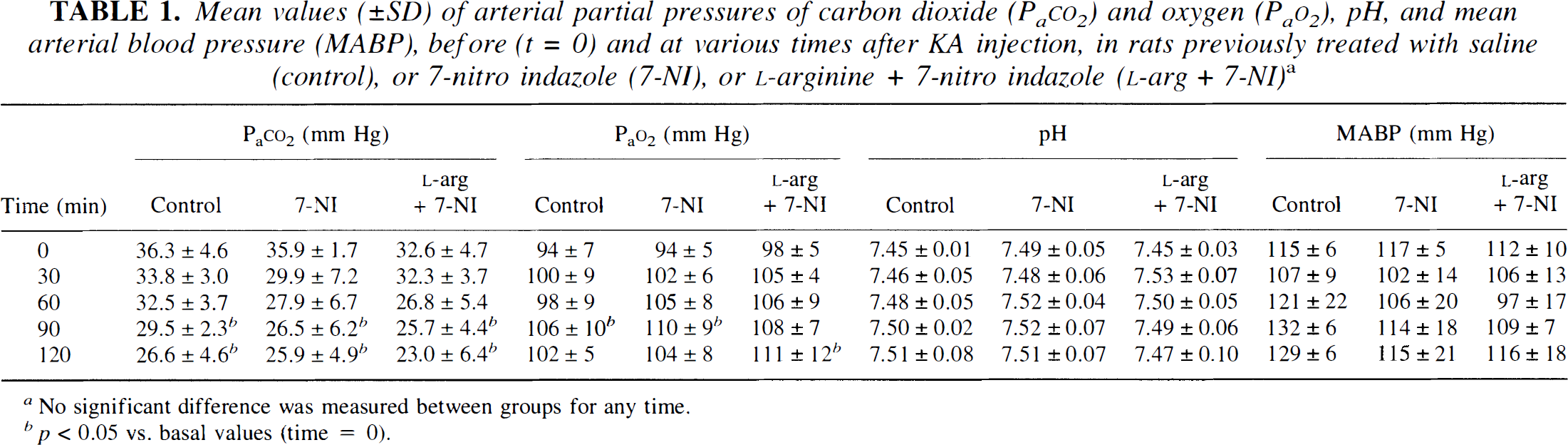
No significant difference was measured between groups for any time.
p < 0.05 vs. basal values (time = 0).
Under kainate.
All saline-treated and 7-NI-treated rats displayed typical limbic seizures under KA. In contrast, only eight of 12 L-arg + 7-NI–treated rats displayed seizure activity after KA administration. The remaining four rats (not displaying seizures) were not included in the study.
The delay between KA injection and onset of spike activity varied only slightly within each group. However, the intergroup comparison revealed that seizure activity occurred significantly sooner in 7-NI-treated rats (9 ± 3 min after KA injection) than in saline-treated or L-arg + 7-NI–treated rats (17 ± 6 min after KA injection). Similarly, status epilepticus started in 7-NI-treated rats significantly sooner (52 ± 7 min) than in saline-treated rats (65 ± 13 min). In L-arg + 7-NI–treated rats, status epilepticus started significantly later (76 ± 12 min) than in saline-treated rats. The maximal seizure activity, as evaluated by EEG, was similar in the three groups of rats. In all groups, spikes were not systematically associated with any behavioral changes. No visible difference in behavior (wet-dog shakes, masticatory movements, myoclonic jerks, or salivation) was observed between groups.
There was no significant difference between groups for changes in any systemic variables throughout the seizures (Table 1). In all groups, Pa
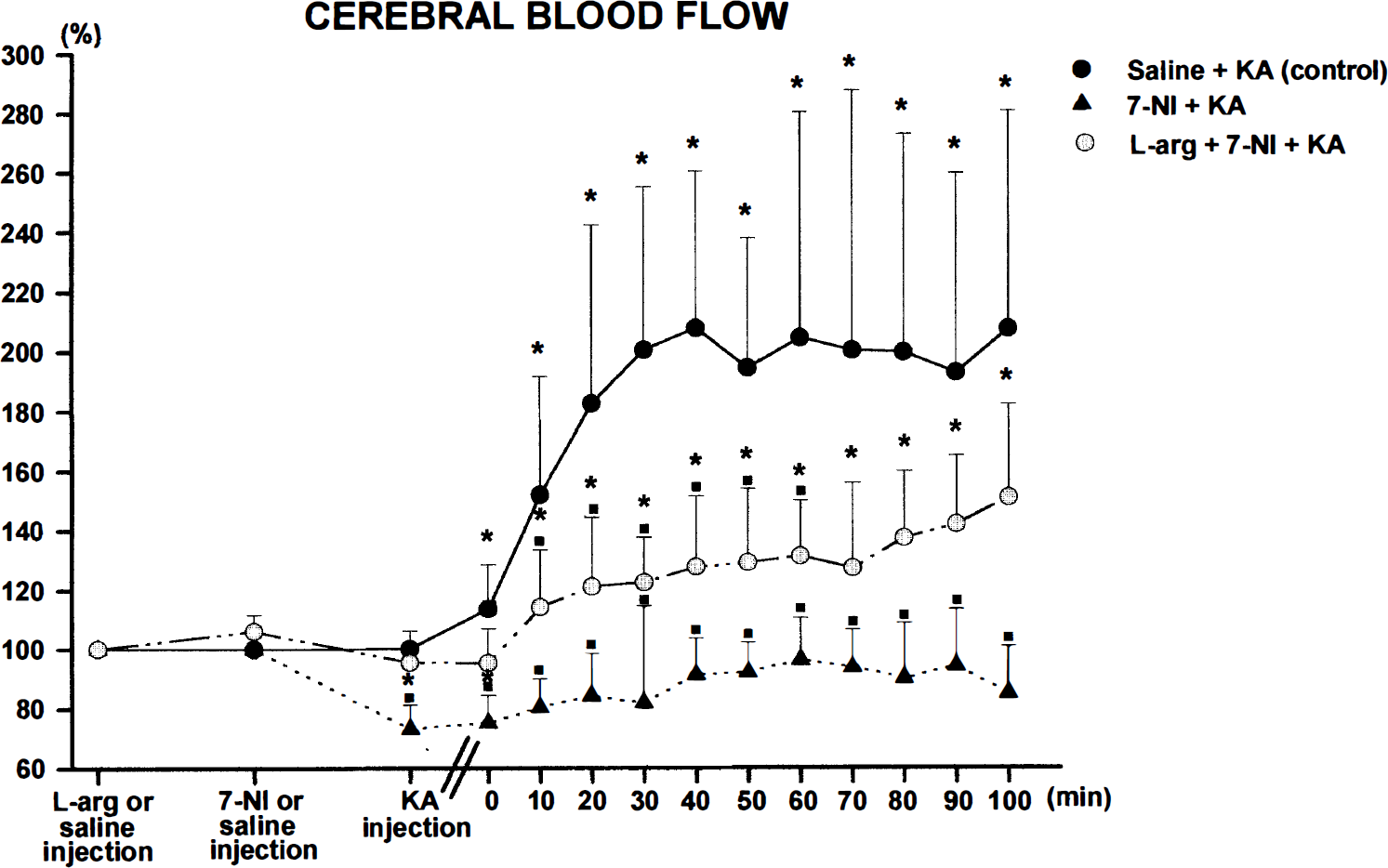
Mean changes in CBF (±SD) before and during seizures induced by KA (10 mg/kg i.p.). Changes are expressed as a percentage of the baseline value, considered 100%. Comparison of three rat groups. The control group (n = 8) was given saline (•), the 7-NI-treated group (n = 8) was given 7-NI (25 mg/kg i.p.) 30 min before KA and every hour thereafter (▴), and the L-arg + 7-NI–treated group (n = 8) was given L-arg (300 mg/kg i.p.) 60 min before KA and 7-NI (25 mg/kg i.p.) 30 min before KA (○). 7-NI injections were repeated hourly, whereas L-arg was continuously infused (5 mg/kg/min) throughout the seizures. CBF was measured by laser Doppler flowmetry in the parietal cortex of awake, freely breathing rats placed in hammocks. The intensity (amplitude, frequency, and duration of spike activity) of the seizures induced by KA was similar in the three groups. The lag between KA injection and onset of spike activity was shorter in 7-NI-treated rats than in control rats and L-arg + 7-NI-treated rats. Time 0 corresponds to the onset of spike activity. The asterisks indicate p < 0.05 vs. basal conditions. The filled square indicates p < 0.05 vs. control group (KA).
In 7-NI-treated rats, there was a transient, insignificant increase in CBF during the seizures when compared with either basal values before 7-NI administration or values under 7-NI. At all times after KA injection, CBF values were significantly different between saline-treated rats and 7-NI-treated rats. In L-arg + 7-NI–treated rats, CBF increased incrementally throughout the seizures, but to a smaller extent than in control rats. CBF values were significantly different between the control group and the L-arg + 7-NI group only during the first 60 min after onset of spike activity.
DISCUSSION
The principal finding of the present study is that the selective inhibitor of neuronal (type I) NO synthase, 7-NI, inhibits the increase in blood flow in the parietal cortex of rats subjected to seizures induced by KA. This cerebrovascular effect of 7-NI is partly reversed by L-arg. The present data also show that 7-NI decreases basal CBF under resting conditions, as reported by others using different techniques of CBF measurement and under different experimental conditions (Kelly et al., 1995; Kovach et al., 1995). In the present study, rats were unanesthetized and relatively free to move, owing to a proprietary device allowing laser probe fixation (Bonvento et al., 1994). This choice was motivated by the fact that anesthesia not only modifies CBF (Goldman and Sapirstein, 1973; Lindauer et al., 1993) but also interferes with the vascular effect of NO. In particular, volatile anesthetics inhibit brain NO synthase activity (Tobin et al., 1994). Halothane interferes with the relaxation caused by NO (Hart et al., 1993), and it selectively and “noncompetitively” inhibits the vasoconstrictor effects of NO synthase inhibitors such as L-NAME (Wang et al., 1993). In addition, anesthetics antagonize excitatory amino acid receptor functions (Carla and Moroni, 1992), precluding the induction of seizures by KA.
Our previous results showed a reduction of the increase in hippocampal blood flow under L-NAME (Rigaud-Monnet et al., 1994) in limbic seizures induced by KA. This reduction was of lower amplitude than that measured in the cortex under 7-NI in the present study, but some major differences between the two studies may well provide an explanation: the inhibition of cerebral NO synthase after acute injections of L-NAME was lower than after 7-NI, the increase in blood flow was much higher in the hippocampus than in the cortex (Pinard et al., 1987), and L-NAME gave rise to a large increase in MABP, whereas 7-NI did not modify MABP.
Unlike L-NAME or nitro L-arginine (Rigaud-Monnet et al., 1994; Maggio et al., 1995), 7-NI had no visible effect on the severity of seizures induced by KA, confirming results of previous studies in rats and mice (Penix et al., 1994) but differing from those of a study using a higher dose of 7-NI (Mülsch et al., 1994). However, in the present study, the delay of onset of status epilepticus after KA injection was shortened by 7-NI and lengthened by L-arg, which may suggest that neuronal NO is an endogenous anticonvulsant. This suggestion is reinforced by the fact that one third of rats treated with L-arg + 7-NI did not exhibit seizure activity after KA injection.
The actual effectiveness of 7-NI regarding NO production was evidenced by the 73% inhibition of cortical NO synthase activity in our experimental conditions. This measurement in brain tissue includes a small fraction of microvessels. However, the parameters of centrifugation made the vascular fraction precipitate, owing to its higher density. Furthermore, microvessels are known to represent maximally 3% of brain tissue (Lasbennes and Gayet, 1984). The comparison of brain cortical NO synthase activity measured in the present study with that reported in other studies is rather difficult. Actually, the experimental protocols are not strictly comparable, and most studies report only the percentage of baseline NO synthase activity under various antagonists (Faraci and Brian, 1995; Wang et al., 1995) or report values in noncomparable units (Bredt and Snyder, 1989). However, the present value obtained in cerebral cortex exclusively (1.51 + 0.38 pmol/mg protein/min) is below that found by Clavier et al. (1994) but above that found by Mackenzie et al. (1994).
The most important point of the NO synthase activity data is that the amplitude of the blockade by 7-NI (73%) is comparable to that found by Mackenzie et al. (1994) in the cerebral cortex of conscious rats at the same time point after 7-NI injection, that is, 30 min. Such a confirmation is meaningful, since the extent of brain NO synthase inhibition produced by 7-NI depends on the timing of its administration, with maximal effect occurring 30 min after treatment. The transient inhibition of cerebral NO synthase by 7-NI, described previously in the mouse (Moore et al., 1993) and rat (Mackenzie et al., 1994), justifies our protocol of several 1-h-interval injections.
When considering constitutive NO synthase, the presumption of the specificity of 7-NI for neuronal NO synthase is based upon results from different investigators. 7-NI alters acetylcholine-induced dilation in neither isolated rabbit aortic rings (Moore et al., 1993) nor in situ rat pial vessels (Faraci and Brian, 1995); neither does it modify the rat CBF response to oxotremorine, a blood-brain barrier permeant muscarinic agonist (Wang et al., 1995). The fact that 7-NI inhibits endothelial NO synthase in crude homogenates of bovine aortic endothelial cells (Babbedge et al., 1993) does not preclude its specificity for neuronal NO synthase in other animal species under physiological conditions. In the present work, 7-NI did not increase systemic blood pressure, unlike L-NAME and other nonspecific NO synthase inhibitors, which shows that 7-NI does not inhibit endothelial NO synthase, thus confirming previous studies (Moore et al., 1993; Faraci and Brian, 1995; Kelly et al., 1995; Wang et al., 1995).
Consequently, the present data indicate that NO of neuronal origin is responsible for the basal cerebral vasodilatory tone and for the CBF response to seizure activity after KA systemic injection. This result is also supported by the fact that there are no significant differences in systemic variables (arterial pH, Pa
The reversal by L-arg of the 7-NI cerebrovascular effect was complete in basal conditions, but only partial and insignificant in seizure conditions. To our knowledge, this result does not detract from the finding that neuronal NO synthase activation is implicated in the increase in CBF induced by KA, since 7-NI binds to the prosthetic heme group of NO synthase in an L-arg-competitive manner, but the binding of 7-NI additionally affects the pteridine site of the enzyme in a noncompetitive manner (Klatt et al., 1994). A partial reversal by L-arg of the 7-NI effect has previously been described, but in terms of its antinociceptive influence (Moore et al., 1993).
Although endothelium-derived vasoactive factors play a major role in the control of blood flow in many physiological and pathological conditions, the present data, indicating a role for neuronally derived NO in the regulation of CBF in seizures induced by KA, are consistent with the sustained release of NO measured in vivo during seizures induced by KA (Balcioglu and Maher, 1993; Mülsch et al., 1994). The release of NO may result from the direct activation of KA receptors (Garthwaite et al., 1989) distributed in many brain regions, including the neocortex (Unnerstall and Wamsley, 1983), and from the involvement of glutamatergic pathways by a presynaptic action of KA (Ferkany and Coyle, 1983). Glutamate acting at its receptor sites induces NO synthesis from L-arg (Bredt and Snyder, 1989; Garthwaite et al., 1988; East and Garthwaite, 1991). In addition, topical application of N-methyl-
Footnotes
Acknowledgment:
This study was supported by CNRS and the University of Paris 7 and by a grant from DRET (no. 95 2515 A). We thank Robert Charbonné for technical assistance.