
Abstract
Blood–brain barrier damage has been implicated in the pathogenesis of cerebrovascular white matter lesions. This type of lesion is responsible for cognitive impairment in the elderly and can be induced by permanent ligation of the bilateral common carotid arteries in the rat. Because it is unclear whether the blood–brain barrier is impaired, we examined whether vascular permeability to horseradish peroxidase is altered using this model. According to light microscopic results, the reaction product of horseradish peroxidase was most intensely localized to the paramedian part of the corpus callosum in the brain, occurring to a small degree at 3 hours, day 1, markedly on day 3, but reduced on days 7 and 14. By electron microscopic study of the same area, the reaction product of horseradish peroxidase was localized to the plasmalemmal vesicles in the endothelial cells 3 hours after ligation, but appeared in the cytoplasm on days 1 and 3, suggesting a diffuse leakage of horseradish peroxidase. In addition, the reaction product was dispersed into the cytoplasm of glial cells in the perivascular regions on day 3. The luminal surface of the endothelial cell cytoplasm appeared irregular on day 7, suggesting a conformational change of the endothelial cells. Collagen fibrils proliferated in the thickened basal lamina and mitochondria degenerated in the pericyte on days 7 and 14. Perivascular glial endfeet were swollen throughout the survival period. In sham-operated rats, the reaction product of horseradish peroxidase was not observed at any time interval, except in vesicular structures. These findings indicate that chronic cerebral hypoperfusion induces blood–brain barrier damage with subsequent morphologic changes of the vascular structures in the corpus callosum. An extravasation of macromolecules, such as proteases and immunoglobulins, may contribute to the pathogenesis of white matter lesions.
Diffuse areas of hypodensity on computed tomography, or hyperintensity on T2-weighted magnetic resonance imaging, are often encountered in the periventricular or subcortical white matter (WM) in the elderly (Pantoni and Garcia, 1995). This finding, often referred to as leukoaraiosis (Hachinski et al., 1987), may correspond to rarefaction of myelin and axons in the deep WM of autopsied brains. Although the exact mechanism by which the WM lesions occur is unknown, the trigger may be chronic cerebral ischemia subsequent to fibrohyalinosis of the long, penetrating arteries in the deep WM (Brun and Englund, 1986;Lin et al., 2000). The diffuse WM lesions may also be related to a blood–brain barrier (BBB) dysfunction, which is likely caused by chronic cerebral ischemia, lacunar infarction, or arterial hypertension (Feigin and Popoff, 1963; Tomimoto et al., 1996; Akiguchi et al., 1998).
White matter lesions can be induced in rats by ligating the bilateral common carotid arteries (Wakita et al., 1994, 1995, 1998). After ligation, 86% of these animals survive, and the myelin becomes rarefied with proliferation of astroglia and activation of microglia in the WM. These animals also exhibit behavioral disturbances and permit us to estimate the effect of long-standing cerebral hypoperfusion. Therefore, they can serve as a good experimental model for the study of vascular dementia (Wakita et al., 1994; Tanaka et al., 1996), despite that this model estimates only the effect of chronic cerebral ischemia irrespective of aging and hypertension, which are closely associated with WM lesions in the humans. Previous studies on human specimens have implicated dysfunction of the BBB and microglial activation as the mechanisms underlying the WM lesions (Tomimoto et al., 1994, 1996, Akiguchi et al., 1998); however, it remains unclear whether there are BBB alterations in this model.
Recently, Rosenberg et al. have shown an increase in matrix metalloproteinase (MMP)-3 in microglia/macrophages in human cerebrovascular WM lesions (Rosenberg et al., 2001) and MMP-9 in focal cerebral ischemia (Rosenberg et al., 1998). Furthermore, in our study, MMP-2 was upregulated in microglia and vascular endothelium in a rat model of chronic cerebral hypoperfusion (Ihara et al., 2001). These increases in MMPs may impair the BBB and transform the vascular structures by degrading the extracellular matrix. Because BBB dysfunction is indicated to be the cause of WM lesions, MMPs may play a triggering role in their pathogenesis. However, no comprehensive information is available regarding the integrity of the BBB in chronic cerebral hypoperfusion.
In this study, we examined the temporal profiles of BBB permeability and ultrastructural features of the vessels in the corpus callosum using a sensitive method to examine the BBB permeability to an exogenous substance, horseradish peroxidase (HRP) (Mesulam, 1978; Balin et al., 1986; Broadwell and Sofroniew, 1993; Ueno et al., 2000, 2001). This protein is a macromolecular tracer (molecular weight, 40,000) that is widely used for light microscopic and ultrastructural investigations of BBB (Reese and Karnovsky, 1967).
MATERIALS AND METHODS
Male Wistar rats (Japan CL, Suita, Japan) weighing 200 to 250 g were used. The procedure for the model of chronic cerebral hypoperfusion was used as described by Wakita et al. (1994) with minor modifications. Briefly, the animals were anesthetized with intraperitoneal sodium pentobarbital (50 mg/kg) and were allowed to spontaneously respire throughout the surgical procedure. Through a midline cervical incision, the bilateral common carotid arteries were exposed and double ligated with silk sutures. The sham-operated animals were treated in a manner similar to that of the operated ones, except that the common carotid arteries were not occluded. After the operation, the rats were allowed to survive for 3 hours or 1, 3, 7, or 14 days in animal quarters with food and water ad libitum. For each period, we examined three rats in the operated group and two rats in the sham-operated group.
After a predetermined period of survival, these animals were anesthetized with sodium pentobarbital and treated with intraperitoneal diphenhydramine (5 mg/kg) 30 minutes before injection of HRP. The animals were injected intravenously with HRP (50 mg per rat, type VI; Sigma Chemical Co., St Louis, MO, U.S.A.) 5 minutes before the perfusion fixation. These rats were perfused transcardially with 0.01 mol/L phosphate-buffered saline for 1 minute and subsequently with a fixative containing 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4). The brain was removed and immersed in 1.25% glutaraldehyde and 1% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) at 4°C for 12 hours (Ueno et al., 2000). The brain was then placed in a sucrose-buffer solution consisting of 10% sucrose in 0.1 mol/L phosphate buffer (pH 7.4) at 4°C for 12 to 24 hours, and was sectioned on a microslicer into coronal 50 μm-thick sections and collected in 0.1 mol/L phosphate buffer (pH 7.4). Some sections were transferred to an incubation medium composed of 0.01 mol/L acetate buffer (pH 3.3), tetramethyl benzidine (0.05 mg/mL) and 0.003% hydrogen peroxide, as reported previously (Mesulam 1978, Ueno et al., 2000), and the rest of the sections were transferred to an incubation medium of 0.05 mol/L Tris buffer (pH 7.6), diaminobenzidine tetrahydrochloride (0.75 mg/mL), and 0.02% hydrogen peroxide (Reese and Karnovsky, 1967; Brightman and Reese, 1969; Ueno et al., 2001).
For light microscopic observation, the sections incubated with tetramethyl benzidine solution were mounted on gelatin-coated glass slides and some were stained with hematoxylin-eosin. For electron microscopic observation, the serial sections incubated in diaminobenzidine tetrahydrochloride solution were postfixed in 1% osmium tetroxide in 0.1 mol/L phosphate buffer (pH 7.4) for 1 hour, stained en block in 1% uranyl acetate, dehydrated in ethanol, and embedded in Epon 812. One micrometer-thick sections were taken from each block and stained with 1% toluidine blue. Ultrathin sections were cut, placed on grids, and observed with a JEM-1200EX electron microscope (Japan Electron Optics Laboratory Co., Tokyo, Japan).
RESULTS
Light microscopic findings
There was no reaction product of HRP with tetramethyl benzidine in the brain parenchyma in sham-operated rats at any time interval, except in BBB-free areas such as the median eminence and choroid plexus (Fig. 1F). At 3 hours and 1 day after bilateral ligation of the common carotid arteries (Figs. 1A and 1B), the reaction product of HRP was seen in the corpus callosum to a small degree. Three days after ligation (Fig. 1C), HRP leakage was marked in the corpus callosum and also in the cerebral cortex. At 7 and 14 days (Figs. 1D and 1E), HRP leakage was not found in the corpus callosum. Because the reaction product of HRP was most intensely localized to the paramedian portion of the corpus callosum facing the dorsal part of the lateral ventricle, electron microscopic investigation was focused on this area.
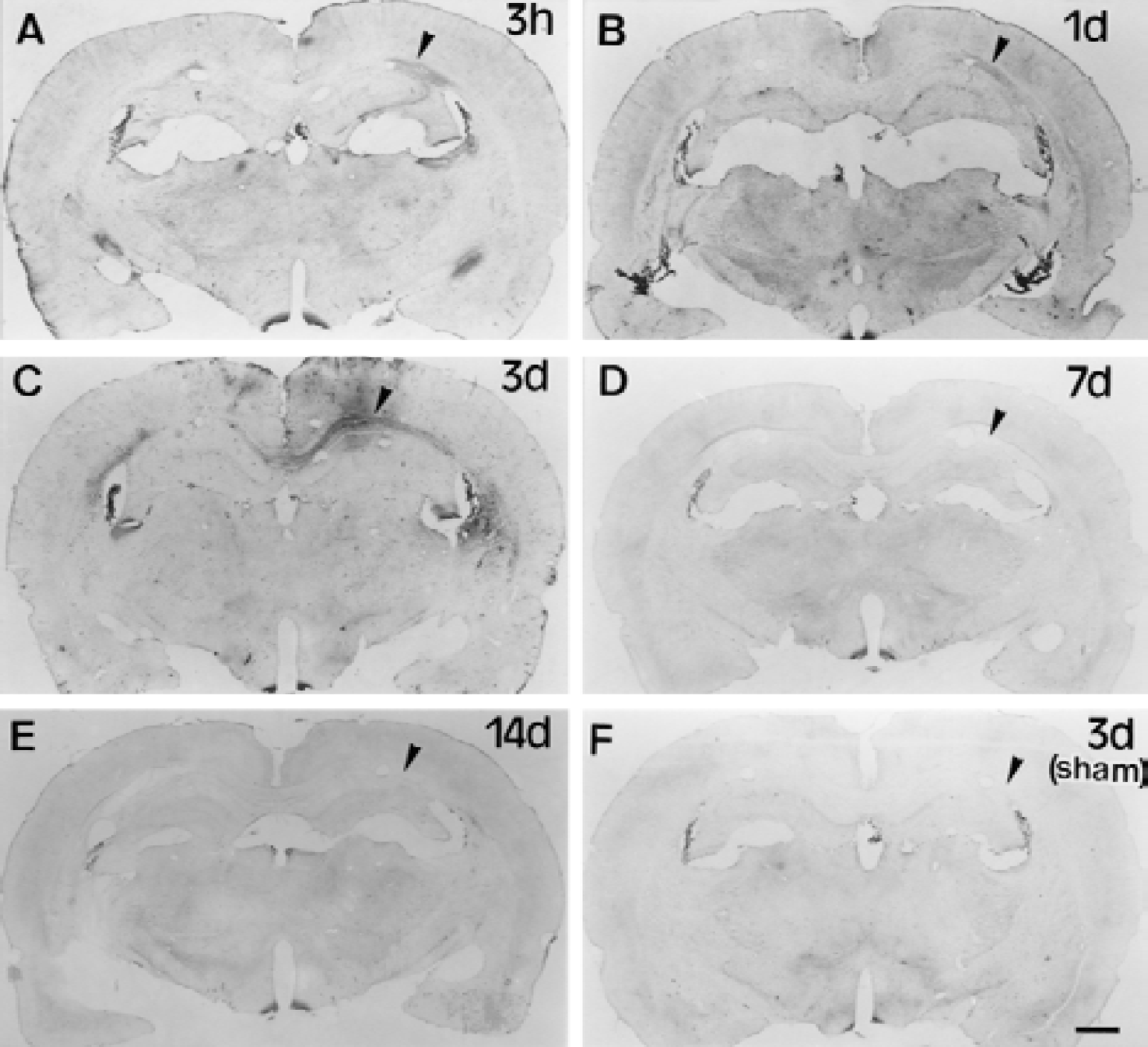
Light microscopic photographs showing the reaction product of horseradish peroxidase (HRP) with tetramethyl benzidine after 3 hours
Electron microscopic findings in the corpus callosum
Three hours after ligation, the reaction product of HRP was observed in plasmalemmal vesicles of the endothelial cells. Swelling of perivascular glial endfeet was rarely observed (Figs. 2A and 2B). On day 1, endothelial cells with the HRP reaction product were found diffusely in the cytoplasm in addition to the plasmalemmal vesicles, though their occurrence was only occasional (Figs. 2C and 2D). On day 3, the endothelial cells with electron-dense cytoplasm were observed more frequently, especially in the corpus callosum facing the lateral ventricle (Fig. 3). In addition, the HRP reaction product also appeared in the cytoplasm of perivascular glial cells.
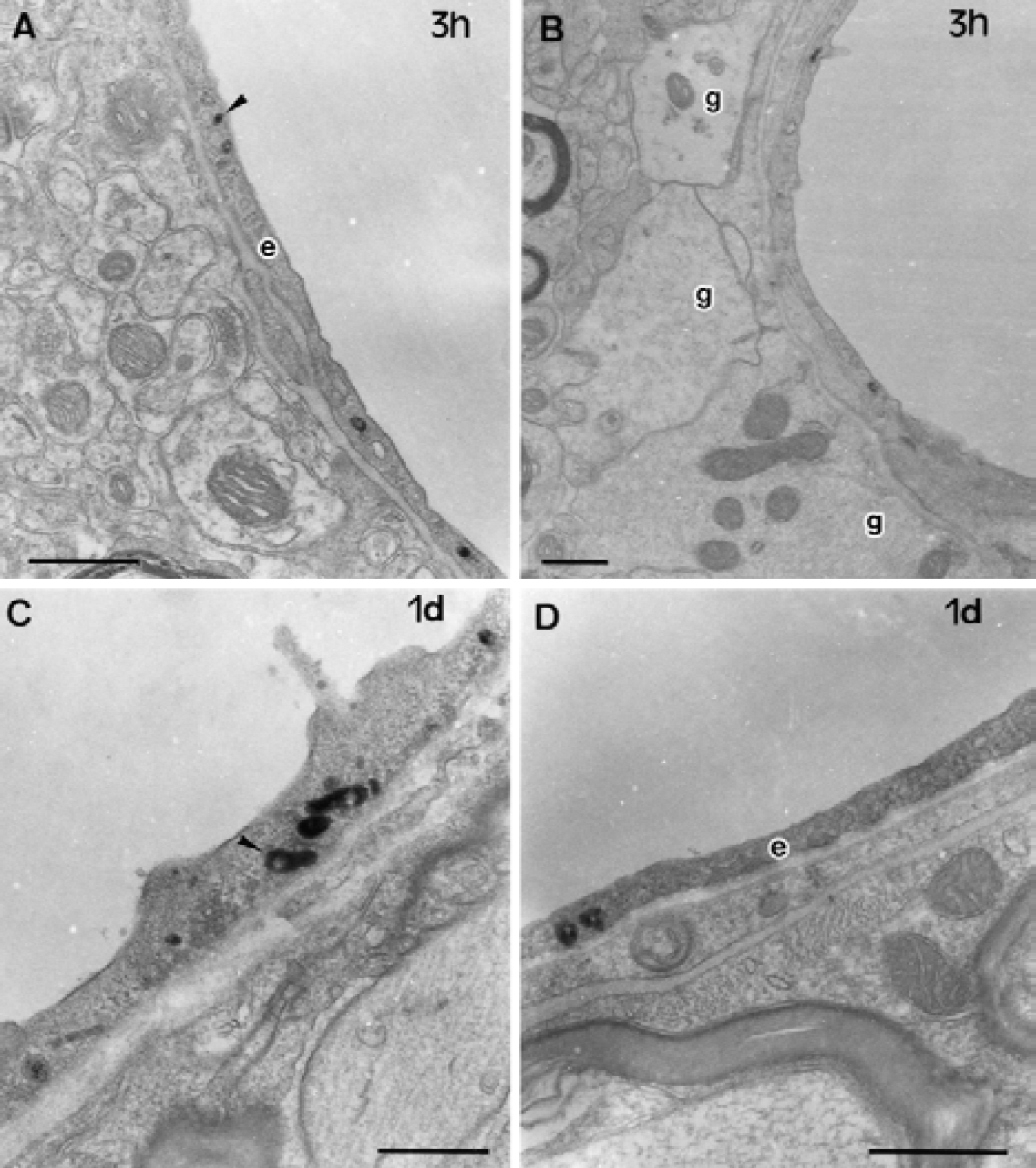
Electron microscopic photographs of the microvessels in the corpus callosum after 3 hours
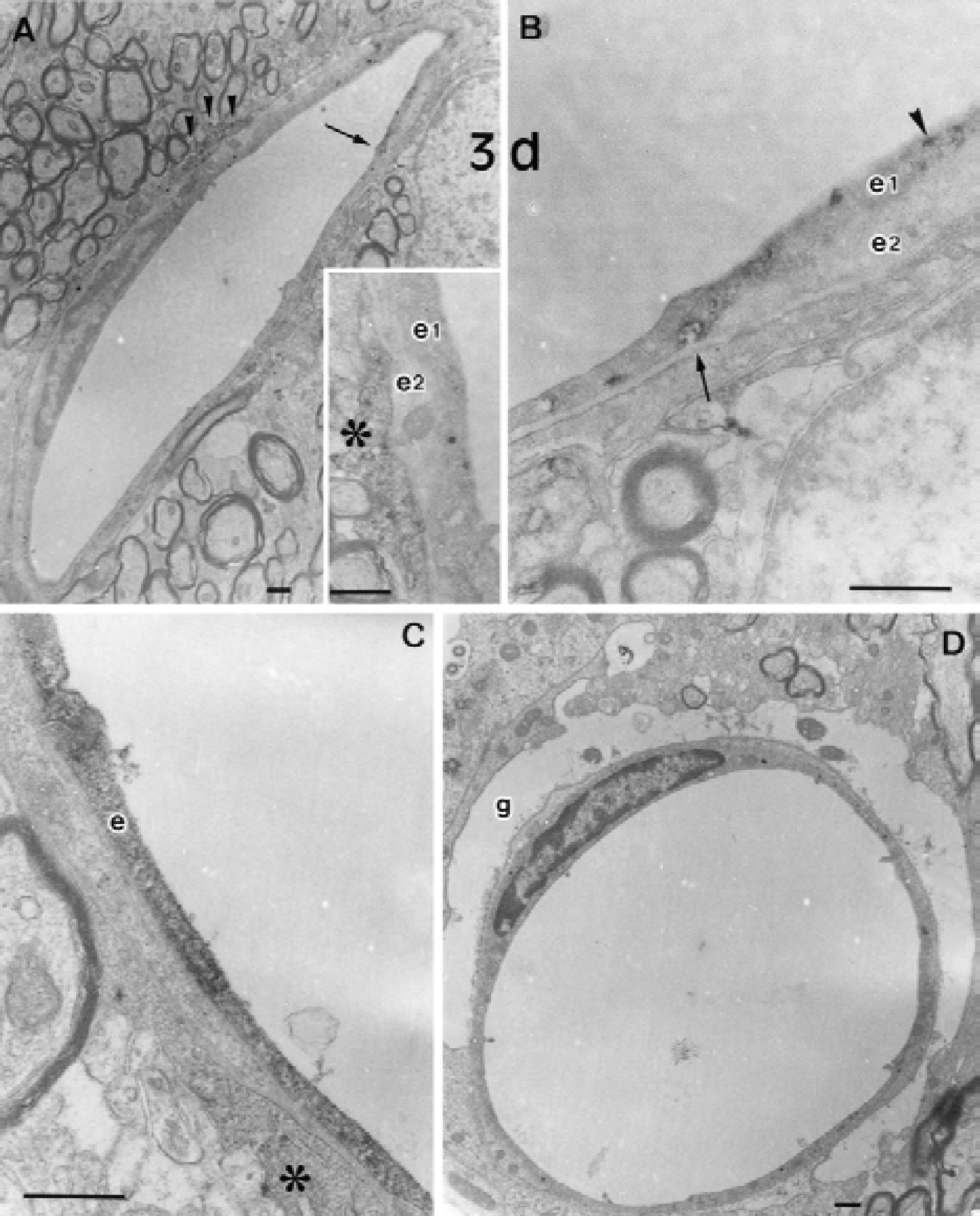
Electron microscopic photographs of microvessels in the corpus callosum after 3 days of chronic cerebral hypoperfusion
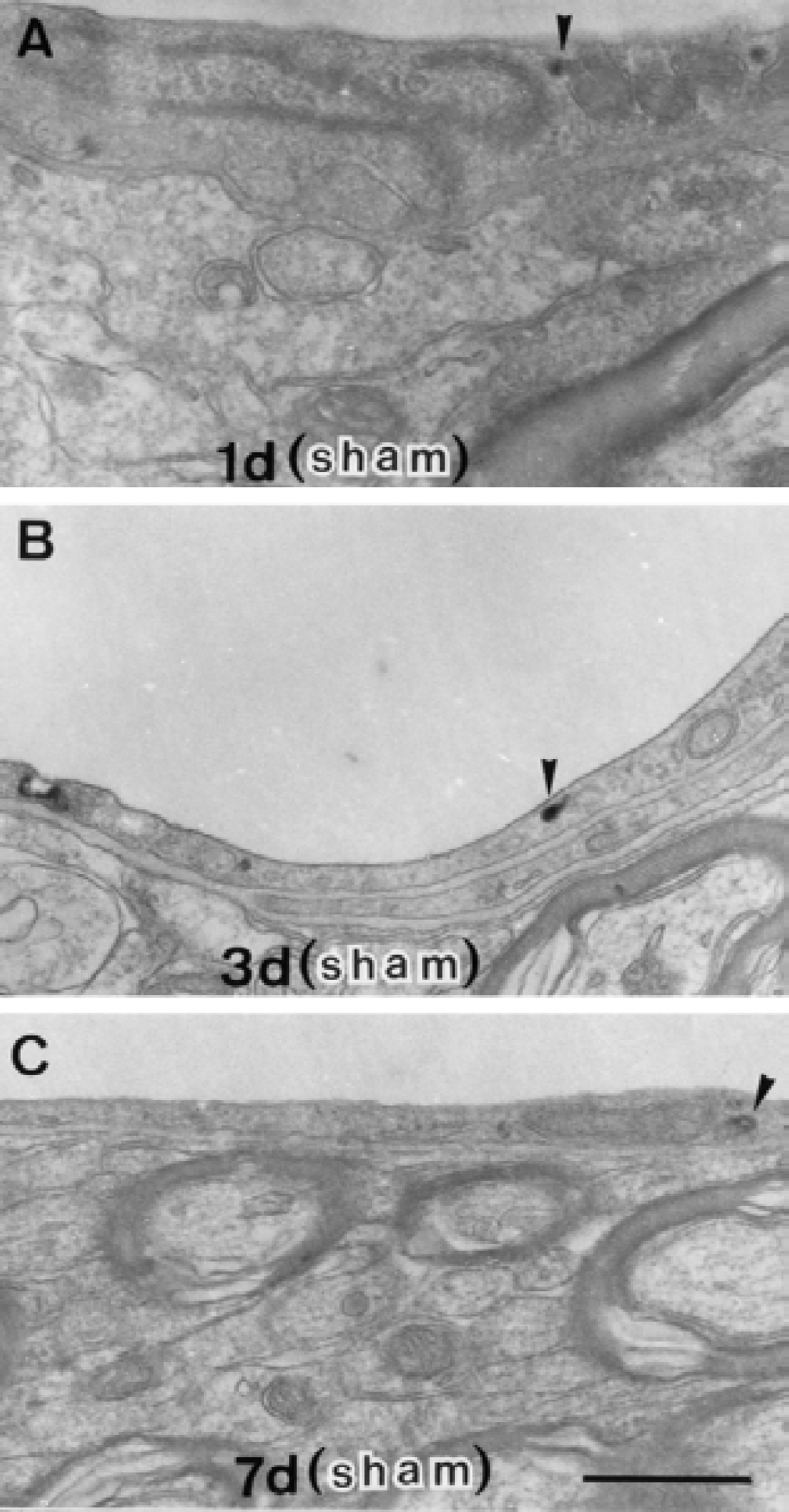
On day 7, the reaction product of HRP was associated with vesicular structures in the endothelial cells, but the diffuse cytoplasmic staining subsided (Fig. 4). The luminal surface of the endothelial cell cytoplasm became rough in some vessels. In addition, collagen fibrils were occasionally deposited in the thickened basal lamina. On day 14, these vesicular structures in the endothelial cell cytoplasm retained the HRP reaction product (Fig. 5). Collagen deposition was also found between the inner and outer basal lamina. The cristae of the mitochondria in the pericyte located near the perivascular collagen were occasionally disintegrated on days 7 and 14 (Fig. 4E, Figs. 5B and 5C). The reaction product of HRP appeared only in endothelial cell vesicular structures at any time interval after the sham operation (Fig. 6).
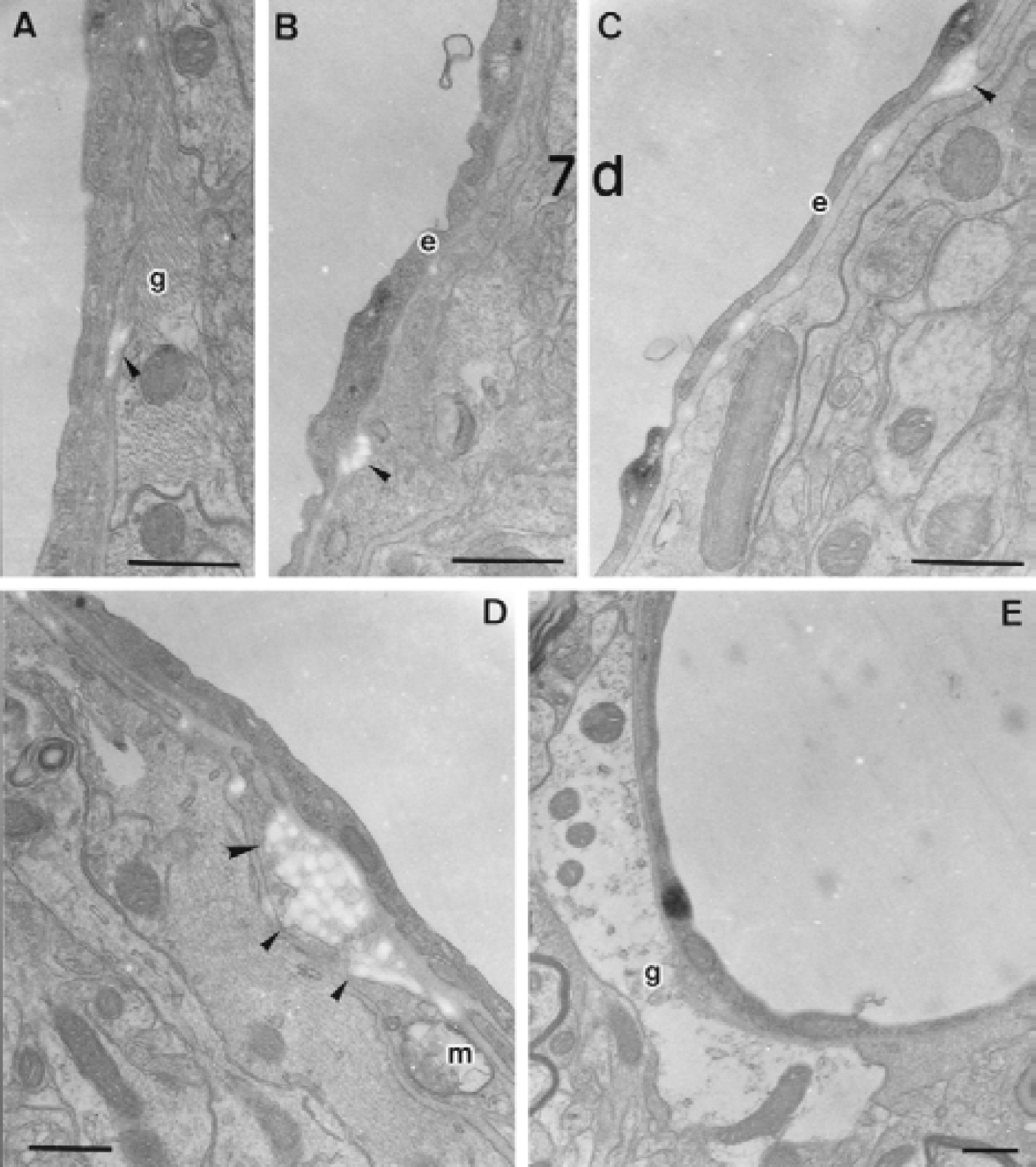
Electron microscopic photographs of microvessels in the corpus callosum on day 7 of chronic cerebral hypoperfusion
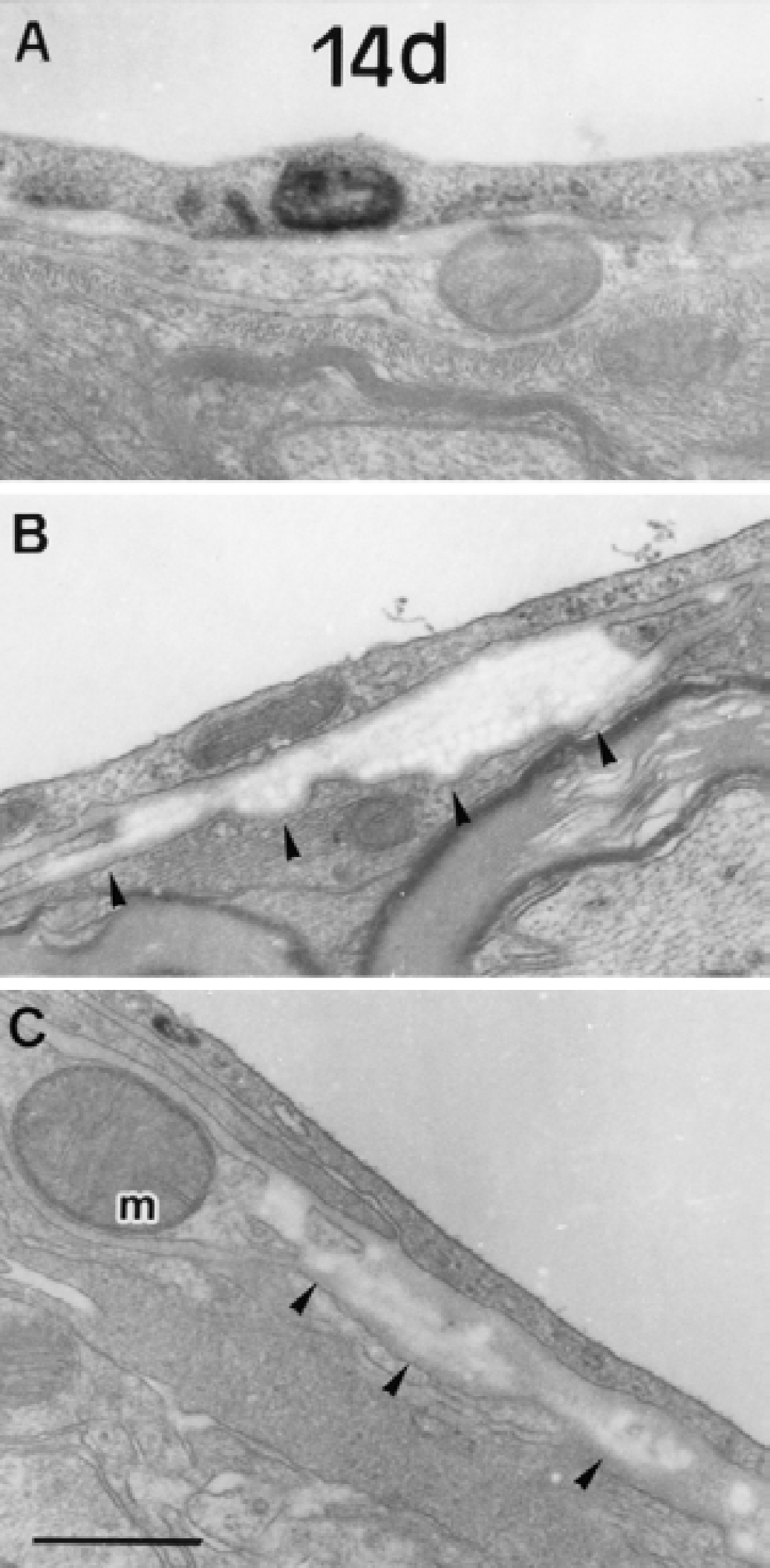
Electron microscopic photographs of the microvessels in the corpus callosum on day 14 of chronic cerebral hypoperfusion
Electron microscopic photographs of the microvessels in the corpus callosum at 1 day
DISCUSSION
Induction of an anaphylactic response (Cotran and Karnovsky, 1967) and stimulation of endocytosis and damage to the cerebral endothelium (Houthoff et al., 1982) have been reported after injection of type II HRP. Balin et al. (1986) reported that cerebral extravasation of type VI HRP delivered intravenously to the rat did not occur, and that type VI HRP did not appear to adversely affect the cerebral endothelium. Therefore, we used type VI HRP as a tracer to examine vascular permeability. With the present method, extravasation of blood-borne HRP is absent in normal conditions, and the reaction product of HRP is seen only in a few vesicular structures of the endothelial cytoplasm (Brightman et al., 1970; Broadwell, 1989). However, these structures increase in number in the endothelial cells, and the reaction product of HRP appears in the basal lamina and the surrounding neuropil in response to pathological insults (e.g., acute hypertension, cerebral ischemia, cerebral edema) (Baker et al., 1971; Westergaard et al., 1977; Petito et al., 1982; Shinnou et al., 1998).
Therefore, our findings indicate that chronic cerebral hypoperfusion induced an increase in BBB permeability to HRP, which appeared by day 1, peaked at day 3, and appeared to recover by day 7 in the corpus callosum. On day 14, BBB permeability had almost returned to the baseline level, but showed abnormalities such as electron-dense endosomes or multivesicular bodies. Obviously, this temporal profile of BBB breakdown is not chronic. However, in acute ischemic models with more marked reductions in cerebral blood flow, BBB damages occur more promptly in the foci of ischemic damages. This finding is also true of WM lesions, which develop immediately in acute focal ischemia (Pantoni et al., 1996) but occur after a latent period in chronic cerebral hypoperfusion. Thus, a prolonged time course of pathological changes clearly differentiates this model from those of acute ischemia.
Electron microscopic examination of the corpus callosum revealed that the HRP reaction product was distributed not only in the vesicular structures of the endothelial cells, but also diffusely in the endothelial cell cytoplasm on days 1 and 3 of chronic cerebral hypoperfusion. The diffuse distribution of reaction product suggests an uncontrolled passage of intravascularly injected HRP through the endothelial cell body. A similar leakage has been observed in transient global ischemia (Petito et al., 1982; Shinnou et al., 1998) and cerebral edema (Baker et al., 1971; Houthoff and Go, 1980; Houthoff et al., 1984). Such uncontrolled passage of macromolecular tracers was interpreted as evidence of endothelial injury or partial degeneration (Nagy et al., 1983; Vorbrodt et al., 1993).
The transport of HRP through junctional clefts between the endothelial cells was not observed. Therefore, it is likely that increased vascular permeability to HRP is attributable to increased vesicular transport and diffuse leakage through the endothelial cell cytoplasm, as seen in transient global ischemia (Petito et al., 1982). The diffuse leakage of HRP through the endothelial cell cytoplasm may indicate endothelial cell damage. Extravasation of HRP, which was most marked in the paramedian portion of the corpus callosum facing the lateral ventricle, may indicate a vulnerability of the BBB in this area, or may suggest the evacuation of HRP through the ependymal cell layer of the lateral ventricle. Interestingly, this portion of the corpus callosum showed the most intense demyelinating changes consisting of vacuoles, redundancy of myelinated fibers, and a decrease of oligodendrocytes (Wakita et al., 1994; Tomimoto et al. 1997; Ihara et al., 2001).
Recovery of the damaged BBB on day 7 of chronic cerebral hypoperfusion corresponded with the tendency toward normalized cerebral blood flow in this model (Tsuchiya et al., 1992, 1993; Tomimoto et al., 1997). During this period, collagen fibrils proliferated in the thickened basal lamina. Proliferation of collagen fibrils was similarly observed in the small arteries, arterioles and even around the capillaries in human cerebrovascular WM lesions (Lin et al., 2000). Although the trigger for proliferation of collagen fibrils remains unclear, an increase of collagen production or endothelial cell-derived tissue inhibitor of metalloproteinase, an endogenous inhibitor of MMPs, may have been involved. In contrast, MMP-2 seems to counteract this factor by degrading the vascular basement membrane and the extracellular matrix (Hamann et al., 1995). This mechanism is especially true in chronic cerebral hypoperfusion because the level of MMPs (type IV collagenases) is upregulated in the endothelial cells (Ihara et al., 2001). The irregular luminal surface of the endothelial cytoplasm on day 7 is similarly observed in spontaneously hypertensive rats, in which the cell surface was rough with protrusions and concavities (Hazama et al., 1979). Therefore, this structural transformation may reflect regressive changes of the endothelial cells. Nelson et al. (1975) reported balloons and craters in endothelial surface of the rabbit common carotid artery after 30-minute occlusion and regarded these changes as reproducible responses of endothelial cells to a variety of injurious stimuli.
These findings suggest that chronic cerebral hypoperfusion induces an increase in BBB permeability in the periventricular WM. The compromised BBB may allow entry of macromolecules and other blood constituents such as proteases, immunoglobulins, complements, and cytokines into the perivascular WM tissues. These serum components may have deleterious effects on the myelin directly (Silberberg et al., 1984) or may enhance the phagocytic activity of microglia through opsonization (Akiyama et al., 1994). The regional concordance between BBB damage and WM lesions in the paramedian portion of the corpus callosum may further indicate a vulnerability to chronic cerebral ischemia in this region, which likely appears as a periventricular lucency in computed tomography in elderly and hypertensive patients.
Footnotes
Acknowledgments:
The authors thank Mr. T. Nakagawa (Research Equipment Center, Kagawa Medical University) for his valuable help with electron microscopic observation, Ms. C. Ishikawa for technical assistance, and Ms. Y. Fujiwara for editorial assistance.