
Abstract
Slowly progressive degeneration of the hippocampal CA1 neurons was induced by 3-minute transient global ischemia in gerbils. Sustained degeneration of hippocampal CA1 neurons was evident 1 month after ischemia. To investigate the effects of an 18-mer peptide comprising the hydrophilic sequence of the rat saposin C domain (18MP) on this sustained neuronal degeneration, an intracerebroventricular 18MP infusion was initiated 3 days after ischemia. Histopathologic and behavior evaluations were conducted 1 week and 1 month after induction of ischemia. When compared with the vehicle infusion, 18MP treatment significantly increased the response latency time in a passive avoidance task. Increased neuronal density was also evident, as was the number of intact synapses in the hippocampal CA1 region at 1 week and 1 month after ischemia. 18MP treatment also significantly decreased the number of TUNEL-positive CA1 neurons 1 week after ischemia. Subsequent in vitro experiments using cultured neurons demonstrated that the 18MP at optimal extracellular concentrations of 1 to 100 fg/mL prevented nitric oxide–induced neuronal damage as expected and significantly up-regulated the expressions of bcl-xL mRNA and its translated protein. These results suggest that the gerbil model of 3-minute ischemia is useful in studying the pathogenesis of slowly progressive neuronal degeneration after stroke and in evaluating effects of novel therapeutic agents. It is likely that the 18MP at low extracellular concentrations prevents neuronal apoptosis possibly through up-regulation of the mitochondrial antiapoptotic factor Bcl-xL.
Occlusion of the common carotid arteries for 5 minutes without maintaining the brain temperature at 37°C ± 0.2°C induces classic delayed neuronal death in the hippocampal CA1 area within 4 to 7 days after ischemic insult (Kirino, 1982). Although this ischemia model is useful for studying the molecular basis of selective vulnerability of hippocampal CA1 neurons to ischemia, it does not mimic the human condition in which long-term neuronal degeneration is evident after mild ischemic episodes. This is caused by concurrent degeneration of nearly all CA1 neurons within 7 days after a 5-minute ischemic insult. Previously, the authors have demonstrated that numbers of peptide growth factors, cytokines, and nonpeptide agents can protect hippocampal CA1 neurons against neuronal death at 7 days after a 3-minute ischemic insult in normothermic gerbils (Sakanaka et al., 1998; Wen et al., 1998). In this model, approximately one half of the hippocampal CA1 neurons degenerated by the seventh day after a 3-minute period of forebrain ischemia. In addition, some of the CA1 neurons exhibited TUNEL staining at 7 days after ischemia, indicating that the neuronal degeneration induced by forebrain ischemia of 3 minutes persists for longer than 7 days (Wen et al., 1998). This long-term degeneration of the hippocampal CA1 neurons differs from the 7-day degeneration of nearly all hippocampal CA1 neurons in gerbils induced by conventional 5-minute forebrain ischemia (Kirino, 1982). It would be worthwhile to investigate whether the 3-minute ischemic gerbil model shows reproducible long-term neuronal degeneration as is evident in humans after mild ischemic episodes.
Prosaposin, the precursor of saposins A, B, C, and D that activate sphingolipid hydrolyzes in lysosomes, is a 517-amino acid glycoprotein (O'Brien and Kishimoto, 1991). It was demonstrated that prosaposin possesses a strong protective action against ischemic damage to hippocampal neurons when infused intracerebroventrically before ischemia (Sano et al., 1994), suggesting that prosaposin may be neurotrophic in action. It is further suggested that the in vitro and in vivo trophic actions of prosaposin on hippocampal and cortical neurons are related to a linear 18-mer prosaposin-derived peptide (18MP: LSELIINNATEELLIKGL) comprising the hydrophilic sequence of the rat saposin C domain (Kotani et al., 1996). However, the role of 18MP in molecular mechanisms of ischemia-induced neuronal degeneration remains to be elucidated.
The first series of studies demonstrated that hippocampal CA1 neurons subjected to a 3-minute ischemic insult developed sustained degeneration between 3 and 28 days after ischemia. Thereafter, CA1 neurons did not exhibit any apparent pathologic morphology up to 1 year after a 3-minute ischemic insult. Therefore, studies were conducted to investigate whether 18MP can prevent neuronal death in the hippocampal CA1 area between postischemic days 3 and 28. In a second series of investigations, infusion of 18MP was initiated 72 hours after 3-minute ischemia for 4 weeks. Effects were evaluated by counting CA1 neurons and the number of synapses and conducting passive avoidance tests. Because the 18MP was shown to prevent persistent neuronal degeneration 3 to 28 days after 3-minute ischemia, a third series of experiments was conducted to elucidate molecular mechanisms underlying the neuroprotective action of 18MP in vitro. Results from these studies demonstrated that the 18MP prevents sodium nitroprusside (SNP)-induced neuronal apoptosis (Toku et al., 1998) through up-regulation of a mitochondrial antiapoptotic factor, Bcl-xL.
MATERIALS AND METHODS
All experimental procedures were conducted according to the Guide for Animal Experimentation at Ehime University School of Medicine. Animals were housed in an animal room with a temperature range between 21°C and 23°C and a 12-hour light/dark cycle (7 am to 7 pm). Animals were allowed access to food and water ad libitum until the day of the experiment. Anesthesia was induced with 1.5% halothane and maintained with 1% halothane in 70% N2 O and 30% O2. Brain and rectal temperatures were maintained between 37.0°C ± 0.2°C while occluding the common carotid arteries. After recovering from anesthesia, animals were maintained in an air-conditioned room at approximately 22°C. An 18-mer peptide (18MP: LSELIINNATEELLIKGL), comprising the hydrophilic sequence of rat saposin C, was synthesized by Sawady Technology (Tokyo, Japan).
Animal preparation
Adult male Mongolian gerbils (Meriones unguiculatus) weighing 60 to 80 g were used in this study. Both common carotid arteries were exposed by making an anterior midline cervical incision and occluding it with miniclips to produce forebrain ischemia. After 3 minutes of occlusion, the clips were removed to restore circulation. A sham operation was performed as the control, in which both common carotid arteries were exposed but were not occluded.
Osmotic minipump implantation
An osmotic minipump (model 2001; Alza, Palo Alto, CA, U.S.A.) was implanted subcutaneously into the back of each animal. A needle from the minipump was placed into the left lateral ventricle approximately 1.5 mm anterior, 1.0 mm lateral, and 2.7 mm ventral to the bregma as demonstrated in the atlas of Thiessen and Yahr (1977).
Postischemic infusion of 18MP
18MP was dissolved in 0.05 mol/L phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (Sigma Chemical, St. Louis, MO, U.S.A.). Three days after the ischemic insult, 18MP (20 ng/day) or the vehicle (PBS with 0.1% bovine serum albumin) was continuously infused for 4 or 28 days into the left cerebral ventricle (n = 7 per group). 18MP (4 ng/day) also was continuously infused for 28 days as described above (n = 7). Sham-operated animals received the infusion of vehicle only for 28 days (n = 7).
Passive avoidance tests and histopathologic evaluations
After reperfusion periods of 1 week, 1 month, 3 months, 6 months, and 12 months, gerbils were trained and tested in a conventional step-down passive avoidance apparatus (Sakanaka et al., 1998; Wen et al., 1998). Response latency was measured as an index of learning ability.
One hour after the passive avoidance tests, each animal was anesthetized with pentobarbital and perfused transcardially with 4% paraformaldehyde/2.5% glutaraldehyde in 0.1 mol/L phosphate buffer (pH 7.4) for light and electron microscopy. A brain section including the dorsal hippocampus from 0.5 to 1.5 mm posterior to bregma was removed and placed in fixative overnight at 4°C. Four serial coronal sections (50-μm-thick) at the level 1.0 to 1.2 mm posterior to bregma were cut with a microslicer (Dosaka EM, Tokyo, Japan) for electron microscopy. The remaining dorsal hippocampus was embedded in paraffin, and 5-μm serial frontal sections were stained with 0.1% cresyl violet. All neurons with intact morphologic appearance along a 1-mm linear length of the CA1 field in 6 serial coronal sections (1.20 to 1.23 mm posterior to bregma) were counted. The mean number of neurons per section was calculated for each animal. Specimens were processed for electron microscopic evaluations as described by Wen et al. (1998). Electron micrographs of the central area (15 μm × 18.75 μm = 280 μm2) of the strata moleculare, radiatum, and oriens of the CA1 region were taken, and the number of intact synapses were counted.
In situ detection of DNA fragmentation (TUNEL staining)
Three days after a 3-minute transient forebrain ischemia, either vehicle (n = 7) or 18-MP(20 ng/d, n = 7) was continuously infused for 4 days into the cerebral ventricles of the gerbils. One week after the ischemic insult, animals were perfused with 4% paraformaldehyde in 0.1 mol/L phosphate buffer (pH 7.4) under pentobarbital anesthesia. Two paraffin sections from each animal were processed for TUNEL staining (ApopTag, Intergen, NY, U.S.A.) to estimate the number of degenerating CA1 neurons at 7 days after the 3-minute forebrain ischemia. All TUNEL-positive neurons along a 1-mm linear length of the CA1 field were counted in two serial coronal sections.
Cortical neuronal cultures and Alamar blue assays
Cerebral cortices of 17-day rat embryos were aseptically dissected, and cortical neurons were dissociated from the tissues by the method of Igase et al. (1999). Three days later, the 18MP at graded concentrations of 1 fg/mL to 100 pg/mL were added to the medium. Neurons were incubated in the medium for 1 day followed by incubation with 100 μmol/L SNP (Wako, Osaka, Japan), a nitric oxide (NO) donor, for 10 minutes at 37°C (Tanaka et al., 1999). The medium was replaced with serum-free Dulbecco's modified Eagle's medium (DMEM) containing the 18MP at graded concentrations of 1 fg/mL to 100 pg/mL, 20 mmol/L N-(2-hydroxyethyl)piperazine-N′,2-ethanesulfonic acid, 4.5 mg/mL glucose, 1 mg/mL insulin, 5 nmol/L sodium selenite, and 5 mg/mL transferrin. Sixteen hours after NO treatment, neuronal cultures were incubated with Alamar blue dye (10% v/v; Alamar Biosciences, Sacramento, CA, U.S.A.) for 2 hours (Tanaka et al., 1998) to assess neuronal viability and metabolic activity. Light absorbance of the reduced form of the dye was measured at 570 nm. The oxidized form was measured at 600 nm. Metabolic activity of cells was expressed by ΔOD = (OD570 − OD600). A total of seven procedures were conducted.
Reverse transcription–polymerase chain reaction
Cultured cortical neurons were seeded on 35-mm-diameter dishes (Sumitomo, Tokyo, Japan) at a density of 1 × 105 cells/cm2. Neurons were cultured at 37°C in DMEM (0.1% glucose; Nipro, Osaka, Japan) supplemented with 10% fetal calf serum (Life Technologies, Rockville, MD, U.S.A.) under a humidified atmosphere containing 5% CO2 for 24 hours. Then the medium was replaced with the serum-free DMEM containing the 18MP at graded concentrations (1 fg/mL to 100 pg/mL), as described above. Six hours later, total RNA was extracted from the cultured cortical neurons by using Isogen (Nippon Gene, Tokyo, Japan). Oligo dT primers together with 3 μg DNase-treated total RNA and Moloney leukemia virus reverse transcriptase (Life Technologies) were used to obtain single strand DNA. Polymerase chain reaction was conducted as previously described (Wen et al., 1998) by using the following primers: rat bcl-xL 5′ primer (5′-GTAGTGAATGAACTCTTTCGGGAT-3′), rat bcl-xL 3′ primer (5′-CCAGCCGCCGTTCTCCTGGATCCA-3′), rat β-actin 5′ primer (5′-AGAAGAGCTATGAGCTGCCTGACG-3′), and rat β-actin 3′ primer (5′-TACTTGCGCTCAGGAGGAGCAATG-3′).
Immunoblot analysis
Cortical neurons were cultured in the presence of 18MP (1 fg/mL to 100 pg/mL) as previously described for immunoblotting assays. Forty-eight hours later, neurons were solubilized with Laemmli's sample solution containing 2% sodium dodecyl sulfate (Laemmli, 1970). An equal amount of protein (40 μg) from the homogenates was electrophoresed to individual lanes on a 6% polyacrylamide gel. Electrophoretic bands were transferred to nitrocellulose sheets and were immunoblotted with a mouse monoclonal antibody against Bcl-xL protein (Transduction Laboratories, Lexington, KY, U.S.A.). The density of each Bcl-xL –positive band was measured by NIH image software (National Institutes of Health, Bethesda, MD, U.S.A.).
Statistics
All values are presented as mean ± SD. The number of TUNEL-positive neurons in the vehicle- and 18MP-treated groups were analyzed statistically by the two-tailed Mann–Whitney U test. Neuronal density, response latency, the number of intact synapses, neuronal viability, and Bcl-xL immunoreactivity were analyzed by analysis of variance followed by Bonferroni's multiple comparison test. P < 0.05 was considered statistically significant.
RESULTS
Long-term occurrence of ischemia-induced neuronal death
When compared with the CA1 field of sham-operated animals stained with cresyl violet (Fig. 1A and 1G), the CA1 regions of the brains of ischemic gerbils exhibited a loss of nearly one half of the pyramidal neurons at 1 week (Fig. 1B and 1H) and a marked (approximately 70%) reduction in the number of pyramidal neurons at 1 month (Fig. 1C and 1I) after 3 minutes of ischemia. After that no changes were evident at 3 (Fig. 1D and 1J), 6 (Fig. 1E and 1K), or 12 months (Fig. 1F and 1L) after 3-minute ischemia (Table 1).
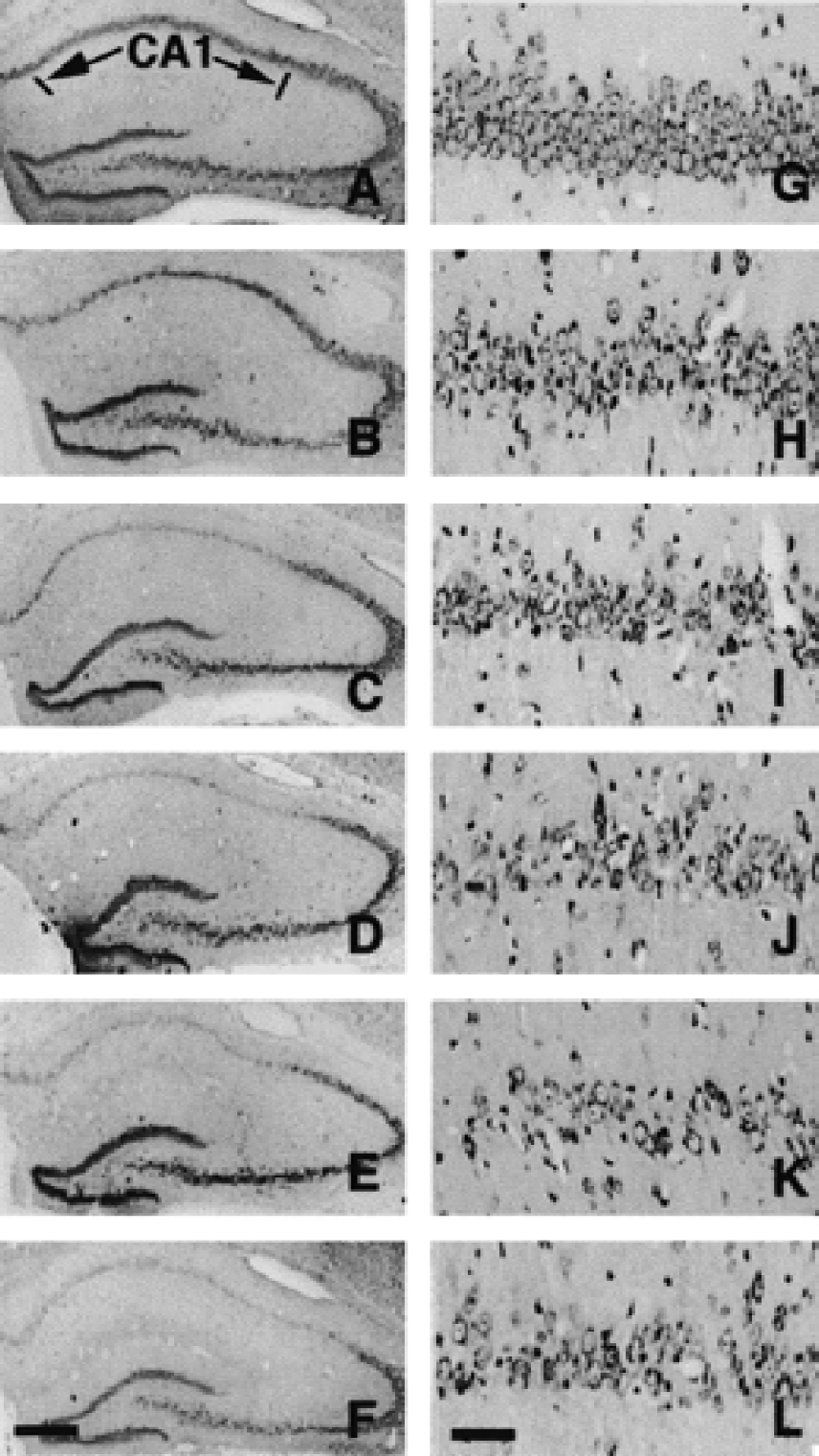
Representative photomicrographs of the dorsal hippocampus from a sham-operated gerbil
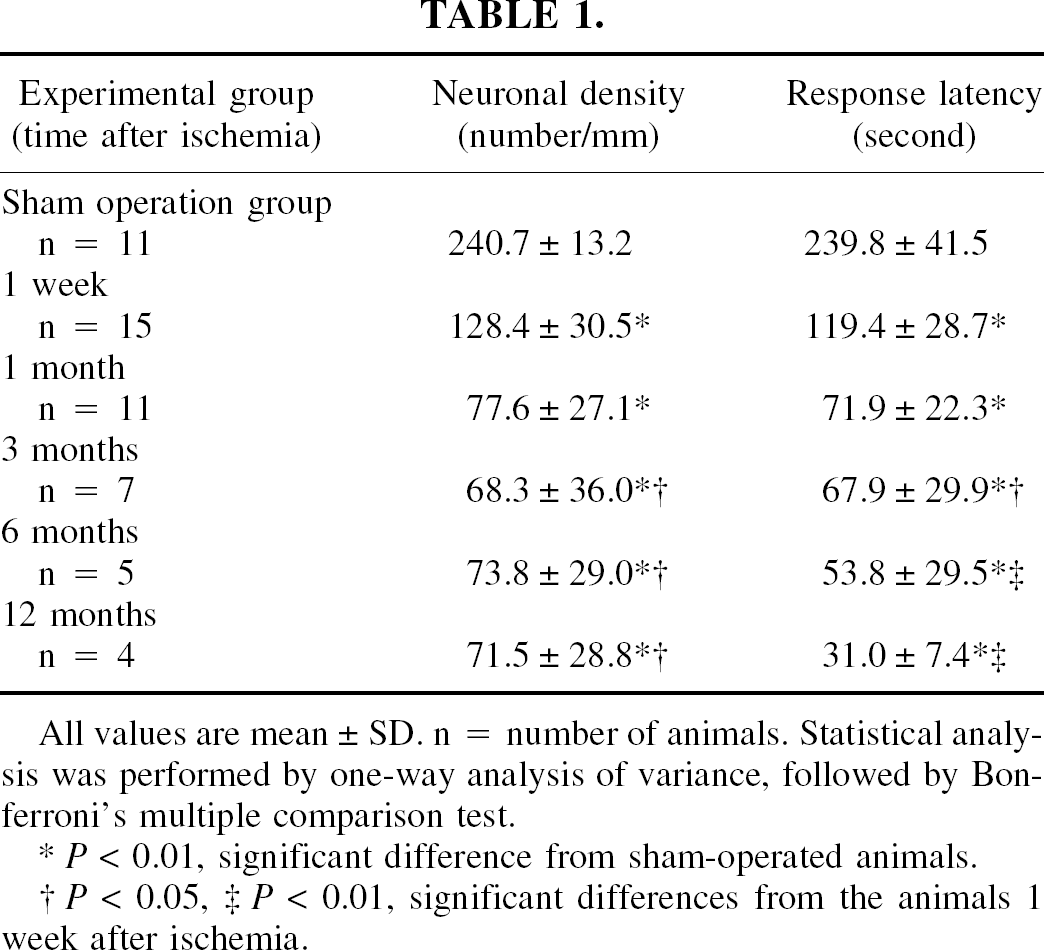
All values are mean ± SD. n = number of animals. Statistical analysis was performed by one-way analysis of variance, followed by Bonferroni's multiple comparison test.
P < 0.01, significant difference from sham-operated animals.
P < 0.05,
P < 0.01, significant differences from the animals 1 week after ischemia.
Passive avoidance tests conducted 1 week after ischemia revealed that the mean response latency of the ischemic animals was significantly shorter than that of the sham-operated animals. The mean response latency gradually decreased to 13% of the sham-operated control at 12 months after 3-minute ischemia (Table 1). Therefore, CA1 neuronal death in gerbils with 3-minute forebrain ischemia appeared to occur within 1 month after the ischemic insult. Based on these results, an additional study was conducted to identify a neuroprotective agent that prevents CA1 neuronal death 1 month after 3-minute forebrain ischemia in gerbils.
Effect of the 18MP on CA1 neuronal density, response latency, and intact synapses
A 4- or 28-day 18MP infusion beginning 3 days after 3-minute forebrain ischemia prevented neuronal death in the hippocampal CA1 field and ameliorated ischemia-induced learning disability (Fig. 2A and 2B). The infusion of the 18MP at a dose of 20 ng/day into the lateral ventricle for 4 or 28 days, starting 3 days after ischemia, rescued many neurons that would have degenerated without 18MP infusion (Fig. 2C to 2F).
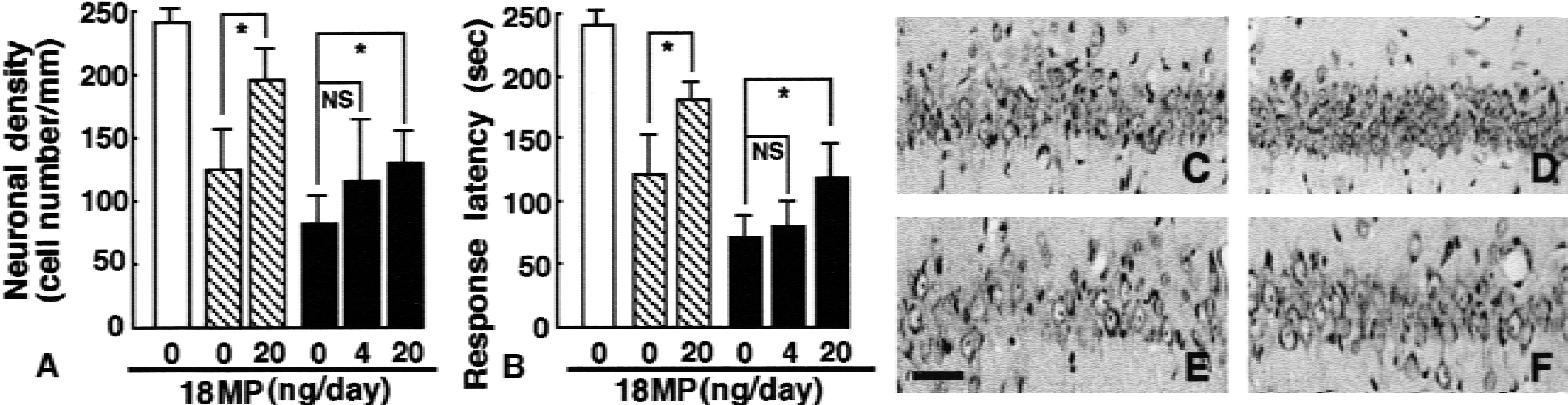
Effects of 18MP on the CA1 neuronal density
As expected from the results of neuron counts, the 4-day infusion of the 18MP at 20 ng/day into the lateral ventricle, starting 3 days after ischemia, caused a significant prolongation in the response latency of the step-down passive avoidance test (Fig. 2B). The 28-day infusion of 18MP at 20 ng/day, starting at the same postischemic period, also caused a significant prolongation in the response latency. It was evident from electron microscopic evaluations that the number of intact synapses within the stratum moleculare, stratum radiatum, and stratum oriens of the hippocampal CA1 region were significantly more numerous in 18MP-treated gerbils (20 ng/day, 28 days) than in vehicle only treated ischemic gerbils (Fig. 3). Electron microscopy also revealed that a significant number of neurons were in various stages of degeneration (Fig. 4A and 4B). In addition, the nuclei of vehicle only treated ischemic neurons at early stages of degeneration had partially condensed chromatin and low electron dense euchromatin (Fig. 4A). Conversely, most of the surviving neurons in the CA1 fields of 18MP-treated gerbils retained normal morphology even 7 days after ischemia (Fig. 4C).
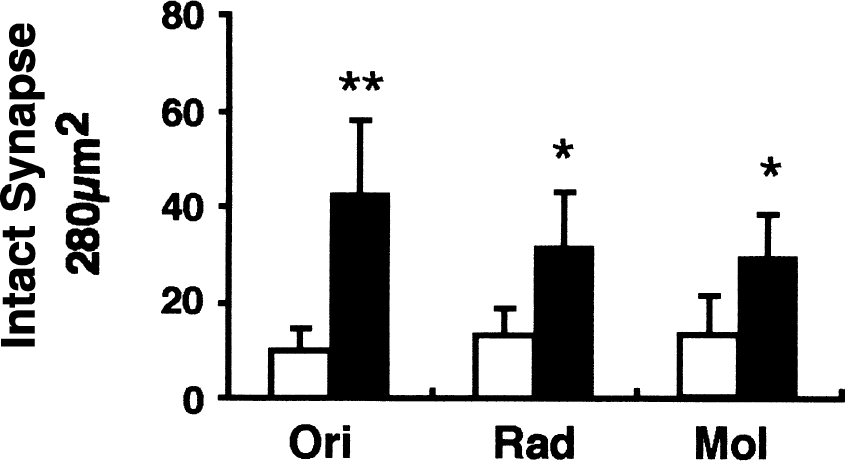
Effect of the 18MP on the number of intact synapses in three strata of the hippocampal CA1 region. Continuous infusion of the 18MP or vehicle was initiated 3 days after ischemia and maintained for 28 days. Intact synapses in the strata oriens (Ori), radiatum (Rad), and moleculare (Mol) of the 18MP-infused ischemic gerbils were significantly more numerous than those in the vehicle-infused ischemic gerbils. Each value represents mean ± SD (n = 7 in each group). Statistical analysis was conducted with one-way analysis of variance, followed by Bonferroni's multiple comparison test. * P < 0.05, ** P < 0.01, significantly different from ischemic animals with vehicle infusion.
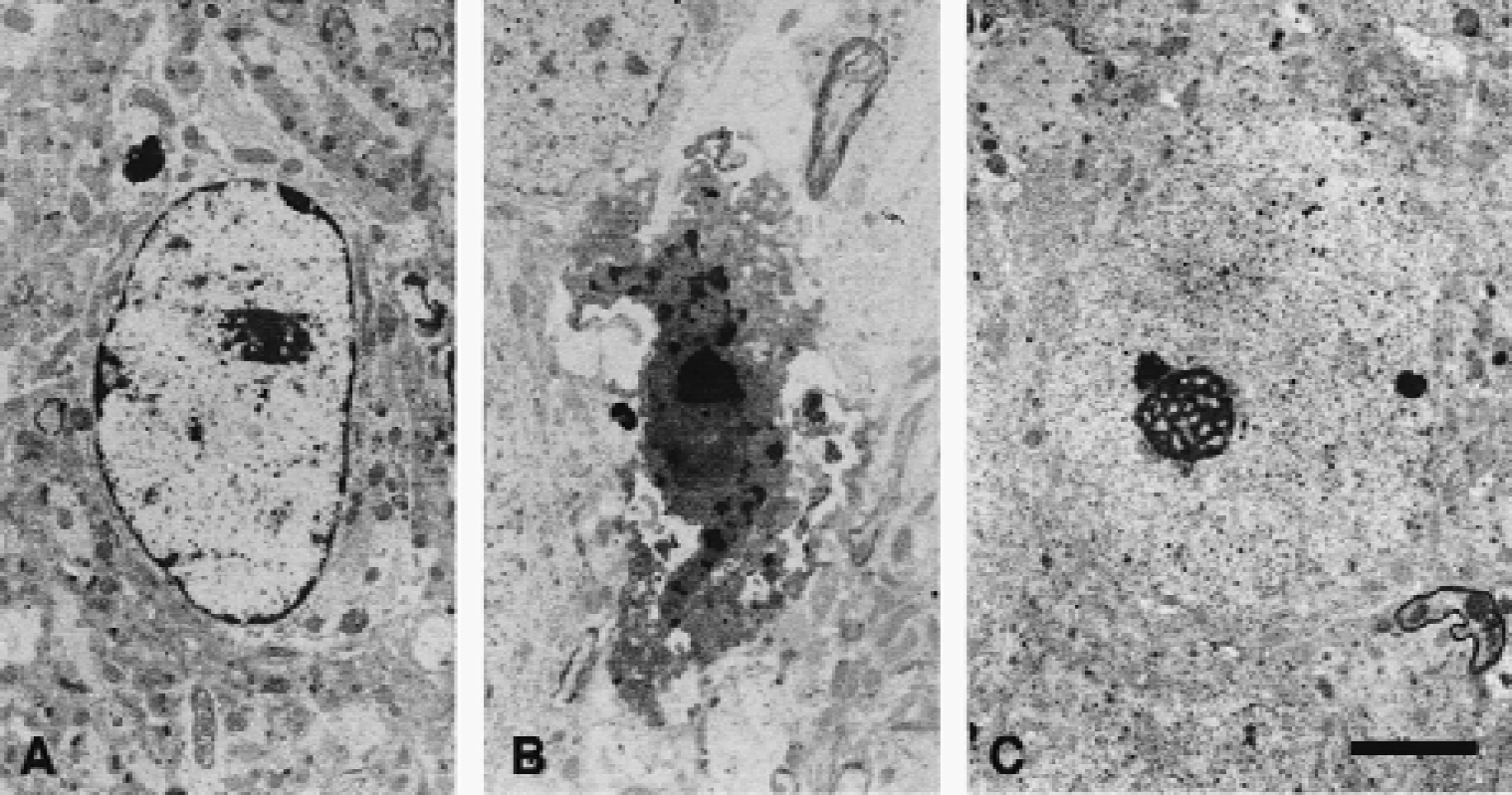
Representative electron micrographs of the soma of ischemic hippocampal CA1 neurons treated with vehicle
Effect of 18MP on the number of TUNEL-positive neurons in the hippocampal CA1 field
To demonstrate the effect of 4-day 18MP infusion starting at 3 days after 3-minute forebrain ischemia, TUNEL staining was performed on paraffin sections taken from the hippocampal CA1 area infused with 18MP or vehicle only. Numerous TUNEL-positive neurons were evident in the hippocampal CA1 area of vehicle-infused gerbils 7 days after 3-minute ischemia, suggesting that irreversible neuronal degeneration was still in progress. (Fig. 5A). The administration of 18MP (20 ng/day), which was initiated 3 days after ischemia, reduced the number of TUNEL-positive neurons detected at 7 days after 3-minute ischemia (Fig. 5B). Consequently, 18MP treatment significantly prevented the late onset of ischemia-induced neuronal degeneration. Approximately 36.7% of neurons were rescued by 18MP treatment (Fig. 5C). It is likely that neurons exhibiting partially condensed chromatins and low electron dense euchromatin as seen in Fig. 4A were visible by TUNEL staining techniques (Wen et al., 1998).
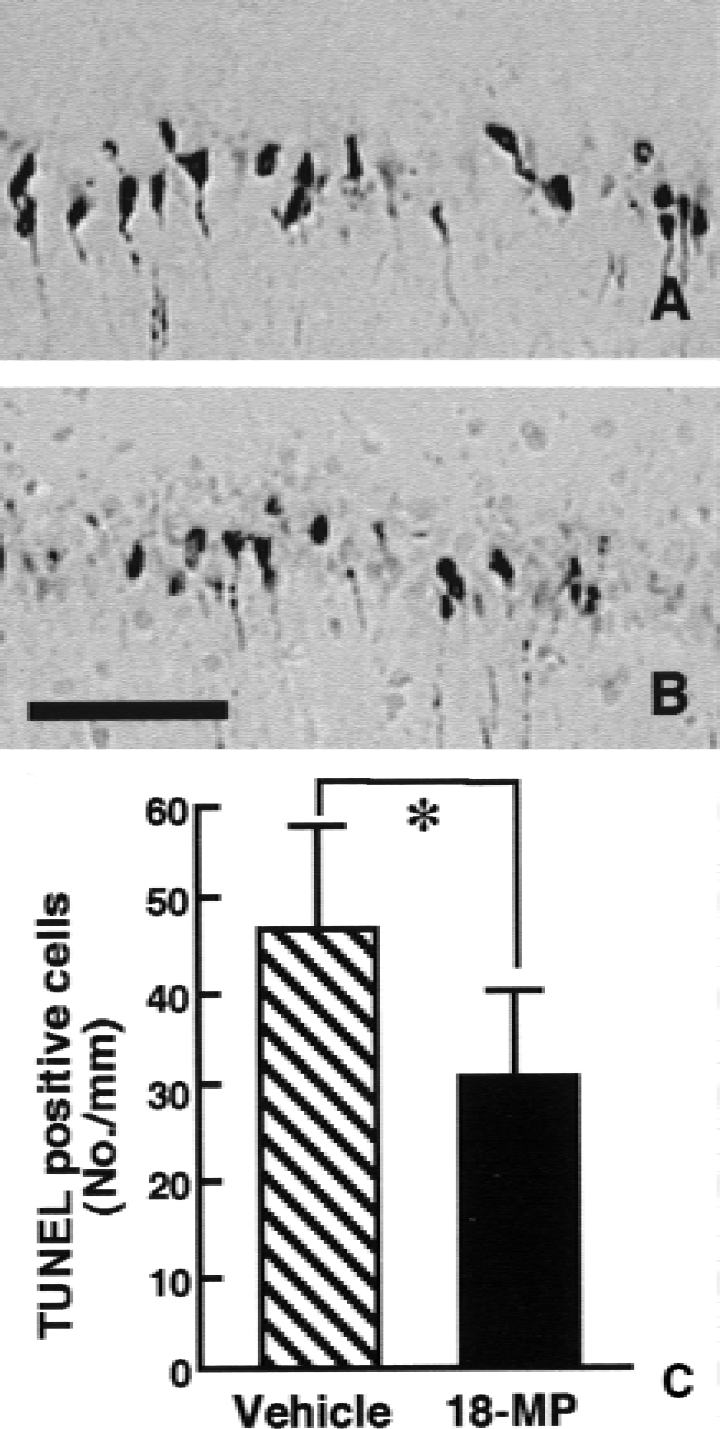
Representative photomicrographs of TUNEL-positive neurons in the hippocampal CA1 fields from gerbils subjected to 3-minute global ischemia. The cerebroventricular infusion of either vehicle
Protective effect of the 18MP on cultured cortical neurons exposed to SNP
The above mentioned results suggest that 18MP treatment if begun 3 days after ischemia prevents ischemia-induced neuronal death. Tissue culture experiments were conducted to determine whether 18MP prevented NO-induced apoptotic cell death of cortical neurons (Toku et al., 1998). As shown in Fig. 6A, 18MP at graded concentrations of 0 to 100 pg/mL did not affect neuronal viability. When cultured neurons were exposed to the NO donor, SNP, without 18MP treatment, they underwent apoptosislike cell death and exhibited decreased viability as determined by Alamar blue assays (Fig. 6B). Treatment of cultured neurons with 18MP at concentrations of 1 to 100 fg/mL, starting either before or after SNP exposure, significantly reduced neuronal apoptosis (Fig. 6B).
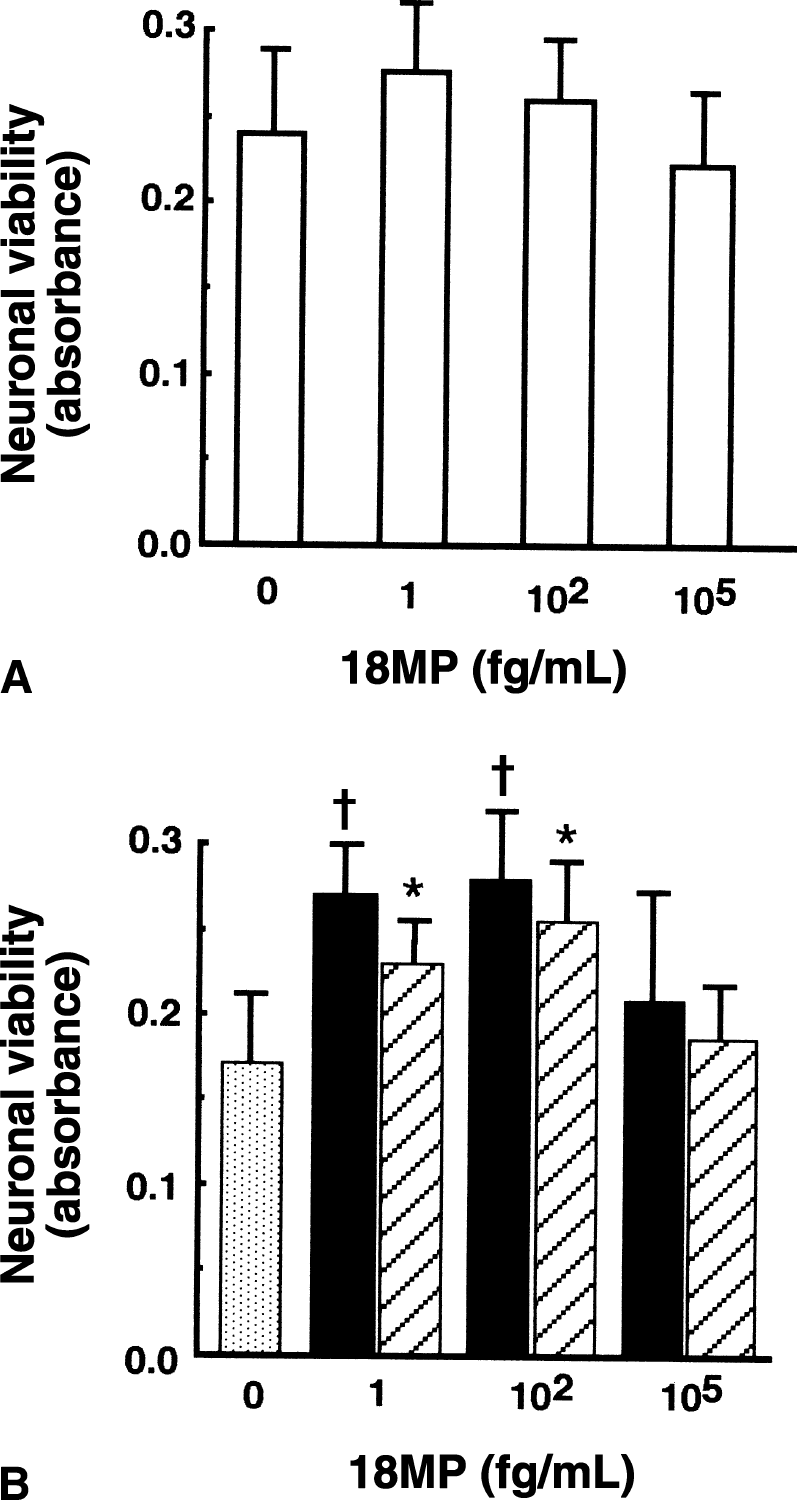
Effect of 18MP on the viability of cortical neurons cultured without sodium nitroprusside (SNP)
Up-regulation of Bcl-xL mRNA and protein by 18MP in vitro
The molecular mechanism by which 18MP prevents neuronal apoptosis then was investigated. Causative factors of cell death that may be affected by 18MP treatment were investigated by evaluating, with reverse transcription–polymerase chain reaction, 18MP-induced changes in the expressions of Bcl-2, Bcl-w, Bcl-xL, Bcl-xS, Apaf-1, Akt, Bax, Bak, Bad, caspase-1, caspase-3, caspase-9, Fas, Fas-legand, P53, P21, and GADD45 mRNA in neurons. Among the factors evaluated, Bcl-xL mRNA expression was markedly up-regulated by treatment with 18MP at concentrations of 1 to 100 fg/mL, the optimal concentrations for 18MP required to produce antiapoptotic effects on cultured neurons (Fig. 7A).
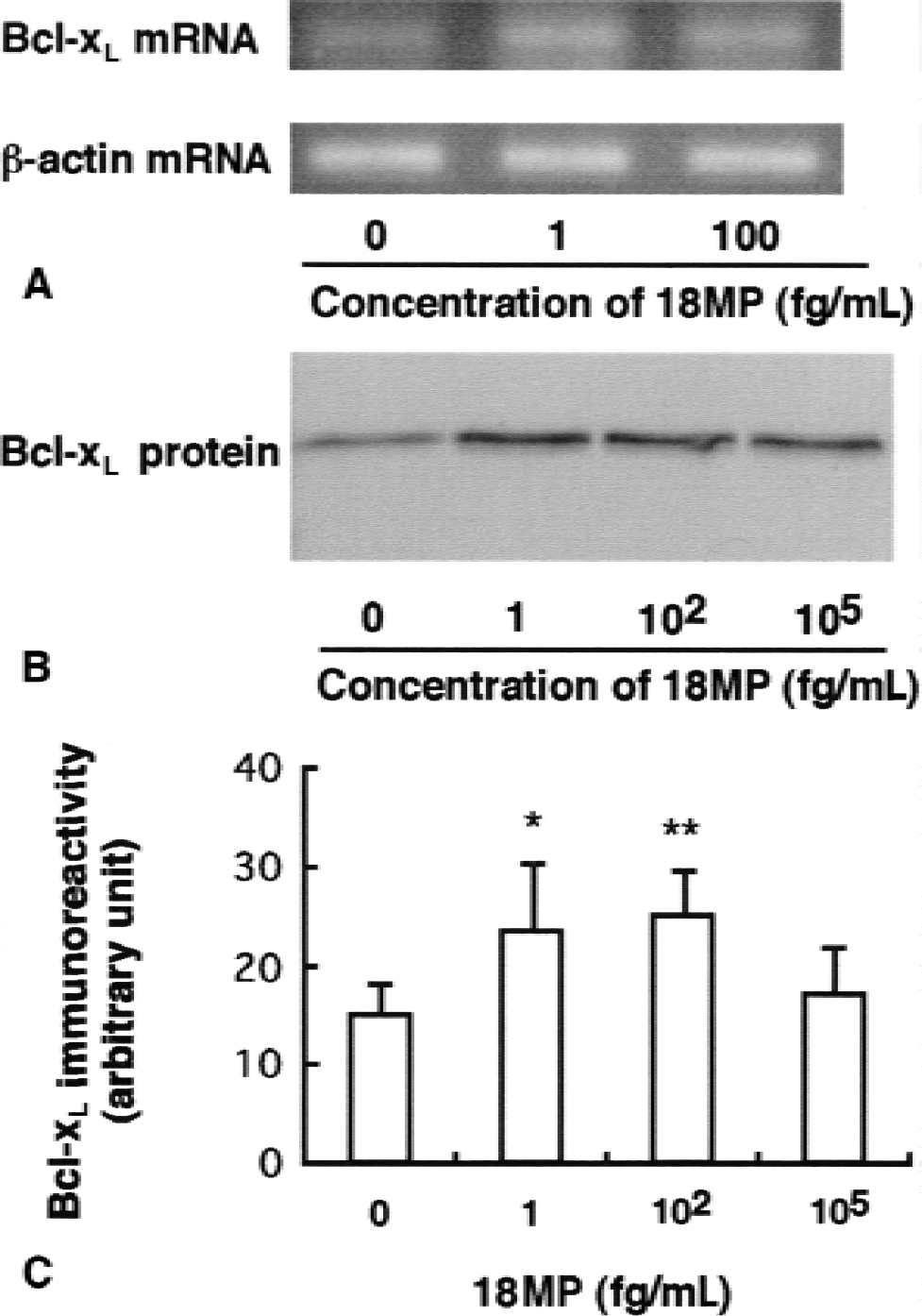
Reverse transcription–polymerase chain reaction analysis of bcl-xL mRNA in cortical neurons cultured in the presence of 0, 1, 100 fg/mL 18MP
As shown in Fig. 7B, Bcl-xL protein with a molecular weight of approximately 29 kD was consistently expressed in cultured neurons. Treatment of cultured neurons with 18MP at concentrations of 1 to 100 fg/mL for 2 days significantly increased Bcl-xL protein expression. These findings suggest that 18MP prevents neuronal apoptosis through up-regulation of the antiapoptotic factor, Bcl-xL.
DISCUSSION
This study demonstrated that 3-minute transient global ischemia in gerbils results in a slower progression of neuronal CA1 damage than 5-minute ischemia. In this model, ischemic neuronal damage was detected at 7 days and did not progress beyond 1 month after reperfusion. Conversely, the step-down passive avoidance tests revealed that the mean response latency gradually decreased up to 12 months after ischemia, indicating that the cognitive ability was still deteriorating. This persistent deterioration of cognitive function may be because of age-dependent changes in the animals (Riekkinen et al., 1991).
It was demonstrated previously that 3-minute transient global ischemia in gerbils, as well as a 5-minute period of ischemia, results in a selective loss of CA1 pyramidal neurons in the hippocampus (Sakanaka et al., 1998; Wen et al., 1998). A similar process is defined as “delayed neuronal death” (Kirino, 1982). The precise mechanism underlying this delayed neuronal death remains unclear. However, the most common explanation is the glutamate-Ca2+ theory. Specifically, the neurotoxicity associated with overstimulation of N-methyl- d -aspartate receptors is believed to be mediated by an excessive influx of Ca2+ into the cytoplasm, leading to a series of cascading, potentially neurotoxic events (Love, 1999). The Ca2+ -mediated activation of nitric oxide synthase can generate nitric oxide (NO) (Dawson et al., 1991). Stimulation of phospholipase A2 or Ca2+ overload of mitochondria can lead to the generation of superoxide anions (O−2) (Lafon-Cazal et al., 1993). NO can react with O−2 to form peroxynitrite (OONO−) (Beckman et al., 1990), which induces dose-dependent neuronal damage (Lipton et al., 1993). Although this theory can explain the early phases of neuronal response to ischemia, it does not offer a satisfactory explanation for the delayed phases of this process. Recently, increasing evidence suggests that additional changes at the molecular level may be required to complete the process for ischemic neuronal death. Among the changes at the molecular level that should be considered are the down-regulation and/or up-regulation of genes associated with apoptotic pathways. For example, proapoptotic genes including Bax are up-regulated (Chen et al., 1996; Krajewski et al., 1995), and caspase-3 activity is elevated in the hippocampal CA1 region after global ischemia (Ni et al., 1998). Despite extensive information on delayed neuronal death, further studies are necessary to elucidate the mechanism(s) underlying slowly progressive neuronal degeneration after 3-minute forebrain ischemia in gerbils.
Using the current ischemia model, the effects of 18MP on hippocampal CA1 neuronal death after ischemia was investigated. To see whether 18MP prevented slowly progressive neuronal degeneration, 18MP treatment was initiated 3 days after 3-minute forebrain ischemia and effects were evaluated 1 week or 1 month after ischemic insults. The results of this study indicate that protective effects of 18MP on progressive neuronal degeneration after 3-minute ischemia in gerbils is evident. The molecular mechanisms underlying this neuroprotective effect of 18MP is not fully understood. However, it has been suggested that the high-affinity prosaposin receptor is present on the surface of neuroblastoma cells (O'Brien et al., 1994). Prosaposin at extracellular concentrations greater than 1 ng/mL induces a mitogen-activated protein kinase phosphorylation in PC12 cells (Campana et al., 1996). It also activates mitogen-activated protein kinase by a Go-protein associated receptor for enhanced sulfatide synthesis and prevents death of Schwann cells and oligodendrocytes (Hiraiwa et al., 1997). Recently, prosaposin at extracellular concentrations greater than 1 ng/mL has been reported to activate phosphatidylinositol 3-kinase (PI3K)/Akt pathways and facilitate the survival of Schwann cells (Campana et al., 1999). Also, extracellular concentrations of 18MP at less than 100 pg/mL have been reported to attenuate neuronal damage caused by free radicals, which are known to be overproduced during and after cerebral ischemia (Igase et al., 1999). In addition to these reports, the in vitro experiments in this study demonstrated that 18MP at the optimal extracellular concentrations of 1 to 100 fg/mL ameliorated neuronal apoptosis caused by the NO donor, SNP, and up-regulated the antiapoptotic protein, Bcl-xL.
Bcl-xL protein is a member of the Bcl-2 family that contains the BH1-BH4 domains and functions as an antiapoptotic factor. Recently, there is a growing body of evidence suggesting that mitochondria play a crucial role in several types of apoptosis (Green and Reed, 1998). Apoptogenic factors, such as cytochrome c (Kluck et al., 1997) and the apoptosis-inducing factor (Susin et al., 1999), are released from the mitochondrial intermembrane space into the cytoplasm and activate subsequent apoptotic pathways. Cytochrome c directly activates caspases (Liu et al., 1996) by binding to the cytoplasmic protein Apaf-1. Apoptosis-inducing factor is a flavoprotein with a significant homology to bacterial oxidoreductases and has an ability to induce nuclear fragmentation reminiscent of apoptosis in a caspase-independent manner (Susin et al., 1999). Both Bcl-2 and Bcl-xL prevent apoptosis-associated mitochondrial changes, including cytochrome c release and permeability transition characterized by membrane potential (ΔΨ) loss (Narita et al., 1998). Moreover, Bcl-xL has the additional ability to preclude caspase activation by sequestering Apaf-1 (Hu et al., 1998). Currently, the precise mechanism underlying 18MP-mediated Bcl-xL up-regulation remains to be clarified. However, the authors' in vitro studies were consistent with the results of in vivo studies. Although other factors, such as secondary energy depletion caused by DNA repair, angiogenesis, vasospasm, and/or microemboli induced by up-regulation of adhesion molecules, must be taken into consideration, this data demonstrated that the prevention of NO-induced neuronal death by 18MP is mediated, at least in part, by up-regulation of antiapoptotic factor Bcl-xL protein.
Footnotes
Acknowledgment:
The authors are grateful to Dr. Kazumasa Ikoma for his encouragement and meaningful suggestions throughout development of this work.