
Abstract
In previous studies, the authors showed that the nitrone radical scavenger α-phenyl-N-tert-butyl nitrone (PBN) and its sulfo-derivative, 2-sulfo-phenyl-N-tert-butyl nitrone (S-PBN), attenuated cognitive disturbance and reduced tissue damage after traumatic brain injury (TBI) in rats. In the current study, the production of reactive oxygen species (ROS) after TBI was monitored with microdialysis and the 4-hydroxybenzoic acid (4-HBA) trapping method. A single dose of PBN (30 mg/kg) or an equimolar dose of S-PBN (47 mg/kg) was administered intravenously 30 minutes before a controlled cortical contusion injury in rats. Plasma and brain tissue drug concentrations were analyzed at the end of the microdialysis experiment (3 hours after injury) and, in a separate experiment with S-PBN, at 30 and 60 minutes after injury. Traumatic brain injury caused a significant increase in ROS formation that lasted for 60 minutes after the injury as evidenced by increased 3,4-dihydroxybenzoic acid (3,4-DHBA) concentrations in the dialysate. PBN and S-PBN equally and significantly attenuated the posttraumatic increase in 3,4-DHBA formation. High PBN concentrations were found bilaterally in brain tissue up to 3 hours after injury. In contrast, S-PBN was rapidly cleared from the circulation and was not detectable in brain at 30 minutes after injury or at any later time point. The results suggest that scavenging of ROS after TBI may contribute to the neuroprotective properties observed with nitrone spin-trapping agents. S-PBN, which remained undetectable even in traumatized brain tissue, reduced ROS production to the same extent as PBN that readily crossed the blood–brain barrier. This finding supports an important role for ROS production at the blood–endothelial interface in TBI.
Keywords
Reactive oxygen species (ROS) are thought to cause widespread oxidative damage in acute brain injury at several molecular and cellular targets, including lipids, proteins, DNA (Siesjo et al., 1989; Hall, 1997; Shohami et al., 1997; Lewén et al., 2000), membrane structures, and mitochondria (Kuroda et al., 1996; Keller et al., 1998; Hillered and Ernster, 1983; Léwen and Hillered, 1998). Nitrone spin-trapping agents such as α-phenyl-N-tert-butyl nitrone (PBN) have emerged as promising pharmacologic tools because of their robust neuroprotective efficacy and large therapeutic time window in several models of central nervous system injury, such as traumatic brain injury (TBI), stroke, and intracerebral hematoma (Cao and Phillis, 1994; Folbergrova et al., 1995; Peeling et al., 1998; Lewén et al., 2001; Marklund et al., 2001a, 2001b). PBN has a high degree of blood–brain barrier (BBB) penetration and a half-life in plasma of 3 hours (Chen et al., 1990). Pretreatment with a single intravenous (IV) dose of PBN (30 mg/kg 30 minutes before injury) recently has been shown to reduce cognitive deficits and lesion volume in a controlled cortical contusion model in rat (Marklund et al., 2001a). The neuroprotective effect of PBN has been attributed to scavenging of ROS, although alternate mechanisms such as influence on inflammatory mediators (Kotake et al., 1998; Pogrebniak et al., 1992), transmitter systems (Gould and Bickford, 1994; Joseph et al., 1995), or blocking of Ca2+ -channels (Anderson et al., 1993) have been suggested.
Nitrones with a poor penetration of the blood–brain barrier, such as the sulfonated derivatives of PBN, sodium 2-sulfo-phenyl-N-tert-butyl nitrone (S-PBN), and disodium 2,4-disulfo-phenyl-N-tert-butyl nitrone (NXY-059), have shown neuroprotective properties in focal ischemia models (Kuroda et al., 1999; Yang et al., 2000; Marshall et al., 2001). PBN has been shown to have a large therapeutic window in rodent models of focal ischemia, reducing infarct volume when administered up to 12 hours after the onset of stroke (Cao and Phillips, 1994). S-PBN reduced infarct volume when administered 2 hours after the onset of focal ischemia with equal efficacy to PBN (Yang et al., 2000). The therapeutic window for drugs targeting free radicals in TBI models has been suggested to be narrow (Wada et al., 1999), although the time window for nitrones in TBI has not been established. However, the authors recently have shown that PBN or S-PBN equally reduced tissue loss when given 30 minutes after fluid percussion injury in rats, whereas only S-PBN improved functional outcome as assessed by the Morris Water Maze (Marklund et al., 2001b). These findings suggest that radical-mediated mechanisms continue beyond the first few minutes after TBI and that microvascular mechanisms play an important role in acute brain injury.
In the current study, the authors studied the effects of PBN and S-PBN on ROS production using microdialysis and the 4-HBA trapping method after controlled cortical contusion injury in rats. The half-life in plasma and the penetration of the two drugs into injured and noninjured brain tissue also was studied.
MATERIALS AND METHODS
Chemicals
The following chemicals were used: 3,4-dihydroxybenzoic acid (protocatechuic acid; 3,4-DHBA) and p-hydroxy-benzoic acid (both from Sigma, St. Louis, MO, U.S.A.), perchloric acid (Fluka, Milwaukee, WI, U.S.A.), citric acid (Merck, KGaA, Darmstadt, Germany), methanol (p.a., Merck), potassium di-hydrophospate (Merck), sodium citrate dihydrate (Merck), EDTA (Titriplex III; Merck). PBN and S-PBN was generously provided by Centaur Pharmaceuticals (Sunnyvale, CA, U.S.A.). The chemical structures of PBN (MW 177) and S-PBN (MW 279) are shown in Fig. 1. PBN and S-PBN were dissolved in saline (yielding a concentration of 15 mg/mL and 23.5 mg/mL, respectively) before injection. The rationale for injecting PBN and S-PBN 30 minutes before trauma was based on the known peak brain concentration of PBN 30 minutes after IV injection (Chen et al., 1990). The same dosing regimen (30 mg/kg IV) was shown to be neuroprotective (Marklund et al., 2001a) and to reduce energy disturbance and phospholipid degradation (Lewén and Hillered, 1998) in this model of controlled contusion injury. For comparison, an equimolar dose of S-PBN (47 mg/kg) was chosen. After IV injection, PBN has a plasma half-life of approximately 3 hours (Chen et al., 1990) and S-PBN has one of 9 minutes (data from the manufacturer).
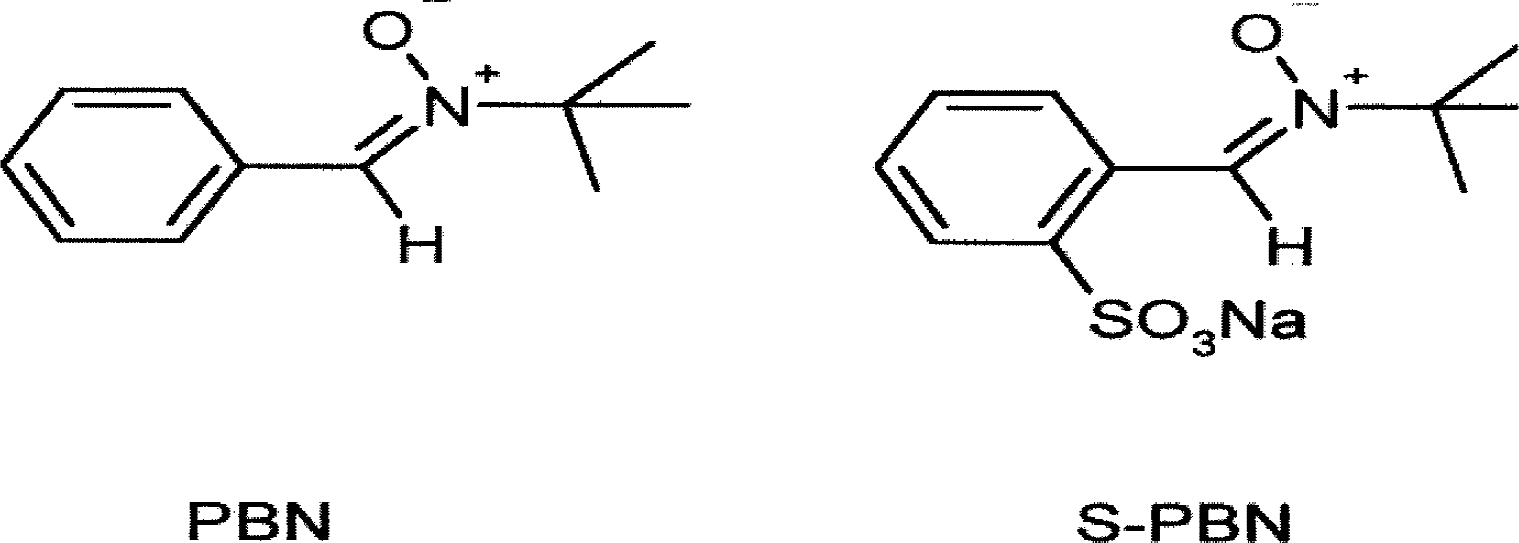
Chemical structures of α-phenyl-N-tert-butyl nitrone (PBN) and 2-sulfo-phenyl-N-tert-butyl nitrone (S-PBN).
Surgical procedure
This study was approved by the local ethics committee for animal research, and all animals were treated according to guidelines from The National Board for Laboratory Animals. Forty-seven male Sprague-Dawley rats (weighing 330 to 420 g) were habituated to the housing conditions for at least 1 week after arrival in the laboratory. Rats were housed at constant temperature (20°C) and humidity (50%) and maintained at a 12-hour light/dark cycle with free access to food and water. After induction of anesthesia with 4.5% halothane in oxygen, the rats were intubated and connected to a ventilator (Rodent Ventilator; Ugo Basile, Varese, Italy). Anesthesia was maintained with a mixture of nitrous oxide and oxygen (30%/70%) and isoflurane (1.2% to 1.4%). The rats were put on a heating pad and a rectal probe was inserted for monitoring of body temperature, which was maintained at 37°C to 38°C throughout the experiment. A catheter (PE 50; Becton Dickinson, MD, U.S.A.) was inserted into the tail artery for monitoring of blood pressure and arterial blood gases. Arterial blood gases were monitored by determination of pH, PCO2, and PO2 during the experiment (IL 1630; ILS Laboratories, Scandinavia) and the ventilator was adjusted as needed. Rats were put in a stereotaxic frame (Kopf Instrument, Tijunga, CA, U.S.A.) and a 6 × 9-mm oval craniotomy was made over the right parietal cortex centered at bregma −3.5 mm and 3.5 mm lateral to the midline using a dental drill. Epidural temperature was measured (TH-5; Physitemp, NJ, U.S.A.) and maintained at 36.5°C to 37.5°C throughout the experiment. A drop in brain temperature was avoided by positioning a heating lamp over the head of the animal. A central venous catheter was inserted in the left external jugular vein for administration of PBN, S-PBN, or saline. A focal contusion was produced by the weight drop method as originally described by Feeney et al. (1981) and modified by the authors' group (Nilsson et al., 1990, 1993, 1996). A 21-g weight was dropped from a height of 35 cm onto a piston with a diameter of 4.5 mm resting on the exposed dura, causing a depression of 2.5 mm, resulting in a severe injury with hemorrhages and a dural tear, ultimately producing a cortical cavity (Marklund et al., 2001a). The device was constructed to prevent bouncing of the weight, thus allowing only a single compression and producing a controlled cortical contusion (Skoglosa et al., 1999).
Immediately after the injury, the probe was reinserted and the heating lamp was repositioned over the animal's head. The experiment ended 3 hours after sham injury or TBI followed by rapid decapitation and collection of trunk blood in heparinized test tubes. Brains were rapidly removed and bilateral tissue samples from the parietal cortices and the cerebellum were taken and put on dry ice. Samples were stored at −70°C awaiting analysis. Four animals were excluded from further analysis because of inadequate trauma (n = 2), inadequate drug delivery (n = 1), and undetectable brain concentrations of 4-HBA around the microdialysis probe (n = 1).
Microdialysis procedure
The 4-HBA free radical trapping method was used as previously described (Ste-Marie et al., 1996; Marklund et al., 2001c). Briefly, a 3-mm microdialysis probe (MicroBiotech, Stockholm, Sweden) was positioned in a vial containing artificial cerebrospinal fluid (aCSF; Na+ 140, K+ 2.7, Ca2+ 1.2, Mg2+ 0.9, Cl− 147 mmol/l) before the experiment. The probe was connected to a microdialysis pump (CMA Microdialysis, Stockholm, Sweden) and perfused with 3 mmol/L 4-HBA dissolved in aCSF for equilibration. A small opening in the dura was made and the probe was inserted into the brain at the central part of the craniotomy. The probe was perfused at 2.0 μL/min and samples were collected every 30 minutes. Samples were stored at − 70°C while awaiting analysis.
Baseline samples were collected for 2 hours before the trauma, and sampling continued for 3 hours after the injury. At the time of injury, the probe was removed and immediately reinserted within 1 minute after the injury using the exact same stereotaxic coordinates (Nilsson et al., 1990). After termination of the experiment, the probe was perfused with Millipore water at 2.0 μL/min for 1 hour and kept in Millipore water until used in 1 to 2 subsequent experiments. The results are presented as absolute dialysate concentrations (ng of 3,4-dihydroxybenzoic acid [3,4-DHBA]/100 μL dialysate) (Fig. 1).
As previously reported (Marklund et al., 2001c), the in vivo probe recovery was 18% for 4-HBA and 12% for 3,4-DHBA, respectively, with no significant differences after trauma. In the current article, microdialysis data are presented without corrections for probe recovery or dead volume of the microdialysis probe system (corresponding to a time lag of 1.2 minutes).
The microdialysis experiment was terminated by rapid decapitation, trunk blood was collected, and bilateral cortical samples and the cerebellum were dissected for analyzing 4-HBA and PBN or S-PBN concentrations.
To determine S-PBN concentrations in brain tissue at early time points after injection, 21 additional rats received a single IV injection of S-PBN (47 mg/kg) and were subjected to either sham injury (n = 9) or TBI (n = 12). At 30 or 60 minutes after TBI or sham injury (60 or 90 minutes after injection, respectively), the rats were decapitated and bilateral cortical and hippocampal samples and the cerebellum were dissected. Trunk blood was collected and centrifuged at 5000 g at 5°C for 5 minutes, and the supernatant was collected and stored at −70°C while awaiting analysis.
High performance liquid chromatography
A metal free high performance liquid chromatography (HPLC) system was used as previously described (Marklund et al., 2001c). Microdialysis samples were analyzed for 3,4-DHBA and 4-HBA by HPLC (pump: LKB 210; Bromma, Sweden) with electrochemical detection (Amperometric Detector LC4B; Bioanalytical Systems, West Lafayette, IN, U.S.A.; reference electrode: RE-4; working electrode: MF = 1046; detection potential: 0.85 mV). A Rheodyne Injector PEEK (Rheodyne, Cotati, CA, U.S.A.) was connected to a 4.6 × 150-mm C-18 reversed phase column (Discovery, Supelco, Bellafonte, PA, U.S.A.), preceded by a precolumn (Discovery S5ODS2), using PEEK tubing (Victrex CO). The aqueous mobile phase contained 30 mmol/L citric acid, 30 mmol/L sodium citrate, 1.8% acetic acid, 1.5% methanol, and 40 mg/L EDTA. The pH was set at 3.7. The flow rate was 0.8 mL/min. The retention time for 3,4-DHBA was 8.5 minutes. Quantification was accomplished using standard curves with 5 known concentrations of 3,4-DHBA or 4-HBA and with injection volumes as external standardization.
Analysis of 4-HBA in brain tissue samples was performed using an HPLC system (pump; Varian 9012 Solvent Delivery System, Walnut Creek, CA, U.S.A.) with UV detection (Spectromonitor 3200; LDC Analytical, Thermo Separation Products, Riviera Beach, FL, U.S.A.) with the wavelength set at 254 nm. The C-18, 4.6 × 150 mm Discovery column was used together with a precolumn (Pack ODS-A, 120 Å, S-5 μm; YMC, Wilmington, NC, U.S.A.). The mobile phase consisted of 83% 50 mmol/l KH2 PO4 and 17.0% acetonitrile, pH 3.72. The flow was 0.4 mL/min. Standard curves with 5 concentrations of 4-HBA were prepared using m-hydroxyphenylacetic acid as internal standard. Brain tissue was homogenized in 0.1 mol/L perchloric acid (100 mg tissue per mL perchloric acid containing 80 μL 10% EDTA/mL), centrifuged at 20,000 g for 10 minutes, and filtered through paper filter (Munktell IF; Tamro Lab, Mölndal, Sweden) to remove remaining precipitate. Twenty microliters of the filtered supernatant added with internal standard was injected into the system.
PBN and S-PBN in plasma and brain tissue was analyzed by HPLC and UV detection (at 290 nm) using the same instrumentation as for 4-HBA above. The mobile phase (flow = 1 mL/min) for PBN consisted of 83% 50 mmol/L KH2 PO4 and 17% acetonitrile, pH 3.72. For S-PBN, the mobile phase (flow = 0.6 mL/min) consisted of 88% 50 mmol/L KH2 PO4 and 12% acetonitrile to get acceptable separation from the solvent front and confounding peaks. Standard curves with five concentrations of PBN or S-PBN added to rat plasma or brain tissue extracts were constructed with 5-hydroxy-indole-acetic acid as internal standard. Excellent linearity (r = 0.999) was obtained within the concentration range under study. Equal volumes (250 μL) of acetonitrile and methanol were added to 100 μL plasma for precipitation of plasma proteins. After thorough mixing (Vortex) and centrifugation (10 minutes at 20,000 g), the supernatants were moved to new test tubes, evaporated to dryness, and redissolved in 100 μL of 10% acetonitrile in H2 O; 20 μL was injected into the HPLC system. The minimum detectable concentration of PBN and S-PBN in plasma was 0.35 μg/mL and 0.62 μg/mL, respectively. For PBN, brain tissue was homogenized in 0.1 mol/L formic acid (50 to 100 mg tissue per 1 mL formic acid containing 80 μL 10% EDTA/mL), centrifuged (10 minutes at 20,000 g), and filtered. Internal standard (1 μg/100 μL) was added to 500 μL of the supernatant; 20 μL was injected into the HPLC system. For S-PBN, 50 mg brain tissue was homogenized in 225 μL 0.15 mol/L formic acid added with 15 μL 10% EDTA and 25 μL of internal standard. After homogenization and centrifugation, 20 μL of the supernatant was injected into the HPLC system. The limit of detection was 2.0 μg/g for PBN and 0.3 μg/g for S-PBN.
Statistics
Two-way analysis of variance (ANOVA) was used for group comparisons followed by t-tests for each time point if statistically significant at P < 0.05. Bonferroni corrections for multiple testing were applied. A statistical program (StatView 4.51; Abacus Concepts, Berkeley, CA, U.S.A.) was used for all handling and statistical analysis of the data. All data are presented as mean ± SD. P < 0.05 was considered statistically significant. The significance levels were * P < 0.05, ** P < 0.01, and *** P < 0.005.
RESULTS
Physiologic variables
Results from the physiologic monitoring (mean ± SD), taken at three time points during the experiment, are shown in Table 1. No statistically significant differences between the groups were found. Core and brain temperature were continuously recorded, maintained at 36.5°C to 37.5°C, and adjusted as needed. No changes were found between the treatment groups.
Physiologic variables before and after trauma in the four treatment groups
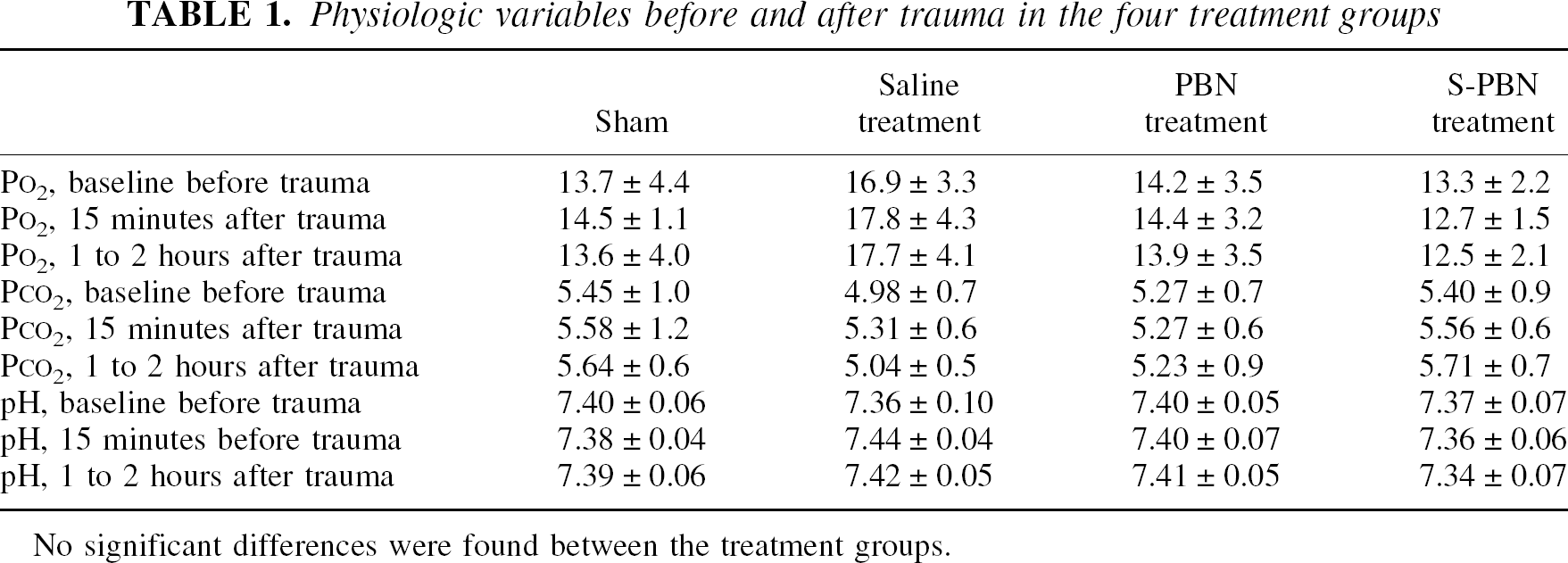
No significant differences were found between the treatment groups.
3,4-DHBA formation after trauma and the effects of PBN and S-PBN
Traumatic brain injury increased ROS formation as compared with sham-operated controls (ANOVA; F = 14.4, P < 0.005) with a 3-to 4-fold increase in 3,4-DHBA formation at 30 (P < 0.005) and 60 minutes (P < 0.01) after trauma (Fig. 2). Pretreatment with PBN significantly attenuated the increase of dialysate 3,4-DHBA (ANOVA; F = 12.0, P < 0.005) although there was no statistically significant difference versus saline-treated animals at any individual time point. There was no statistically significant difference between PBN-treated rats and sham-injured controls (ANOVA; F = 0.05). Pretreatment with S-PBN also attenuated the 3,4-DHBA production (ANOVA; F = 5.4, P < 0.05) as compared with saline-treated animals, with S-PBN-treated animals showing lower 3,4-DHBA levels at 30 minutes after injury (P < 0.05) as compared with saline-treated animals. There was no significant difference between S-PBN treated and sham-injured control animals (ANOVA; F = 2.8, P = NS). In addition, there was no significant difference between PBN-and S-PBN–treated rats (ANOVA; F = 1.6, P = NS).
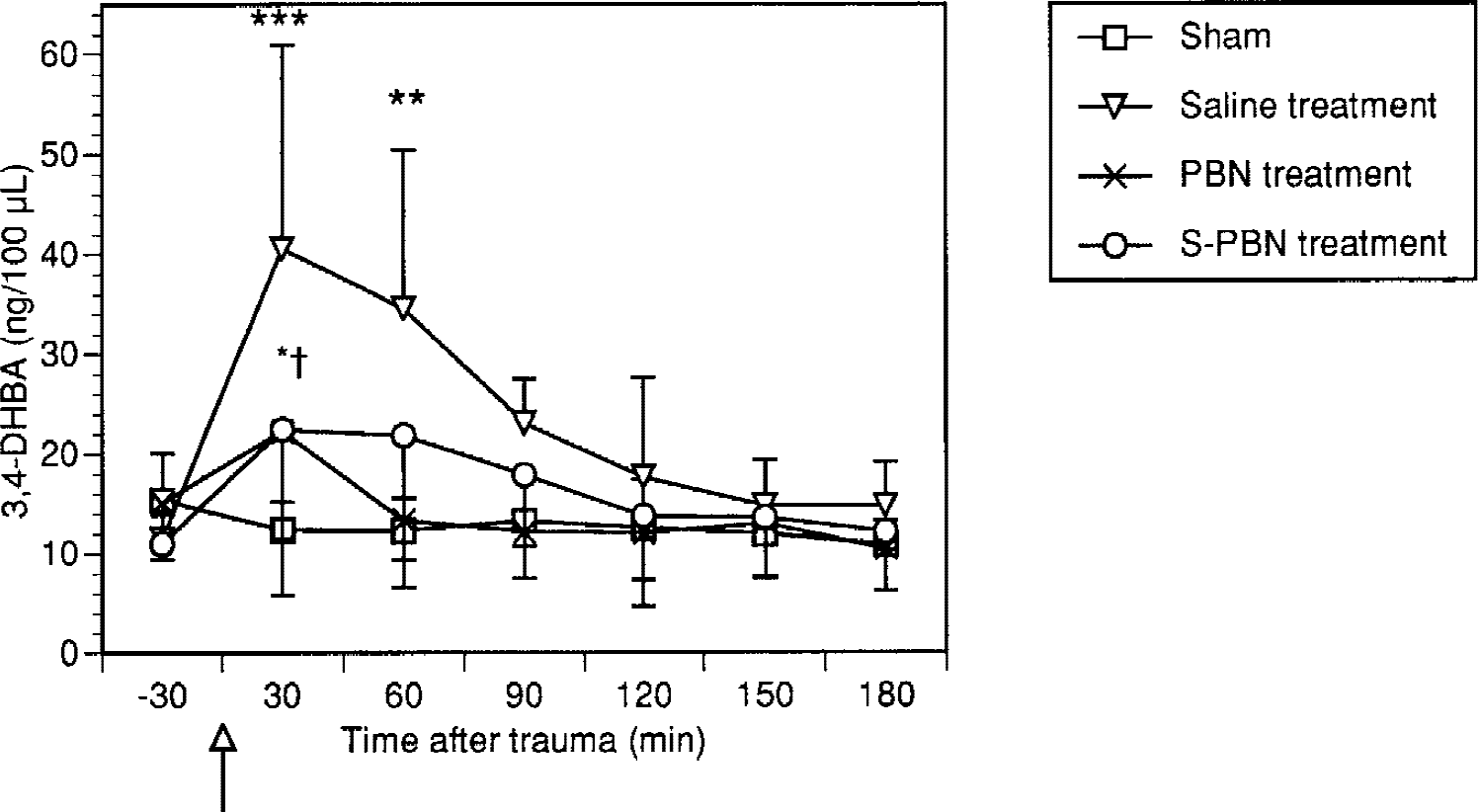
Effect of trauma on 3,4-dihydroxybenzoic acid (3,4-DHBA) formation (mean ± SD). ***(P < 0.005) and **(P < 0.01) indicate statistically significant differences between saline-treated and sham-operated animals. *†Significant difference between saline-treated and 2-sulfo-phenyl-N-tert-butyl nitrone (S-PBN)-treated animals at 30 minutes after trauma. Time for trauma is indicated by an arrow. See text for further explanation of the statistical analysis.
Brain and plasma concentrations of PBN, S-PBN, and 4-HBA
Plasma and brain PBN concentrations remained high throughout the experiment. At the end of the experiment, 210 minutes after injection, high PBN concentrations were found in all brain regions analyzed (Table 2). There were no statistically significant differences between parietal cortices of the injured and the contralateral hemisphere. However, no detectable S-PBN concentrations were found in plasma or any brain region at this time point. In a separate experiment, brain and plasma were analyzed for S-PBN at 30 and 60 minutes after trauma or sham injury, respectively. S-PBN disappeared from plasma with an estimated half-life of 9 minutes. S-PBN concentrations were below the level of detection at both time points in all brain regions, including the injured parietal cortex (Table 2).
Plasma and brain concentrations of PBN and S-PBN
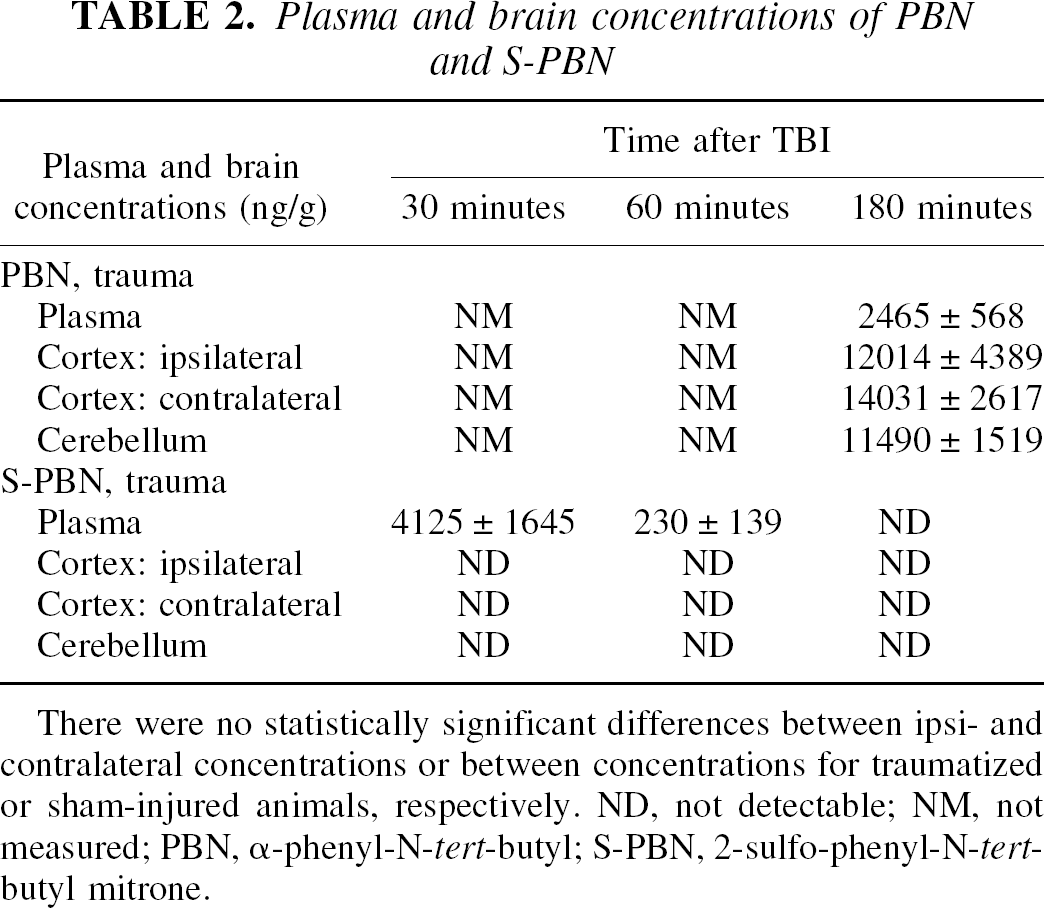
There were no statistically significant differences between ipsi- and contralateral concentrations or between concentrations for traumatized or sham-injured animals, respectively. ND, not detectable; NM, not measured; PBN, α-phenyl-N-tert-butyl; S-PBN, 2-sulfo-phenyl-N-tert-butyl mitrone.
The cortical brain concentrations of 4-HBA taken at the termination of the experiment are shown in Table 3. There were no statistically significant differences between the groups.
4-HBA concentrations taken from the parietal cortex at the termination of the microdialysis experiment (3 hours after injury)
There were no statistically significant differences between the treatment groups. PBN, α-phenyl-N-tert-butylnitrone; S-PBN, 2-sulfo-phenyl-N-tert-butyl nitrone.
DISCUSSION
As previously reported (Marklund et al., 2001c), a cortical contusion injury in rats increased dialysate 3,4-DHBA formation as an indicator of increased reactive oxygen species (ROS) production. The major finding in the current study was that single equimolar IV injections of the nitrone ROS scavengers PBN or S-PBN at 30 minutes before injury equally reduced 3,4-DHBA formation despite the fact that no detectable S-PBN concentrations were found in brain tissue, whereas PBN readily crossed the blood–brain barrier with brain concentrations in the 11 to 14 μg/g range.
Nitrones are well-studied spin-trapping agents in vitro forming stable adducts with carbon-and oxygen-centered radicals (Carney and Floyd, 1991), but their precise action in vivo is not known. Recently, the authors showed that pretreatment with PBN attenuated the cognitive impairment and morphologic damage after contusion injury in the rat (Marklund et al., 2001a). In addition, 24-hour IV infusions with PBN or S-PBN starting 30 minutes after fluid percussion injury reduced tissue damage, and S-PBN attenuated cognitive deficits as assessed in the Morris Water Maze (Marklund et al., 2001b). S-PBN has been found to reduce excitotoxic injury and striatal lesions after injections of NMDA, AMPA, and kainic acid by reducing hydroxyl radical formation (Schulz et al., 1995a) and has been found to attenuate neuronal damage in a model of histotoxic hypoxia (Schulz et al., 1995b).
Although alternative neuroprotective mechanisms have been suggested for nitrones, the current findings suggest that scavenging of ROS may contribute to the beneficial effects observed with PBN and S-PBN after TBI. Scavenging of ROS is an attractive concept for the treatment of TBI because the central nervous system is considered the most sensitive organ in the body to ROS-mediated injury because of its relatively low antioxidant capacity, high utilization rate of oxygen, and high levels of polyunsaturated lipids and iron (Shohami et al., 1997). After TBI, biochemical reactions occur—for example, oxidation of hemoglobin and catecholamines, increased xanthine oxidase activity, increased arachidonic acid metabolism, and mitochondrial dysfunction (Braughler and Hall, 1989)—that are associated with ROS formation. Accordingly, numerous studies have been published showing beneficial effects of pharmacologic compounds with radical scavenging properties on morphologic changes, or cognitive dysfunction, or both, after TBI (for references see McIntosh et al., 1998).
The relative importance of the different sources of ROS formation in acute brain injury is not known, but the intracellular arachidonic acid cascade (Kontos, 1987), mitochondria (Keller et al., 1998), and the microvasculature (Wei et al., 1981) are some of the chief candidates.
The superoxide anion, hydrogen peroxide, and the highly toxic hydroxyl radical may all contribute to tissue damage within the central nervous system (Siesjo et al., 1989; Kontos and Wei, 1986; Hall et al., 1994). The superoxide anion may diffuse through cell membranes but it does not react significantly with DNA, proteins, or phospholipids. However, the superoxide anion may cause formation of hydroxyl radicals through the Fenton reaction, which is markedly accelerated in the presence of iron and copper ions (Halliwell and Gutteridge, 1992). Traumatic brain injury causes an early increase of the expression of all three isoforms of nitric oxide synthase (Gahm et al., 2000). The combination of NO and superoxide produces peroxynitrite (Beckman et al., 1996), a powerful oxidant that is more toxic than NO and superoxide themselves, which may increase microvascular permeability (Imaizumi et al., 1996).
According to in vitro work, 3,4-DHBA is formed when 4-HBA reacts with hydroxyl radicals (Ste-Marie et al., 1996). However, the increased formation of extracellular 3,4-DHBA seen in the current study may reflect an attack on the parent compound, 4-HBA, either by hydroxyl radicals or other highly reactive species such as peroxynitrite (Bogdanov et al., 1999). In addition, the total amount of ROS produced after TBI is not known, but the increase seen in the current study correlates well with other reports using other methods of ROS detection after experimental TBI, for example, the salicylate trapping technique (Hall et al. 1994; Globus et al., 1995; Marklund et al., 2001c).
Mechanisms of action of nitrones
Both PBN and S-PBN are spin-trapping agents that reduce salicylate hydroxylation in vitro with similar efficacy (Carney and Floyd, 1991; Maples et al., 2001). The precise mechanism of action for nitrones in vivo is not clear, but PBN has been shown to improve mitochondrial function (Kuroda et al., 1996) and brain energy state (Folbergrova et al., 1995) after focal ischemia and to attenuate lactate formation after TBI (Lewén and Hillered, 1998), in addition to scavenging free radicals. PBN also has been shown to reduce NO formation (Miyajima and Kotake, 1995), but, to the authors' knowledge, there are no data on the effect of S-PBN on NO production. An effect on NO production may decrease 3,4-DHBA formation through a diminished peroxynitrite formation.
S-PBN is more polar than PBN and only trace amounts may cross an intact BBB as was shown for disodium 2,4-disulfo-phenyl-N-tert-butyl nitrone (NXY-059;Kuroda et al., 1999). In contrast, PBN readily penetrates the BBB (Chen et al., 1990). In general, little information exists on the change in drug transport across the BBB after TBI (Boucher and Hanes, 1998). Traumatic brain injury produces a BBB disturbance (Schmidt and Grady, 1993), and in the current study, the authors studied the effect of TBI on transport of PBN and S-PBN into brain tissue. No detectable concentrations of S-PBN were found in brain tissue or plasma in traumatized animals at 3 hours after the injury in contrast with PBN-treated animals that had high drug concentrations in every cortical region analyzed. Because S-PBN is more rapidly eliminated than PBN, the authors analyzed S-PBN in brain and plasma at 30 and 60 minutes after injury. Again, no detectable S-PBN levels were found in any investigated brain region ipsi-or contralateral to the site of injury in either sham-injured or traumatized animals in spite of measurable plasma concentrations. The authors cannot exclude the penetration of trace amounts of S-PBN below the detection limit of their HPLC system. However, scavenging of ROS requires a much higher concentration of the trapping agent in the tissue. These results imply that S-PBN may exert its effects by affecting mechanisms at the blood–endothelial interface. The findings that the two sulfonated derivatives of PBN are neuroprotective in acute brain injury raise the question of whether PBN mainly acts outside rather than inside the BBB. Another possibility is that nitrone degradation products are formed in vivo. According to cell culture work (Atamna et al., 2000), N-t-butyl hydroxylamine, a degradation product of PBN, accumulated intracellularly and was more effective than PBN in delaying senescence in human lung fibroblasts. However, no data exist regarding in vivo formation of degradation products of S-PBN, and the role of N-t-butyl hydroxylamin in acute brain injury is currently unclear.
The current finding that S-PBN, a compound virtually confined to the bloodstream with minimal passage through the BBB into neural tissue, markedly reduced ROS production with similar efficacy as PBN, supports an important role for microvascular ROS production in TBI. In vitro studies have shown a release of superoxide anion from endothelial cell monolayers (Schinetti et al., 1989) and hydroxyl radicals from isolated rat brain microvessels (Grammas et al., 1993). In addition, microvessels have been shown to be a source of peroxynitrite (Althaus et al., 2000) and the poorly penetrating peroxynitrite scavenger penicillamine improved neurologic recovery after severe TBI in mice (Hall et al., 1999). In mice and rat models of TBI, an early rise in hydroxyl radicals was found after injury (Hall et al., 1994) that was attenuated by saline perfusion to remove intravascular blood before brain removal. The lipid peroxidation inhibitor tirilazad mesylate showed neuroprotection in numerous studies of acute brain injuries (Hall, 1995) and reduced radical formation (Althaus et al., 1993) despite a poor BBB penetration. These studies together with the current data support the view that ROS production originating at the blood–endothelial interface is of importance in acute brain injury. This is further emphasized by the neuroprotective efficacy of nonpenetrating nitrones in TBI and focal ischemia models (Marklund et al., 2001b; Yang et al., 2000; Kuroda et al., 1999; Marshall et al., 2001). The known ability of PBN to cross cell membranes enables it to scavenge ROS from any intracellular source. The current results suggest that scavenging of ROS formed at the blood–endothelial interface may be an important action both for PBN and S-PBN. However, it is not clear if 4-HBA as used in the current experiment reacts with ROS intracellularly; therefore, trapping of intracellular ROS by PBN may escape detection.
Brain temperature is known to affect radical production, and induction of hypothermia (30°C) significantly attenuated ROS formation after fluid percussion injury in rats (Globus et al., 1995). Large doses of PBN (100 mg/kg) were shown to reduce body temperature (Pazos et al., 1999), and a reduction of ROS formation through a decrease in brain temperature could contribute to the observed effects seen with nitrones in the current study. However, as shown here and in previous reports (Marklund et al., 2001a; Lewén and Hillered, 1998), PBN (30 mg/kg) did not cause any decrease in epidural or core temperature. Therefore, a reduction of ROS formation explained by nitrone-induced hypothermia is highly unlikely.
It has been suggested that hydroxyl radicals are initiating vascular injury by triggering peroxidation of polyunsaturated fatty acids in endothelial cell membranes (Hall et al., 1994; Kawamata et al., 1997). However, endothelial cells are exposed to free radicals from several possible sources in blood and neural tissue. Cerebral endothelial cells can themselves generate high amounts of superoxide radicals under certain conditions because they are rich in mitochondria, an important source for ROS production, and because they contain the majority of the xanthine oxidase and xanthine dehydrogenase activity in brain (Betz, 1985; Gidday et al., 1999). Endothelial cells are also exposed to white blood cells that contain the enzyme myeloperoxidase, which produce ROS when activated (Royo et al., 1999; Whalen et al., 1998). Although migration of white blood cells into injured brain tissue does not occur in significant amounts until several hours after injury, the increased adhesion of white blood cells early after TBI (Hartl et al., 1997) may cause increased release of ROS from the activated neutrophils. Reactive oxygen species also have been shown to influence neutrophil–endothelial cell interactions through up-regulation of adhesion molecules such as CD11b, ACAM-1, and P-selectin (Yoshikawa et al., 1996). PBN has been shown to influence inflammatory responses (Kotake et al., 1998; Floyd et al., 1999), and an effect of PBN or S-PBN on adhesion of neutrophils could reduce ROS formation outside of the BBB.
Conclusion
The current results show that PBN and S-PBN attenuate ROS production as reflected by a decrease in 3,4-DHBA formation from 4-HBA after severe controlled contusion injury in rats. No detectable brain concentrations of S-PBN were found after injury. Reactive oxygen species scavenging may contribute to the neuroprotective efficacy seen with PBN and S-PBN after TBI. These data implicate an effect of the nitrone spin-trapping agents on mechanisms at the blood–endothelial interface affecting the pathophysiologic process in acute brain injury.
Footnotes
Acknowledgment:
The authors gratefully acknowledge Ms. Anne-Maj Gustafsson for excellent work with bioanalysis of 4-HBA, salicylate, and their adducts.