
Abstract
Hypoxic preconditioning induces tolerance to hypoxic-ischemic injury in neonatal rat brain and is associated with changes in gene expression. Hypoxia-inducible factor-1 (HIF-1) is a transcription factor that is strongly induced by hypoxia or the hypoxia-mimetic compound cobalt chloride (CoCl2). Hypoxia-inducible factor-1 modulates the expression of several target genes including the glycolytic enzymes, glucose transporter-1 (GLUT-1), and erythropoietin. Recently, HIF-1 expression was shown to increase after hypoxic and CoCl2 preconditioning in newborn rat brain. To study the involvement of HIF-1 target genes in neonatal hypoxia-induced ischemic tolerance, the authors examined the brains of newborn rats after exposure to hypoxia (8% O2 for 3 hours) or injection of CoCl2 (60 mg/kg). Preconditioning with hypoxia or CoCl2 24 hours before hypoxia-ischemia afforded a 96% and 76% brain protection, respectively, compared with littermate control animals. Hypoxic preconditioning increased the expression of GLUT-1 mRNA and protein, and of aldolase, phosphofructokinase, and lactate dehydrogenase proteins but not mRNA. This suggests that the modulation of glucose transport and glycolysis by hypoxia may contribute to the development of hypoxia-induced tolerance. In contrast, preconditioning with CoCl2 did not produce any change in HIF-1 target gene expression suggesting that different molecular mechanisms may be involved in the induction of tolerance by hypoxia and CoCl2 in newborn brain.
Tolerance to brain injury can be achieved by pretreatment with a sublethal stress. This initial preconditioning stimulus is believed to trigger a cascade of endogenous adaptive mechanisms resulting in the development of tolerance. Depending on the nature of the preconditioning stimulus, delayed tolerance is initiated for several hours and can persist for several days. In the brain, tolerance to ischemic injury has been demonstrated after sublethal hyperthermia (Ota et al., 2000), hypothermia (Nishio et al., 2000), brief sublethal ischemia (Simon et al., 1993), spreading depression (Kobayashi et al., 1995), and exposure to metabolic inhibitors (Wiegand et al., 1999). Hypoxia also has been shown to induce tolerance to brain injury both in vitro and in vivo. Indeed, prior exposure to hypoxia protects neurons in culture against oxygen-glucose deprivation (Bruer et al., 1997). Hypoxic preconditioning also prevents neuronal damage and seizures associated with kainate administration in adult rats (Emerson et al., 2000) and reduces hypoxic-ischemic brain injury in neonatal rats (Gidday et al., 1994; Vannucci et al., 1998a; Bergeron et al., 2000).
Little is known about the mechanisms that occur in the time period between hypoxic preconditioning and the development of tolerance to brain ischemia. Tissue hypoxia induces a broad range of adaptive responses including electrophysiologic and biochemical modifications (Haddad and Jiang., 1997). In addition, hypoxia stimulates the expression of many genes, some of which are involved in erythropoiesis, angiogenesis, vasodilation, glucose transport, and stimulation of anaerobic glycolysis (Semenza, 2000). Activation of hypoxia-inducible factor-1 (HIF-1) has been proposed as a crucial event in the regulation of hypoxia-inducible gene expression in mammalian cells (Semenza, 2000). Hypoxia-inducible factor-1 is a member of the basic helix-loop-helix family of transcription factors and requires the dimerization of HIF-1α and HIF-1β subunits for its activity (Wang and Semenza, 1995). Hypoxia-inducible factor-1 heterodimer binds to a specific DNA consensus sequence in the enhancer and promoter regions of many hypoxia-inducible genes (Ebert et al., 1996; Semenza et al., 1994, 1996). Under normoxic conditions, HIF-1α protein is negatively regulated by ubiquitination and proteasomal degradation (Huang et al., 1998; Kallio et al., 1999) such that constitutive levels of HIF-1α protein are generally low or undetectable (Wiener et al., 1996; Bergeron et al., 1999b, 2000). During hypoxia, HIF-1α expression and concomitant HIF-1 DNA binding activity are markedly increased in several cell lines ((Wang and Semenza, 1995; Wang et al., 1995; Jiang et al., 1996a) and primary neuronal cultures (Ruscher et al., 1998). In the brain, increased HIF-1α expression has been observed in rats exposed to systemic hypoxia (Wiener et al., 1996; Bergeron et al., 2000) or subjected to global (Jin et al., 2000) and focal ischemia (Bergeron et al., 1999b; Marti et al., 2000). Interestingly, in vitro exposure to the divalent metal cobalt chloride (CoCl2) or the iron chelator desferrioxamine during normoxic conditions triggers transcriptional changes that mimic a hypoxic response that is characterized by increased HIF-1α and HIF-1β expression, up-regulation of HIF-1 binding activity, and consequent HIF-1 target gene expression (Ebert et al., 1996; Semenza et al., 1994, 1996; Zaman et al., 1999). The hypoxia-mimetic effect of these compounds may involve the interaction with a ferroprotein oxygen sensor.
Recent evidence has shown that HIF-1α and HIF-1β mRNA and protein are induced after preconditioning with hypoxia or CoCl2 in neonatal rats, suggesting that HIF-1–regulated gene expression may be involved in the hypoxia-induced tolerance to ischemic injury in immature brain (Bergeron et al., 2000). To further elucidate the role of HIF-1–mediated processes in the development of ischemic tolerance in newborn rat brain, the authors studied the effect of hypoxia and CoCl2 preconditioning on the expression of several HIF-1 target genes including glucose transporter-1 (GLUT-1), phosphofructokinase (PFK), aldolase (ALD), pyruvate kinase (PK), lactate dehydrogenase (LDH), and erythropoietin (EPO).
MATERIALS AND METHODS
Preconditioning treatments
All animal use procedures were performed in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals. Protocols were approved by the Lilly Research Laboratories Animal Care and Use Committee. Hypoxic preconditioning was performed on male and female Sprague-Dawley rat pups on postnatal day 6 (P6) as described previously (Gidday et al., 1994). Briefly, pups were placed in an 8% O2/92% N2 humidified atmosphere in chambers partially submerged in a water bath maintained at the constant temperature of 37°C for 3 hours. This treatment has been shown to protect the newborn rat brain against subsequent hypoxic-ischemic brain injury (Gidday et al., 1994; Vannucci et al., 1998a; Bergeron et al., 2000). Control animals were maintained at 37°C for 3 hours in normoxic conditions. To further investigate the role of HIF-1–mediated processes in tolerance, the authors used the hypoxia-mimetic compound CoCl2, which also has been shown to produce ischemic tolerance in newborn rat brain (Bergeron et al., 2000) and induce the expression of HIF-1 and several HIF-1 target genes in vitro (Ebert et al., 1996; Semenza et al., 1994, 1996). For CoCl2 preconditioning, P6 rat pups received a single subcutaneous injection of CoCl2 (60 mg/kg), whereas control littermates received 0.9% sodium chloride vehicle solution. For Northern blot and immunoblot analyses of HIF-1 target gene expression, preconditioned rat pups along with their control littermates, were anesthetized at different time points (0.5, 1, 2, 4, 6, 8, and 12 hours) after the preconditioning treatments, their brains were quickly removed, dissected on ice, and frozen at −70°C.
Animal surgery and histopathology
To determine the degree of neuroprotection afforded by the two preconditioning treatments against hypoxic-ischemic brain injury in the neonate rat, a separate group of animals was preconditioned with either hypoxia (n = 27) or CoCl2 (n = 32) as described above. Control littermates were subjected to normoxia (n = 35) or vehicle injection (n = 32), respectively. Then 24 hours after control or preconditioning treatments, rat pups were anesthetized with 1.5% isoflurane in 30%O2/70% N2 mixture and underwent unilateral hypoxia-ischemia (HI) as described previously (Rice et al., 1981). Briefly, the left common carotid artery was exposed through a ventral midline neck incision and was permanently occluded by electrocoagulation. The wound was sutured and animals were returned to their mother for 1.5 hours. Pups then were placed in an 8% O2/92% N2 humidified chamber at a constant temperature of 37°C for 2.5 hours. This combined procedure is known to produce select neuronal damage or infarction in the hemisphere ipsilateral to the carotid occlusion, whereas hypoxia alone (contralateral hemisphere) does not produce any significant brain injury (Rice et al., 1981; Gidday et al., 1994; Vannucci et al., 1998a; Nakajima et al., 2000). Five days after HI, animals were anesthetized and their brains were rapidly removed and frozen in isopentane on dry ice. Rostro-caudal coronal sections (10 μm) were cut on a cryostat and stained with cresyl violet (Nissl) to assess the extent of HI injury. Histopathologic assessment of injury on each newborn rat brain was performed blindly by two independent observers as described previously (Bergeron et al., 1997, 2000; Vannucci et al., 1998a). The following histologic scoring system was used: 0, no histologic damage; 1, minimal neuronal loss; 2, columnar cortical infarction with moderate neuronal loss; and 3, severe neuronal loss and gliosis associated with extensive brain infarction.
Northern blot analysis
Rats were subjected to either hypoxic or CoCl2 preconditioning as described above, and brains were collected and frozen at −70°C at various times after treatments. Brains were dissected out and immediately frozen at −70°C. The cerebral cortex was chosen for analyses because it is not only protected by hypoxic and CoCl2 preconditioning (Gidday et al., 1994; Vannucci et al., 1998a), but also shows a marked increase in HIF-1 expression after these treatments (Bergeron et al., 2000). Brains from control normoxic or vehicle-treated animals were processed in a similar manner. Poly A+ RNA (mRNA) from pooled samples (n = 2 animals per time point) was isolated using FasTrack 2.0 Kit (Invitrogen, San Diego, CA, U.S.A) and 5 μg of mRNA extracts were separated on 1% agarose gel containing formaldehyde. After mRNA transfer onto nylon membranes (Nytran SuperCharge Plus; Schleicher and Schuell, Keene, NH, U.S.A), blots were incubated in a prehybridization solution containing 20 X SSPE (sodium chloride, sodium phosphate, EDTA), 20% sodium dodecyl sulfate (SDS), 2.5 X Denhardt's solution, 10% dextran sulphate, 125 μg/mL heat denatured salmon sperm DNA for 2 hours at 42°C. Antisense oligonucleotide probes (Table 1) were labeled with 32P-deoxyadenosine triphosphate (DuPont-NEN, Boston, MA, U.S.A) at the 3′ end using terminal deoxynucleotidyltransferase (Gibco-BRL, Gaithersburg, MD, U.S.A). After removal of unincorporated labeling with Mini Quick Spin Oligo columns (Roche Molecular Biochemicals, Indianapolis, IN, U.S.A), the membranes were hybridized with 5 × 105 cpm of 32P-labeled probe per 1 mL of hybridization solution overnight at 42°C. The membranes then were washed as follows: twice in 2 X SSPE and 0.1% SDS for 30 minutes at 42°C; once in 1 X SSPE and 0.1% SDS for 30 minutes at 42°C, and subsequently in 1 X SSPE and 0.1% SDS for 30 minutes at 45°C, 50°C, 55°C, and 60°C to increase the wash stringency as necessary. Blots were exposed to Kodak Biomax MS film (Rochester, NY, U.S.A) with one intensifying screen at −70°C for 1 to 3 days. Membranes were stripped and reprobed with other 32P-labeled oligonucleotide probes as described above. The MCID computer based imaging system (Imaging Research, St. Catherines, Ontario, Canada) was used to measure the band area and density of individual transcripts after subtraction of the film background. Each transcript was normalized with the housekeeping gene cyclophilin. These experiments were repeated three times with similar results.
Antisense oligonucleotide probes

Immunoblot analysis
Rats were exposed to either hypoxic or CoCl2 preconditioning as described above, and brains were collected and frozen at −70°C at different times after treatments (n = 3 to 6 animals per time point, per treatment). Brains from control normoxic or vehicle-treated animals were processed similarly. Protein samples were prepared from the cerebral cortex by homogenization in ice-cold lysis buffer containing 20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EDTA, 0.5% Triton X-100, 0.1% SDS, and 1 tablet of Complete Protease Inhibitor Cocktail (Roche Molecular Biochemicals, Indianapolis, IN, U.S.A). Protein content was determined by the bicinchoninic acid method (Pierce, Rockland, IL, U.S.A). Equal amounts (10 to 40 μg) of protein per sample were separated on 10% SDS polyacrylamide gels (Novex; Invitrogen, San Diego, CA, U.S.A). After transfer to nitrocellulose membranes (0.2 μm; Invitrogen, San Diego, CA, U.S.A), immobilized proteins were stained with Ponceau solution to verify equal protein loading. After incubation in blocking solution containing 5% (w/v) nonfat dry milk, 1% (w/v) bovine serum albumin, and 0.1% Tween-20 in 0.1 mol/L phosphate-buffered saline, pH 7.4, membranes were incubated overnight at 4°C with the appropriate primary antibody diluted in phosphate-buffered saline containing 1% bovine serum albumin and 0.1% Tween-20. Primary antibodies against the following proteins were used: GLUT-1 (rabbit anti-rat; diluted 1:10000; a gift from Dr. S.J. Vannucci, Pennsylvania State University, Hershey, PA, U.S.A.); PFK, ALD, PK, and LDH (all goat anti-rabbit; diluted 1:1000; Rockland Immunochemicals; Gilbertsville, PA, U.S.A.); and EPO (rabbit polyclonal; diluted 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A). After washes, the membranes were incubated with horseradish peroxidase coupled secondary antibodies recognizing antigens from the same host as the corresponding primary antibody (mouse and rabbit IgG from Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.; goat IgG from Sigma, St Louis, MO, U.S.A.; diluted 1:5000 in phosphate-buffered saline containing 1% bovine serum albumin and 0.1% Tween-20). Bands were visualized using Lumiglo chemiluminescence reagents (New England Biolabs, Beverly, MA, U.S.A). The MCID computer-based imaging system was used to measure the area and density of protein bands after subtracting the background of the autoradiographic film.
Data analysis
To determine the neuroprotective effects of preconditioning treatments, median histopathologic score values were analyzed by unpaired two-tailed Mann-Whitney U nonparametric test. Histogram data represent the mean ± SD. For the time-course analysis of protein expression, multiple intergroup comparisons were performed by one-way analysis of variance followed by the Tukey post hoc test. P < 0.05 was considered significant.
RESULTS
Neuroprotective effect of preconditioning with hypoxia or CoCl2
The authors first established that the preconditioning treatments used in the current study effectively protected against HI in newborn rats. The model of neonatal HI (Rice et al., 1981) is associated with damage to the cerebral cortex, striatum, hippocampus, and thalamus in the hemisphere ipsilateral to the carotid artery occlusion (Figs. 1A, 1C, 2A, and 2C). As described previously ((Gidday et al., 1994; Vannucci et al., 1998a; Bergeron et al., 2000), preconditioning with hypoxia 24 hours before HI resulted in tolerance to brain injury, compared with control normoxic animals. Brain injury in animals subjected to hypoxia (median score = 0; Fig. 1B, 1D, and 1E) was significantly less (P = 0.004) than that observed in normoxic control littermates (median score = 2; Fig. 1A, 1C, and 1E). Because the aim of the current study was to investigate the role of HIF-1–mediated mechanisms in the development of tolerance, the authors used the hypoxia-mimetic and HIF-1 inducer CoCl2 to induce tolerance. Consistent with previous studies (Bergeron et al., 2000), brain injury in animals preconditioned with CoCl2 (median score = 1; Fig. 2B, 2D, and 2E) was significantly less (P = 0.0459) than that observed in vehicle-injected control littermates (median score = 3; Fig. 2A, 2C, and 2E). Overall, the protection afforded by CoCl2 preconditioning (∼76%) was less than that observed after hypoxic preconditioning (∼96%), which is consistent with previous data (Bergeron et al., 2000). As reported by others ((Gidday et al., 1994; Vannucci et al., 1998a), the mortality rate associated with the HI procedure was reduced 3.6-and 1.8-fold in animals subjected to hypoxic or CoCl2 preconditioning, respectively, compared with nonpreconditioned rat pups.
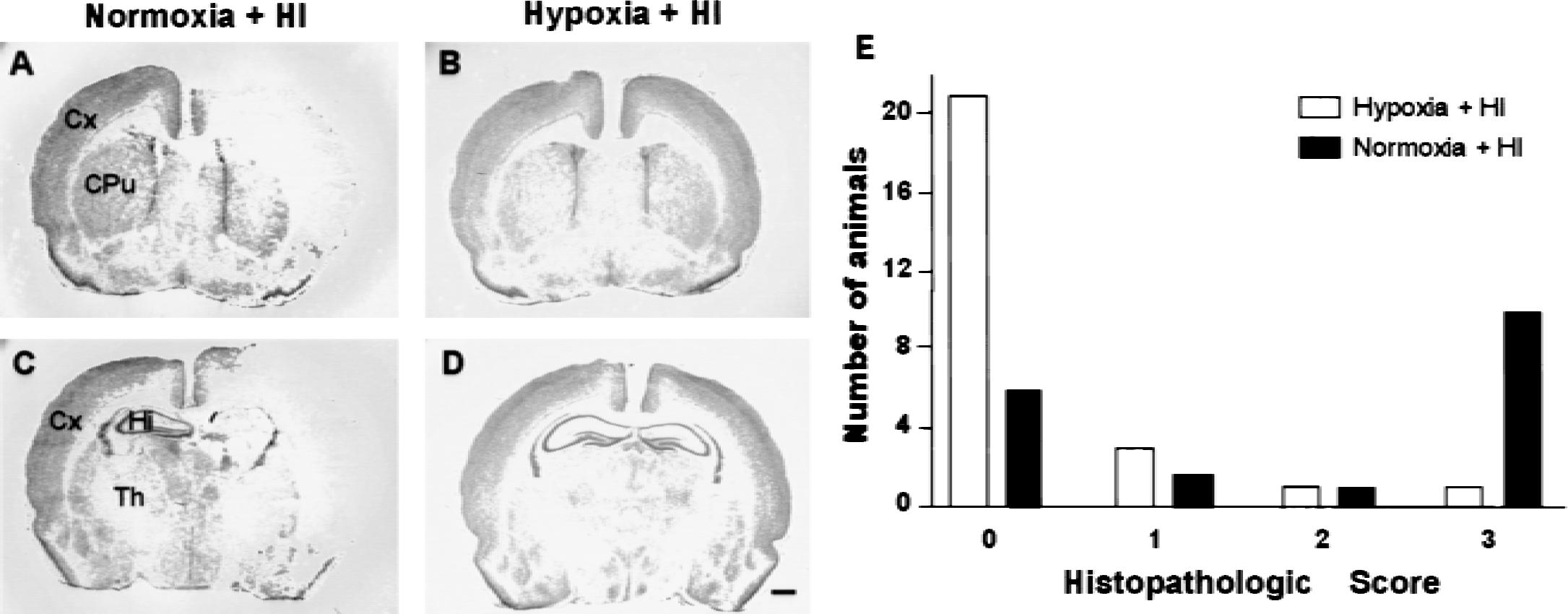
Pretreatment with hypoxia confers protection against hypoxic-ischemic brain injury in newborn rat brain. Rats at postnatal day 6 (P6) were exposed to hypoxia
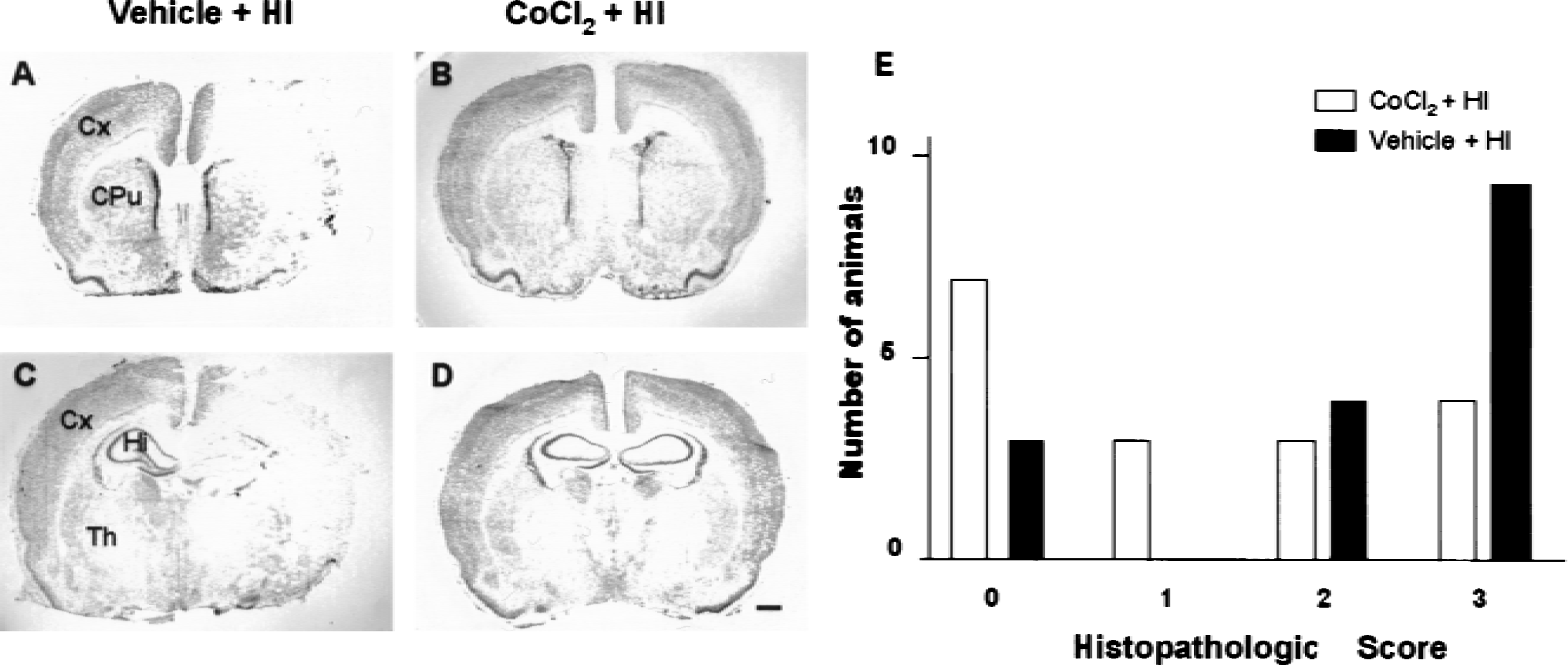
Pretreatment with CoCl2 confers protection against hypoxic-ischemic brain injury in newborn rat brain. Rats at postnatal day 6 (P6) were injected with either vehicle
Constitutive expression of HIF-1 target genes in neonatal rat brain
Northern blot analysis of neonatal rat brain showed that the antisense oligonucleotides (Table 1) used for hybridization all recognized a single transcript of the appropriate size as indicated in Fig. 3. Transcripts for GLUT-1, PFK, ALD, EPO (Fig. 3A and 3, lane C), PK-M, and LDH-A (data not shown) were constitutively expressed in control, nonpreconditioned tissue. Similarly, immunoblot analysis demonstrated constitutive protein expression of all genes studied (Figs. 4 and 6, lane C).
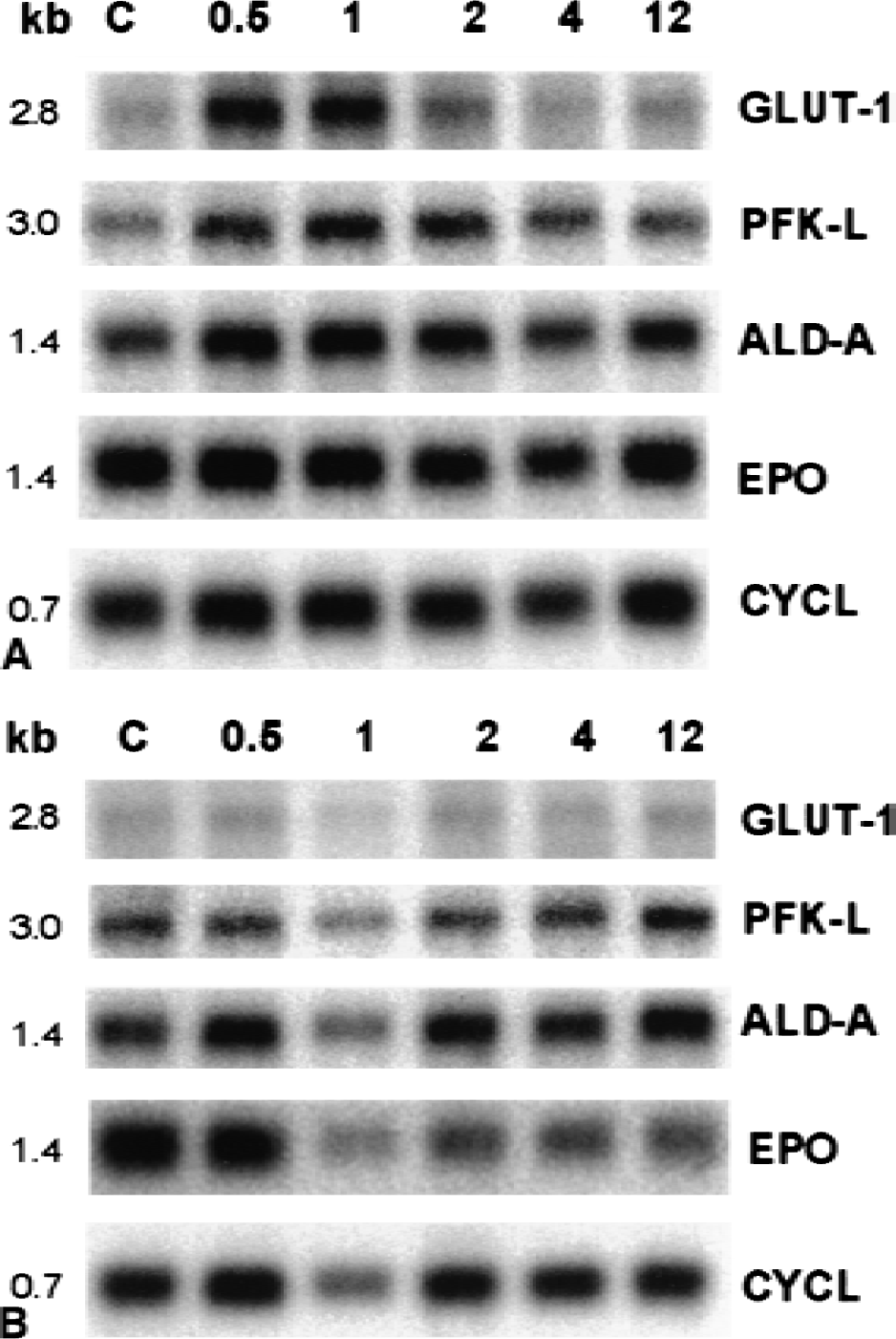
Northern blot analysis of HIF-1 target genes in the cerebral cortex of newborn rats after
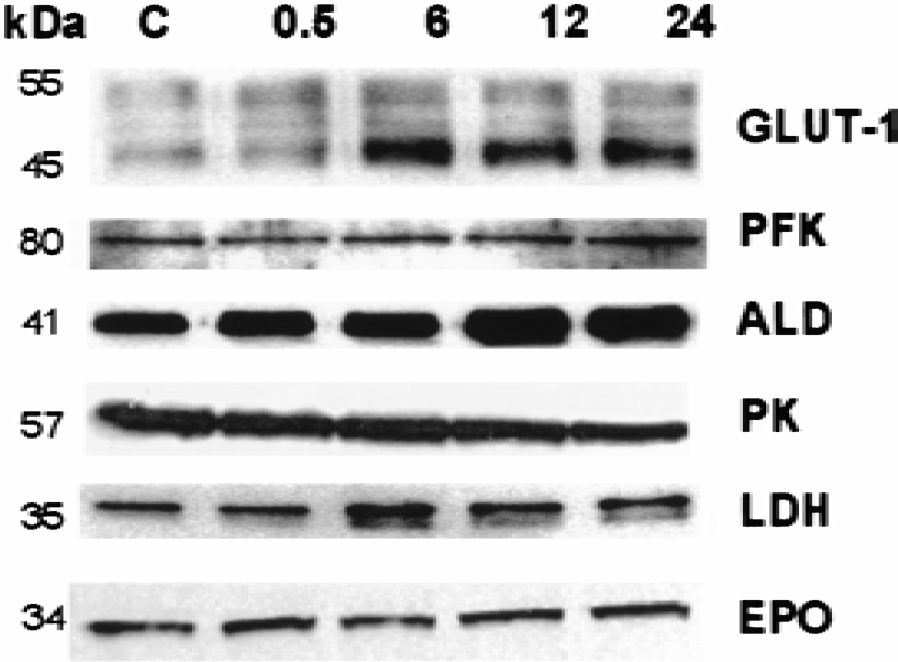
Immunoblot analysis of HIF-1 target genes in the cerebral cortex of newborn rats after hypoxic preconditioning (8% O2 for 3 hours). Equal amounts of protein samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting with antibodies against glucose transporter-1 (GLUT-1), phosphofructokinase (PFK), aldolase (ALD), pyruvate kinase (PK), lactate dehydrogenase (LDH), and erythropoietin (EPO). Brains were analyzed at 0.5, 6, 12, and 24 hours after hypoxic preconditioning. Control (C) normoxic rat brain is shown in lane 1. The molecular weight of each protein (in kilodalton) is indicated on the left.
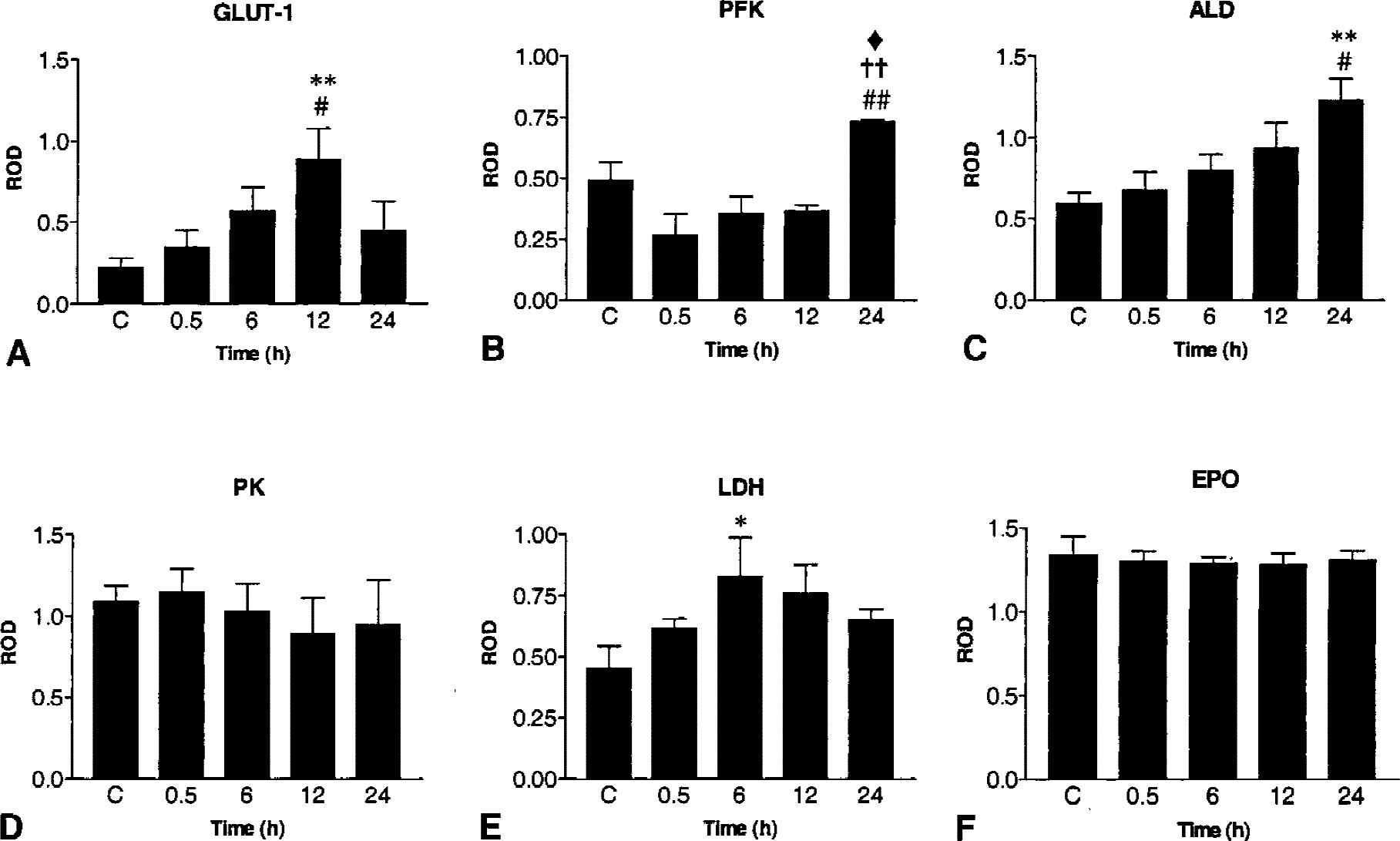
Changes over time in
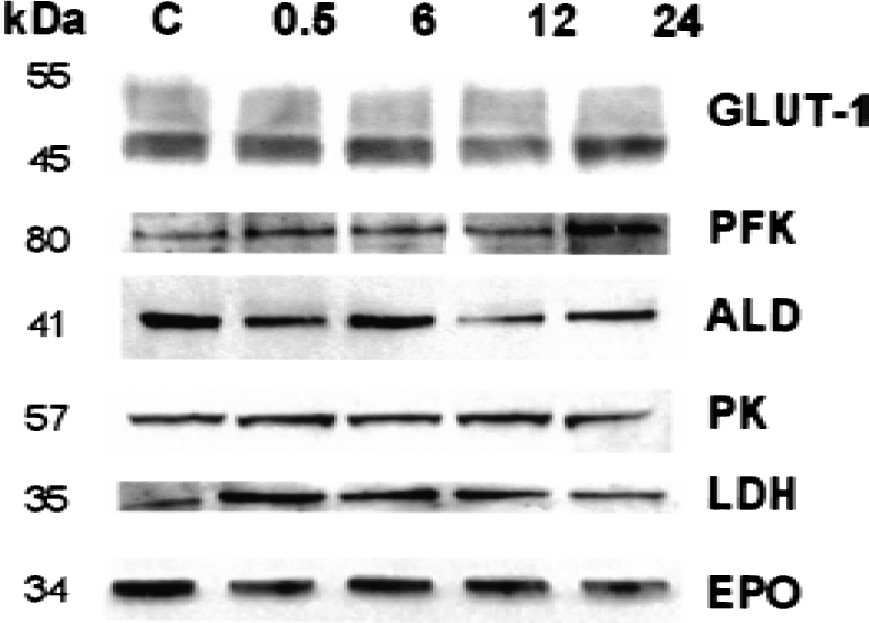
Immunoblot analysis of HIF-1 target genes in the cerebral cortex of newborn rats after CoCl2 (60 mg/kg subcutaneous) preconditioning. Equal amounts of protein samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting with antibodies against glucose transporter-1 (GLUT-1), phosphofructokinase (PFK), aldolase (ALD), pyruvate kinase (PK), lactate dehydrogenase (LDH), and erythropoietin (EPO). Brains were analyzed at 0.5, 6, 12, and 24 hours after CoCl2 administration. Vehicle-treated control (C) rat brain is shown in lane 1. The molecular weight of each protein in kilodalton is indicated on the left.
Effect of hypoxia on HIF-1 target gene expression
Recent studies have shown that hypoxic preconditioning not only induces tolerance to ischemic damage but also increases HIF-1 mRNA and protein expression in newborn rat brain (Bergeron et al., 2000). Because HIF-1 activation may enhance several adaptive mechanisms (Semenza, 2000), the current study investigated the effects of hypoxic preconditioning on the expression of HIF-1 target genes at different time points (0.5, 1, 2, 4, 6, and 12 hours) after treatment. After normalization with cyclophilin, Northern blot analysis showed a ∼1.4-fold increase in GLUT-1 mRNA levels at 0.5 to 2 hours after hypoxia preconditioning (Fig. 3A, lanes 2, 3 and 4), compared with controls (Fig. 3A, lane 1). Similarly, previous studies using the model of neonatal HI in the rat have reported an early increase in GLUT-1 mRNA expression in the hemisphere contralateral to the carotid occlusion (subjected to hypoxia only) (Vannucci et al., 1996). Interestingly, the mRNA levels of PFK-L, ALD-A, EPO (Fig. 3A), PK-M, and LDH-A (data not shown) were unchanged by hypoxia exposure. Because translational regulation of mRNA is an important step in the control of gene function, the level of protein expression of HIF-1 target genes was evaluated by immunoblotting at different time points (0.5, 6, 12, and 24 hours) after preconditioning. Hypoxic preconditioning produced a significant increase in GLUT-1, PFK, ALD, and LDH protein levels (Figs. 4 and 5). In particular, significant differences were observed at 6 hours (LDH), 12 hours (GLUT-1), and 24 hours (PFK and ALD) after hypoxia, compared with controls and earlier time points (Fig. 5). No significant hypoxia-inducible change in the levels of PK and EPO proteins were detected over the time course examined (P > 0.05; Fig. 5).
Effect of CoCl2 on HIF-1 target gene expression
Studies have shown that the hypoxia-mimetic CoCl2 strongly induces HIF-1 expression and activation in vivo (Bergeron et al., 2000) and in vitro (Semenza et al., 1994, 1996; Wang and Semenza, 1995; Wang et al., 1995). In addition, CoCl2 exposure to cell lines and primary cell cultures strongly enhances the expression of several HIF-1 target genes (Semenza et al., 1994; Ebert et al., 1996; Zaman et al., 1999). To further investigate the role of HIF-1–mediated processes in the induction of tolerance, the authors studied the effect of CoCl2 preconditioning on HIF-1 target gene expression. In contrast to hypoxic preconditioning, CoCl2 preconditioning did not have any significant effect on the expression of GLUT-1, ALD, PFK, PK, or EPO mRNA and proteins (P > 0.05; Figs. 3B, 6, and 7). Finally, there was no significant difference in the levels of all mRNA between CoCl2 and hypoxic preconditioning for all time points studied.
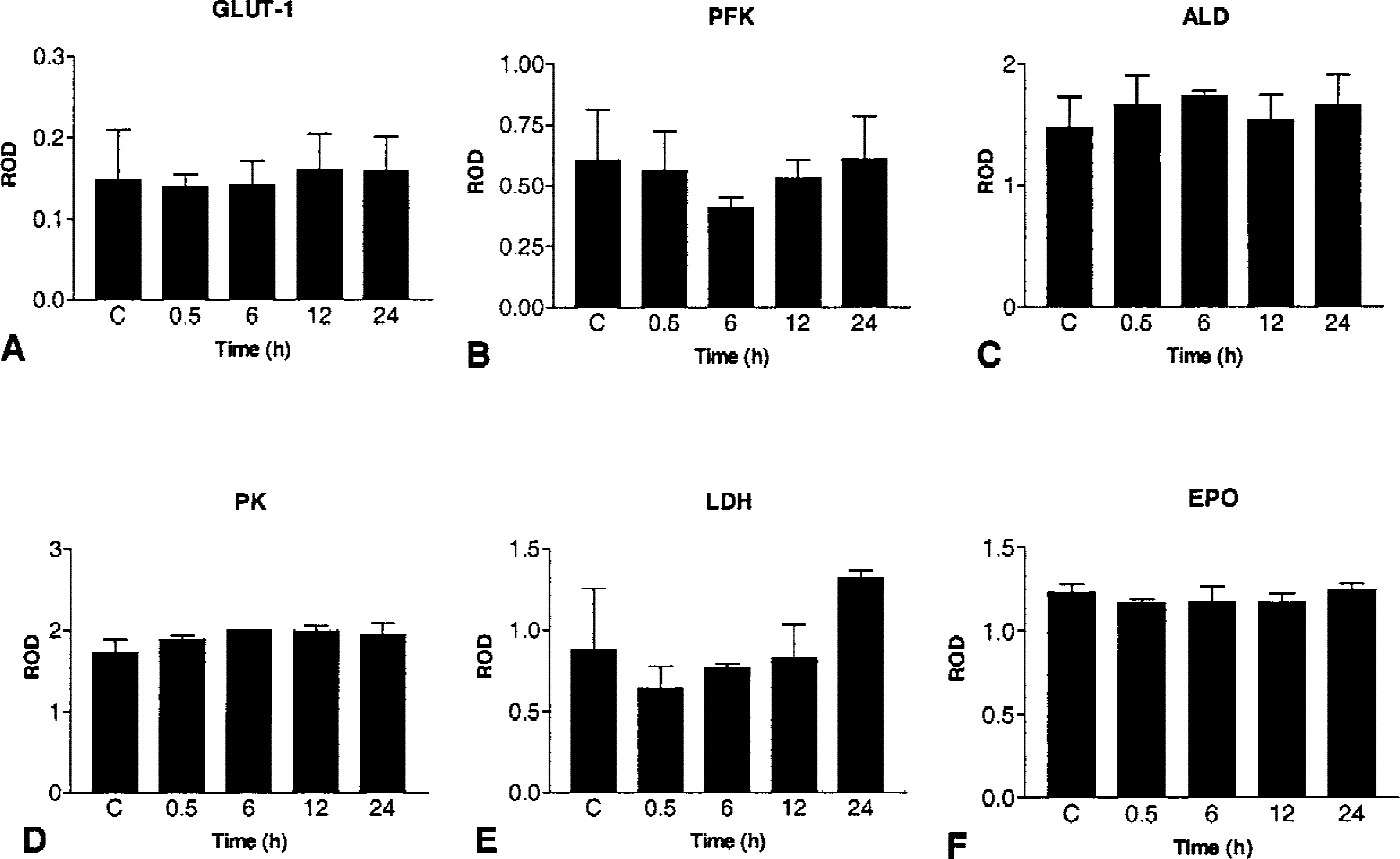
Changes over time in
DISCUSSION
Exposure to hypoxia enhances HIF-1α expression, HIF-1 DNA binding activity, and the expression of several HIF-1 target genes in cell lines (Ebert et al, 1996; Jiang et al., 1996a; Wang et al., 1995), cultured neurons (Ruscher et al., 1998), and rodent brain (Wiener et al., 1996). Because HIF-1α expression is increased throughout the brain after hypoxic preconditioning in newborn rats (Bergeron et al., 2000), it suggests that HIF-1–mediated gene expression may be involved in hypoxia-induced tolerance. In the current study, induction of GLUT-1 mRNA was observed 0.5 to 2 hours afterhypoxic preconditioning, whereas GLUT-1 protein level peaked at 12 hours after hypoxia. An elevated GLUT-1 mRNA level has been reported in the contralateral (hypoxic) hemisphere of neonate rats 1 hour after HI (Vannucci et al., 1998b), whereas increased GLUT-1 protein level was observed as early as 4 hours after the same treatment (Vannucci et al., 1996). Although hypoxia can prolong the half-life of several mRNA, including HIF-1α, GLUT-1, EPO, and vascular endothelial growth factor (Paulding and Czyzyk-Krzeska, 2000), the current study showed that hypoxic preconditioning induces the expression of PFK, ALD, and LDH proteins but not mRNA. This suggests that these changes may be independent of HIF-1 transcriptional regulation. However, it can not be ruled out that the Northern blot method used in the current study may be less sensitive than immunoblots for the detection of small increases of HIF-1 target gene expression in brain tissue. Alternatively, it is also possible that hypoxia increases the stabilization of select proteins by preventing proteolytic degradation and/or ubiquitination much like what has been reported for HIF-1α protein (Kallio et al., 1999).
Increased expression of HIF-1 mRNA and protein, and of mRNA encoding GLUT-1, PFK-L, ALD-A, PK-M, and LDH-A was demonstrated in the area just outside the ischemic infarct after permanent focal ischemia in adult rats (Bergeron et al., 1999b). Increased HIF-1 expression with a concomitant increase of another HIF-1 target gene, vascular endothelial growth factor, also has been shown in the brain after both global (Jin et al., 2000) and focal (Marti et al., 2000) ischemia in adult rats. The difference in the regulation of HIF-1 target gene mRNA expression between the current study and the permanent focal ischemia studies (Bergeron et al., 1999b; Marti et al., 2000) may be because of differences in the duration and/or severity of the hypoxic insult (Jiang et al., 1996b). Indeed, many of the in vitro studies describing the concurrent hypoxic induction of HIF-1 and HIF-1 target genes use more severe hypoxia (1% O2) and examine times after continuous exposure to hypoxia. In hepatoma cells, HIF-1α protein expression peaks after 4 to 8 hours of hypoxia (Wang et al., 1995; Jiang et al., 1996b), whereas expression of the HIF-1 target genes ALD, PK-M, enolase, and phosphoglycerate kinase peaks after 8 to 16 hours of hypoxia (Semenza et al., 1994, 1996). As measured by the EF5 binding method, the 3-hour hypoxic pretreatment used in the current study is associated with mild brain hypoxia (Bergeron et al., 1999a). In contrast, the area surrounding the infarct after permanent focal ischemia is characterized by a moderate but sustained reduction of blood flow from 1 to 24 hours after ischemia (Bergeron et al., 1999b), with intense EF5 binding and tissue hypoxia at 20 hours after ischemia (Marti et al., 2000).
Although HIF-1 DNA binding sites appear necessary for the transcriptional activation of most oxygen-regulated genes, recent evidence suggests that the hypoxic inducibility of these genes is modulated by other factors, including multiprotein complexes involving different transcription factors, coactivator proteins, and other proteins interacting on distant cis-acting elements. A number of coactivator proteins (p300, CBP, p35srj, TIF-1, Ref-1, SRC-1) have been shown to enhance HIF-1– mediated transcriptional activity (Semenza, 2000). Inaddition to HIF-1 DNA binding sites, several other regulatory binding sites (for example, AP-1, CRE, HRE, HSE, NFκB, Sp1) have been found in the promoter region of several hypoxia-inducible genes including those examined in the current study. Therefore, the transcriptional regulation of the target genes examined in the current study may require the simultaneous activation of several coactivator proteins and DNA binding sites. Hypoxic preconditioning in neonatal brain has no effect on neuronal integrity (Rice et al., 1981; Nakajima et al, 2000) and the expression of several genes including the immediate early genes fos and jun (Munell et al., 1994) and HSP72 (Ferriero et al., 1990). Therefore, it is possible that the mild hypoxic stimulus (Bergeron et al., 1999a) used for preconditioning in the current study was not sufficient to activate many of the transcription factors and/or coactivator proteins necessary for target gene mRNA expression. Heme oxygenase-1 (HO-1), a known HIF-1 target gene that has multiple regulatory binding sites in its promoter, is not induced after hypoxic preconditioning in neonate rats (Bergeron et al., 1997). Similarly, although tolerance to kainate-induced lesions is observed in adult rats subjected to hypoxic preconditioning, it is not associated with changes in HO-1 expression (Emerson et al., 2000). Interestingly, HO-1 expression was increased by hypoxia in HIF-1α–deficient Chinese hamster ovary cells (Wood et al., 1998).
To examine further the role of HIF-1 in the induction of tolerance, the authors studied the effect of the hypoxia-mimetic CoCl2 on HIF-1 target gene expression. Hypoxia-inducible factor-1 expression and DNA binding activity, as well as HIF-1 target gene expression, are strongly induced by CoCl2 (Ebert et al., 1996; Semenza et al., 1994, 1996). In addition, CoCl2 application to neuronal cultures not only enhances HIF-1 activation and HIF-1 target gene expression but also protects against neuronal damage induced by glutathione depletion and oxidative stress (Zaman et al., 1999). In agreement with previous reports (Zaman et al., 1999; Bergeron et al., 2000), the current study showed that preconditioning with CoCl2 protected against HI in immature rat brain. As described previously (Bergeron et al., 2000), CoCl2 pretreatment appeared to be less effective at inducing tolerance than hypoxia. In contrast to hypoxic preconditioning, which resulted in enhanced GLUT-1, PFK, ALD, and LDH protein expression, CoCl2 preconditioning did not alter the mRNA or protein expression of all HIF-1 target genes examined. The lower level of HIF-1α expression reported after CoCl2 preconditioning compared with that observed after hypoxic preconditioning (Bergeron et al., 2000) may account, at least in part, for the lack of HIF-1 target gene expression and reduced protection in CoCl2-treated animals. In contrast with the study of Badr et al. (1999), which used chronic administration of CoCl2 in drinking water to induce GLUT-1 expression in adult rat brain, the current study showed that a single injection of CoCl2 to neonate rats did not alter the expression of GLUT-1 or the other HIF-1 target genes examined. As suggested by in vitro data (Ebert et al., 1996; Zaman et al., 1999), prolonged and sustained exposure to CoCl2 may be required to increase HIF-1 target gene expression. Although CoCl2 preconditioning enhances HIF-1 expression in neonate rat brain (Bergeron et al., 2000), the failure to produce changes in HIF-1 target gene expression suggests that CoCl2-induced tolerance is likely to involve HIF-1 independent pathways. For example, the protection afforded by CoCl2 may involve the induction of several stress proteins (Massa et al., 1996), blockade of voltage-dependent Ca2+ channels, and subsequent inhibition of Ca2+-dependent glutamate release (Maeda et al., 1998). Interestingly, preconditioning with another hypoxia-mimetic compound desferrioxamine failed to protect against oxygen-glucose deprivation in cortical neurons, despite HIF-1 DNA binding activity (Sorond et al., 2000).
Proteins that are structurally related to HIF-1α (HIF-2α, HIF-3α) and HIF-1β (ARNT2 and ARNT3) have been shown to form multiple functional HIF dimers (Semenza, 2000). In addition to regulating the expression of several known HIF-1 target genes, these individual complexes also may be involved in the modulation of other genes. Deletion of HIF-1β results in a significant reduction of CoCl2-induced HO-1 and GLUT-1 mRNA expression, but does not affect CoCl2-induced ALD mRNA expression (Tomita et al., 2000), suggesting that different HIF dimers may have distinct roles in mediating CoCl2-induced gene expression. Accordingly, the formation of various HIF complexes may be differentially regulated by hypoxia and CoCl2, which may explain the greater neuroprotection and different pattern of gene expression observed after hypoxia compared with CoCl2.
Taken together, these observations suggest that different mechanisms are involved in the development of tolerance induced by hypoxia and CoCl2 in immature rat brain. However, up-regulation of HIF-1–regulated genes other than those examined in the current study cannot be ruled out as a common mechanism underlying the tolerance to HI induced by both preconditioning treatments.
The current study showed that hypoxic preconditioning induces changes in protein expression of GLUT-1, PFK, ALD, and LDH, which are genes involved in the maintenance of cellular energy supplies. Promoting glucose transport and glycolysis may thus play a role in hypoxia-induced tolerance. Indeed, hypoxic preconditioning has been shown to prevent the secondary depletion of phosphocreatine and adenosine triphosphate that is generally associated with HI in newborn rats (Vannucci et al., 1998a), thus suggesting a greater metabolic capacity and preservation in hypoxia-preconditioned animals. Increasing glucose uptake and glycolytic capacity also protect against focal ischemia in adult rats (Lawrence et al., 1996) and hypoxia in cultured astrocytes (Marrif and Juurlink, 1999). Enhanced glucose transport in the blood–brain barrier may be of particular importance in light of recent evidence demonstrating that ischemic preconditioning has a protective effect in brain vasculature and thus may contribute to decrease edema and cell death (Masada et al., 2001). The increased GLUT-1 expression observed after hypoxic preconditioning may be important for the induction of tolerance by maintaining the integrity of the brain microvasculature. Indeed, a role for endothelial NOS has been proposed in hypoxia-induced tolerance in the neonate (Gidday et al., 1999) supporting the idea that the development of tolerance involves changes in the brain vasculature in addition to the parenchyma.
Footnotes
Acknowledgments:
The authors thank Dr. Susan J. Vannucci (Dept. of Pediatrics, Pennsylvania State University, Hershey, PA, U.S.A.) for kindly providing the GLUT-1 antibody. The authors also are grateful to Dr. James A. Clemens for helpful comments on the manuscript.