
Abstract
Cerebral ischemia results in activation of the mitogen-activated protein kinase pathway and increased tyrosine phosphorylation of proteins associated with postsynaptic densities (PSDs). The authors investigated the possible relation between these events by determining the effect of ischemia on tyrosine phosphorylation of the brain-specific, PSD-enriched, Ras-GTPase activating protein, SynGAP. Transient (15 minutes) global ischemia was produced in rats by 4-vessel occlusion and PSDs prepared from forebrains immediately after ischemia or at 20 minutes, 1 hour, or 24 hours of reperfusion. Tyrosine phosphorylation of SynGAP was elevated relative to sham-operated controls by 20 minutes of reperfusion and remained elevated for at least 24 hours. Tyrosine phosphorylation of SynGAP also increased in CA1 and CA3/DG subfields of the hippocampus. Enhanced tyrosine phosphorylation of SynGAP was not accompanied by a change in PSD RasGAP activity. SynGAP bound to the SH2 domains of Src and Fyn in a tyrosine phosphorylation-dependent fashion, and this interaction increased after ischemia. SynGAP binds to the PDZ domains of PSD-95/SAP90 and coimmunoprecipitated with PSD-95. The coimmunoprecipitation of SynGAP with PSD-95 decreased after ischemia. The results indicate that changes in the properties and interactions of SynGAP may be involved in the neuropathology of ischemia.
Transient cerebral ischemia is followed by a series of pathophysiologic changes that lead to the degeneration of neurons in vulnerable brain regions several days after the ischemic episode. The specific biochemical events that initiate ischemia-induced cell death remain unknown and the identification of changes that occur early in the postischemic period is an important objective. The initiation of reperfusion after an ischemic episode results in the transient activation of the mitogen-activated protein (MAP) kinase signaling pathway (Campos-Gonzalez and Kindy, 1992; Hu et al., 2000; Hu and Wieloch, 1994; Shamloo et al., 1999). Activation of the MAP kinase pathway has been shown to be linked to N-methyl- d -aspartate (NMDA) receptor activation and changes in intracellular calcium, most likely by a pathway involving the GTP-binding protein, Ras (Bading and Greenberg, 1991; English et al., 1999; English and Sweatt, 1997; Gonzalez-Zulueta et al., 2000; Xia et al., 1996). GTP-bound Ras recruits Raf kinase to the membrane, which in turn initiates a sequence of phosphorylation reactions leading to MAP kinase activation (Cobb and Goldsmith, 1995). Ras has slow intrinsic GTPase activity that catalyzes the conversion of active, GTP-bound Ras to the inactive, GDP-bound form. GTPase activating proteins (GAPs) enhance the rate of GTP hydrolysis and catalyze the rapid inactivation of Ras (Downward et al., 1990; Wigler, 1990). The mechanism by which activation of the NMDA receptor is linked to the MAP kinase cascade is not known, but a possible candidate for this role was suggested by the recent identification of the brain-specific, synaptic enriched, Ras-GTPase activating protein, SynGAP (Chen et al., 1998; Kim et al., 1998).
SynGAP interacts with the PDZ domains of the postsynaptic anchoring protein PSD-95/SAP90 and is concentrated in postsynaptic densities (PSDs) of excitatory synapses (Chen et al., 1998; Kim et al., 1998). SynGAP contains a sequence that is 30% homologous to p120 RasGAP and exhibits Ras-GTPase activating activity (Chen et al., 1998; Kim et al., 1998). The additional presence of regions that show homology to the PH domain, a phospholipid-binding module that binds polyphosphatidylinositides (Shaw, 1996), and to the C2 domain of p120 GAP that is involved in the binding of Ca2+ in a phospholipid-dependent manner (Luo and Weinstein, 1993; Yamaguchi et al., 1993), suggest that SynGAP may be regulated by changes in Ca2+ and phospholipid second messengers. SynGAP contains a number of potential phosphorylation sites for CaMK II in its C-terminal domain (Kim et al., 1998) and is inhibited by CaMK II (Chen et al., 1998). Potential sites for tyrosine phosphorylation are also present in the C-terminal domain, but it is not presently known if SynGAP is phosphorylated by tyrosine kinases.
The tyrosine phosphorylation of proteins present in isolated PSDs is increased after transient global ischemia (Cheung et al., 2000). As part of the authors' continuing studies on the effect of ischemia on the tyrosine phosphorylation of PSD proteins they reported that ischemia induces a marked and sustained increase in the tyrosine phosphorylation of SynGAP in the forebrain and hippocampus of the rat brain.
MATERIALS AND METHODS
Animal surgical procedure
All procedures using animals were approved by the Animal Care Committee of the University of Toronto and were in accordance with the guidelines established by the Canadian council on Animal Care. Male Wistar rats, weighing between 200 and 250 g, were given free access to food and water before surgery. Transient global ischemia (15 minutes) was induced as described previously (Shinno et al., 1997; Takagi et al., 1997; Zhang, 1997), using a modified version of the 4-vessel occlusion method (Kirino, 1982; Pulsinelli and Brierley, 1979). The current experiments were performed only if animals showed a completely flat bitemporal electroencephalogram, maintenance of a dilated pupil, and the absence of a corneal reflex during the duration of the carotid artery occlusion. Sham-operated animals received exactly the same surgical procedures without cauterization of the vertebral artery or occlusion of the carotid artery.
Tissue preparation
Animals were allowed to recover from the ischemic challenge for periods up to 24 hours. Animals were killed by decapitation, and the heads were quickly frozen in −42°C isopentane and stored at −72°C until use. For dissection, brains were warmed to −5°C to 0°C and the forebrain and hippocampus removed. For dissection of the CA1 and CA3/dentate gyrus regions, heads were rapidly cooled, but not frozen, by brief exposure to −42°C isopentane, and the dissection was performed immediately. Dissected hippocampal subfields were rapidly frozen and stored at −72°C. Tissue samples were homogenized in ice-cold 0.32 mol/L sucrose containing 0.1 mmol/L sodium orthovanadate, 0.1 mmol/L phenylmethylsulfonyl fluoride, and 5 μg/mL each of antipain, aprotinin, and leupeptin. Homogenates were centrifuged at 800 gAv for 10 minutes at 4°C and the supernatants were centrifuged at 100,000 gAv at 4°C for 60 minutes to obtain the membrane pellet. This pellet was resuspended in 0.32 mol/L sucrose containing phosphatase and protease inhibitors as described above. Postsynaptic densities were prepared from forebrains pooled from two ischemic or two sham-operated animals as described (Cheung et al., 2000), according to the method of Cho et al. (1992), using one extraction with Triton X-100 to obtain the final PSD preparation. Postsynaptic densities were suspended in 0.32 mol/L sucrose. Protein was determined by the method of Lowry et al. (1951). Postsynaptic densities were solubilized in 1N NaOH before protein determination. All fractions were stored at −72°C until use.
Immunoprecipitation and immunoblotting
For the immunoprecipitation of tyrosine-phosphorylated proteins or SynGAP, 0.02 mL protein A/G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) was precoated with 2 μg of antibodies against phosphotyrosine (PY20; Transduction Laboratories, Lexington, KY, U.S.A.) or SynGAP (Upstate Biotechnology, Lake Placid, NY, U.S.A.) for 4 hours at 4°C in 50 mmol/L Tris-HCl buffer, pH 7.4, containing 1% (v/v)NP-40, 0.25% (w/v) Na-deoxycholate (DOC), 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 5 μg/mL each of aprotinin, leupeptin, and antipain, 0.1 mmol/L sodium orthovanadate, and 1 mmol/L NaF (immunoprecipitation buffer). Proteins to be immunoprecipitated were solubilized in 1% sodium dodecyl sulfate (SDS) containing 1% β-mercaptoethanol for 5 minutes at 100°C, diluted 10-fold with immunoprecipitation buffer, and incubated with 0.02 mL of protein A/G PLUS-Agarose for 1 hour at 4°C to remove any proteins that adhered nonspecifically to the protein A/G PLUS Agarose. After centrifugation, supernatants were added to the precoated protein A/G PLUS-Agarose and incubated at 4°C for 4 hours or overnight. The immune complexes were isolated by centrifugation, washed 5 times with immunoprecipitation buffer and proteins eluted by heating at 100°C for 5 minutes in 62.5 mmol/L Tris-HCl, pH 6.8, containing 2% SDS, 1% β-mercaptoethanol, and 5% glycerol (sample buffer). For coimmunoprecipitation studies, membranes were extracted at 37°C for 30 minutes with immunoprecipitation buffer containing 1% (v/v) NP40, 0.1% (w/v) SDS, and 0.5% (w/v) DOC. Insoluble proteins were removed by centrifugation at 100,000 gAv for 10 minutes at 4°C, and the supernatant was used for immunoprecipitation with antibodies against SynGAP or PSD-95 as described above.
For immunoblotting, proteins were separated on 8% polyacrylamide gels and transferred to nitrocellulose as described (Gurd et al., 1992). Immunoblotting was performed using anti-phosphotyrosine antibody from Upstate Biotechnology (Clone 4G10), SynGAP antibodies that were provided by Dr. R. Huganir (Yale University, New Haven, CT, U.S.A.), or PSD-95 antibodies (Affinity Bioreagents, Golden, CO, U.S.A.). Bound antibodies were detected by enhanced chemiluminescence (Pierce Super Signal, Rockford, IL, U.S.A.) and exposed x-ray film was scanned using a BioRad GS 700 gel scanner (Hercules, CA, U.S.A.). In some cases, immunoblots that had been reacted with anti-phosphotyrosine antibodies were reprobed for SynGAP after removing bound phosphotyrosine antibodies by heating the blots for 30 minutes at 55°C in 0.1 mol/L glycine, pH 2.7. The efficacy of the stripping procedure was confirmed by reacting the stripped blot with secondary antibody only; under these conditions no bound antibodies were detected. Statistical comparison between ischemic and sham-operated samples was performed using the unpaired Student's t-test.
SH2 binding
For the preparation of SH2 fusion proteins, cDNA encoding the SH2 domain of Src or the SH2 domain of Fyn was subcloned into pGEX expression vectors. Bacterial cells containing the vectors (provided by Dr. T. Pawson, Mt. Sinai Hospital Research Institute, Toronto, Canada) were induced with isopropylthiogalactoside, and the expressed GST–SH2 fusion proteins were isolated by binding to glutathione-sepharose (Pharmacia). The resulting beads, containing the SH2 domain of Src or Fyn, are referred to as Src SH2-sepharose and Fyn SH2-sepharose, respectively. Glutathione-sepharose beads containing only bound GST, without an associated SH2 domain (GST-sepharose), served as control. The binding of SynGAP to the SH2-domains of Src and Fyn was determined by incubating PSD proteins that had been solubilized by heating at 100°C for 5 minutes in 1% SDS containing 1% β-mercaptoethanol with SH2-sepharose as described previously (Gurd and Bissoon, 1997). In some experiments, the SH2-binding reaction was performed in the presence of the high affinity Src SH2-binding peptide, EPQpYEEIPIA (50 × 10−6 mol/L; provided by Dr. M. Salter, Hospital for Sick Children, Toronto, Canada), or its nonphosphorylated homologue, EPQYEEIPIA (100 × 10−6 mol/L). To dephosphorylate tyrosine residues, PSDs were suspended in 50 mmol/L Tris-HCl pH 7.0, 100 mmol/L NaCl, 2 mmol/L EDTA, 5 mmol/L dithiothreitol (DTT), 0.01% Brij35, 1 mg/mL bovine serum albumin, 5 μg/mL each of antipain, aprotinin, and leupeptin, and protein tyrosine phosphatase (40 units, recombinant from Yersinia enterocolitica; Boehringer Mannheim, Germany) was added. After 30 minutes at 37°C, fresh enzyme (40 units) was added and the incubation continued for an additional 30 minutes. The dephosphorylation reaction was stopped by the addition of one-tenth volume of 10% SDS containing 10% β-mercaptoethanol and heating at 100°C for 5 minutes. To determine the effect of tyrosine phosphorylation by exogenous Src, endogenous protein kinases were first inactivated by heating PSDs at 60°C for 30 minutes (Suzuki and Okumura-Noji, 1995). Postsynaptic densities then were incubated in 50 mmol/L HEPES buffer, pH 7.1 containing 0.1% Triton X-100, 10 mmol/L MgCl2, 2 mmol/L adenosine triphosphate (ATP), 100 × 10−6 mol/L each of phenylmethylsulfonyl fluoride and sodium orthovanadate, 5 μg/mL each of antipain, leupeptin, and aprotinin (phosphorylation buffer), and 3 units of recombinant c-Src (Upstate Biotechnology). Phosphorylation was performed for 30 minutes at 30°C and the reaction was stopped by the addition of one-tenth volume of 10% SDS containing 10% β-mercaptoethanol and heating at 100°C for 5 minutes.
Determination of Ras-GTPase activating protein activity
Ras-GTPase activating protein activity was determined as described elsewhere (Kim et al., 1998; Settleman et al., 1992) with some modifications. Briefly, 0.5 × 10−6 M GST-Ras (Calbiochem) was incubated with 0.2 × 10−6 M [γ 32 P]GTP (3000 Ci/mmol/L; ICN, Pharmaceuticals, Irvine, CA, U.S.A.) in 50 mmol/L Tris HCl buffer, pH 7.5, containing 50 mmol/L NaCl, 5 mmol/L EDTA, 1 mg/mL bovine serum albumin, and 1 mmol/L DTT for 10 minutes at 25°C. 3 × 10−3mL of this reaction, containing [γ −32P]GTP-bound GST-Ras, was added to 50 mmol/L Tris HCl buffer, pH 7.5, 10 mmol/L MgCl2, 1 mg/mL bovine serum albumin, 1 mmol/L DTT, 0.1% Triton X-100 (incubation buffer), and 40 μg PSD protein in a total volume of 0.03 mL. After 7.5 minutes at 25°C, the reaction was stopped by the addition of 0.5 mL ice-cold 50 mmol/L Tris HCl buffer, pH 7.5, 10 mmol/L MgCl2, 150 mmol/L NaCl, 1 mmol/L DTT, and 0.1% Triton X-100 and centrifugation at 12,000 g for 10 minutes to remove PSDs. The supernatant then was added to 0.08 mL of a 50% suspension of glutathione-agarose beads (Amersham Pharmacia Biotech AB, Baie d'Urfé, Québec, Canada) and incubated at 4°C for 45 minutes. Beads were washed three times with incubation buffer and nucleotides dissociated from GST-Ras by boiling for 5 minutes in 0.2 mL 1% SDS. The supernatant was spotted on hemagglutinin (HA) membranes (Millipore), membranes were washed 3 times with incubation buffer and radioactivity determined. To measure the effect of phosphorylation by CaMK II on GAP activity, PSDs were prephosphorylated for 3 minutes at 30°C in 50 mmol/L Tris HCl buffer, pH 7.5 containing 10 mmol/L MgCl2, 5 mmol/L β-mercaptoethanol, 0.3 mmol/L CaCl2, 4 × 10−6mol/L calmodulin, and 0.1 mmol/L ATP. Controls were incubated under similar conditions but in the absence of ATP. Phosphorylation was stopped by the addition of 2 mmol/L ethylene glycol-bis(β-aminoethyl ether) N,N,N′,N′-tetraacetic acid (EGTA) and 10 mmol/L NaPPi, and PSDs then were used for the determination of RasGAP activity as above.
RESULTS
Transient global ischemia results in increased tyrosine phosphorylation of SynGAP
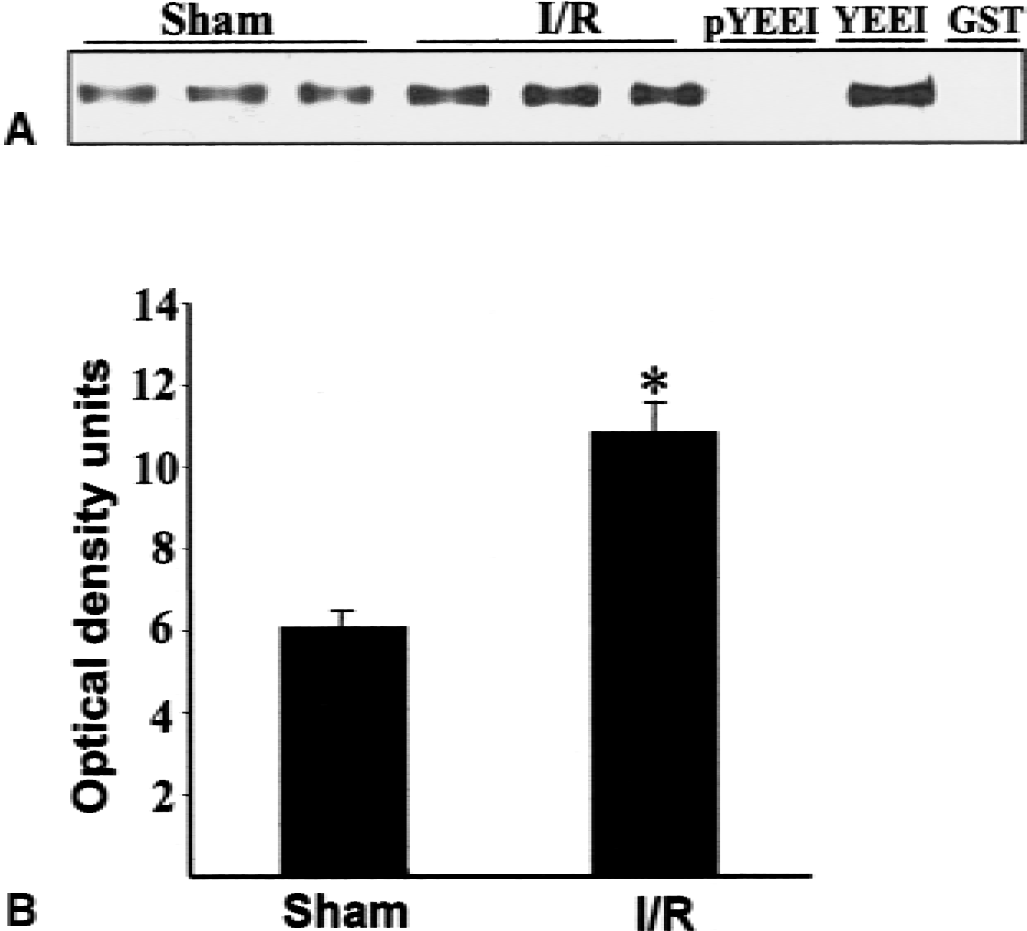
Transient global ischemia followed by reperfusion induces a marked increase in the tyrosine phosphorylation of a number of proteins associated with the PSD (Cheung et al., 2000), most of which remain to be identified. Because SynGAP is enriched in PSDs and its C-terminal domain contains a number of potential tyrosine phosphorylation sites (Chen et al., 1998; Kim et al., 1998), the authors investigated the effect of ischemia on its tyrosine phosphorylation. Postsynaptic densities were isolated from the forebrains of sham-operated rats and rats that had been subjected to 15 minutes of global ischemia followed by 1 hour of reperfusion, a time at which tyrosine phosphorylation of PSD proteins is elevated (Cheung et al., 2000). SynGAP was immunoprecipitated from the PSDs and tyrosine phosphorylation was assessed by probing blots of the immunoprecipitate with anti-phosphotyrosine antibodies. Low levels of tyrosine-phosphorylated SynGAP were present in PSDs from sham-treated animals and tyrosine phosphorylation was markedly increased after ischemia (Fig. 1A). Similar amounts of SynGAP were present in the immunoprecipitates from sham and ischemic samples, indicating that the increase in phosphotyrosine immunoreactivity was not because of an increase in the concentration of SynGAP in PSDs from the ischemic animals (Fig. 1A). In agreement with these results, anti-phosphotyrosine antibodies immunoprecipitated more SynGAP from ischemic than from sham PSDs (Fig. 1B). The ischemia-induced increase in tyrosine phosphorylation of SynGAP was apparent after 20 minutes of reperfusion and was sustained for at least 24 hours (Fig. 2).
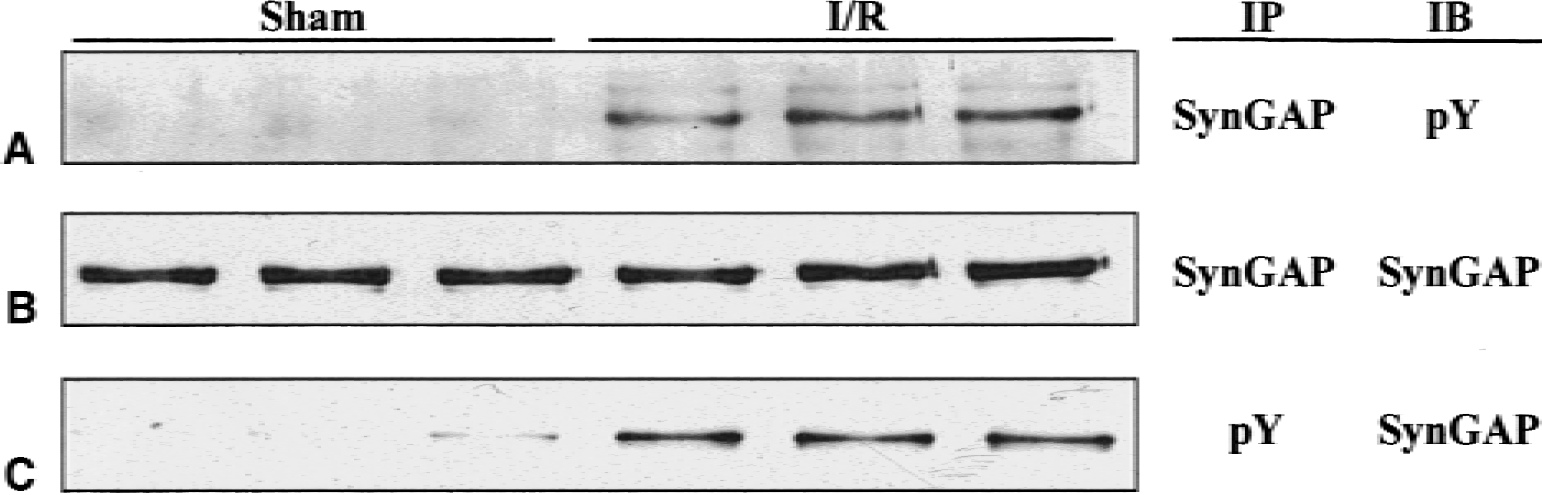
Effect of transient cerebral ischemia on tyrosine phosphorylation of SynGAP.
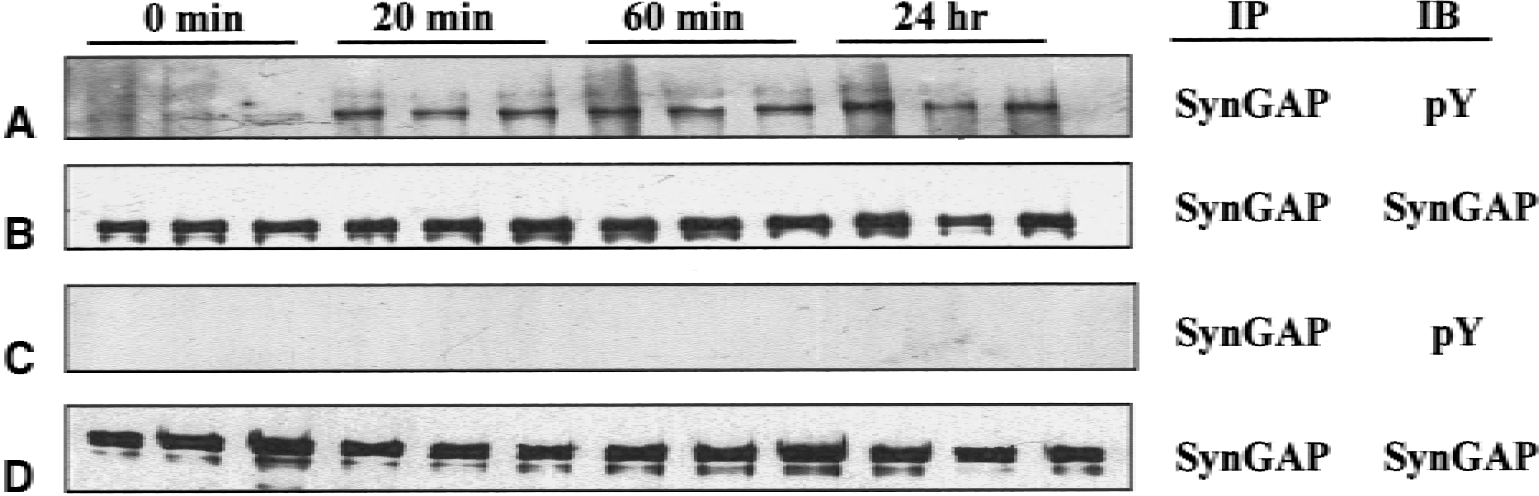
Effect of time of reperfusion on the tyrosine phosphorylation of SynGAP. Postsynaptic densities (PSDs) were prepared from ischemic animals at various times of reperfusion
Ischemia-induced increase in tyrosine phosphorylation of SynGAP occurs in both CA1 and CA3/dentate gyrus
The above results were obtained using PSDs prepared from rat forebrain. The authors next investigated the effect of ischemia on tyrosine phosphorylation of SynGAP in the hippocampus, which is more vulnerable to ischemia-induced cell damage than the forebrain (Kirino, 1982; Petito et al., 1987; Pulsinelli et al., 1982). SynGAP was immunoprecipitated from membranes prepared from the hippocampus and the immunoprecipitates analyzed by immunoblotting with anti-phosphotyrosine antibodies. Similar to the results obtained with forebrain PSDs, low levels of tyrosine-phosphorylated SynGAP were present in hippocampal samples from sham animals, and ischemia resulted in an increase in tyrosine phosphorylation levels (Fig. 3A). Ischemia was reported to result in differential increases in tyrosine phosphorylation in CA1 and CA3/DG regions (Hu and Wieloch, 1994), and the authors next compared tyrosine phosphorylation of SynGAP in these two hippocampal subfields. The results presented in Fig. 3B and 3C show that ischemia induced an increase in the tyrosine phosphorylation of SynGAP in both regions, with a trend toward a greater increase in the more vulnerable CA1 region (Fig. 3B and 3C).
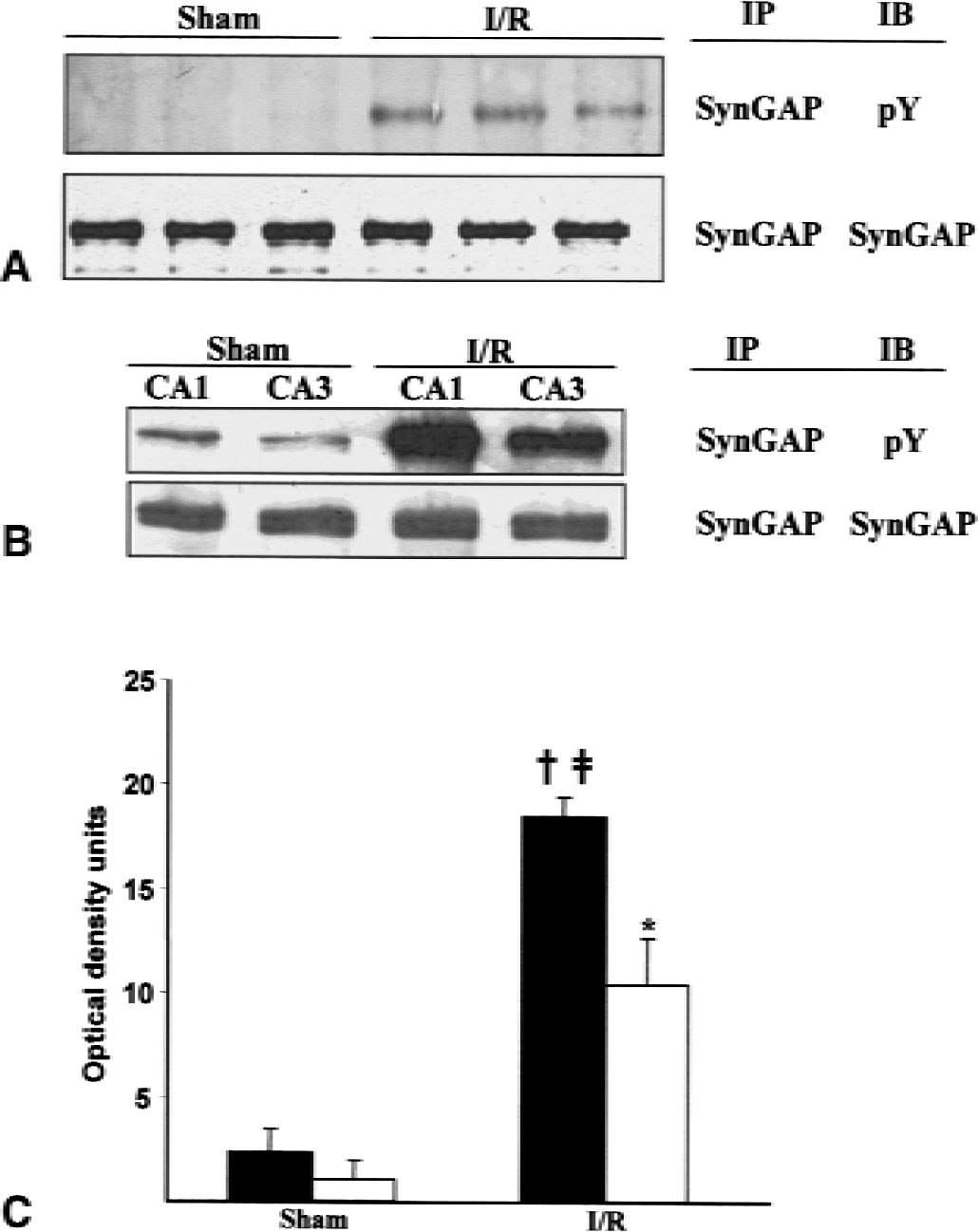
Effect of ischemia on tyrosine phosphorylation of SynGAP in rat hippocampus. Membrane proteins from the hippocampus
PSD RasGAP activity is not altered after ischemia
To determine the possible functional consequences of the ischemia-induced increase in tyrosine phosphorylation of SynGAP, the authors first assessed the effect of ischemia on the RasGAP activity of PSDs isolated after ischemia and 1 hour of reperfusion. Initial experiments showed that RasGAP activity was proportional to the amount of PSD protein added to the assay mixture up to at least 60 μg PSD protein, and that the release of 32P from [γ −32P]GTP Ras increased with time of incubation (Fig. 4A and 4B). In contrast to the marked effect of ischemia on tyrosine phosphorylation of SynGAP, ischemia did not alter the total RasGAP activity associated with isolated PSDs (Fig. 4C). SynGAP is inhibited after phosphorylation by Cam kinase II (Chen et al., 1998). The authors therefore determined the effect of prephosphorylation of sham and ischemic PSDs in the presence of calcium and calmodulin on RasGAP activity. In agreement with previous results (Chen et al., 1998), PSD GAP activity was decreased after phosphorylation under these conditions. The inhibitory effect of the prephosphorylation reaction was similar with PSDs from sham and ischemic animals (Fig. 4C).
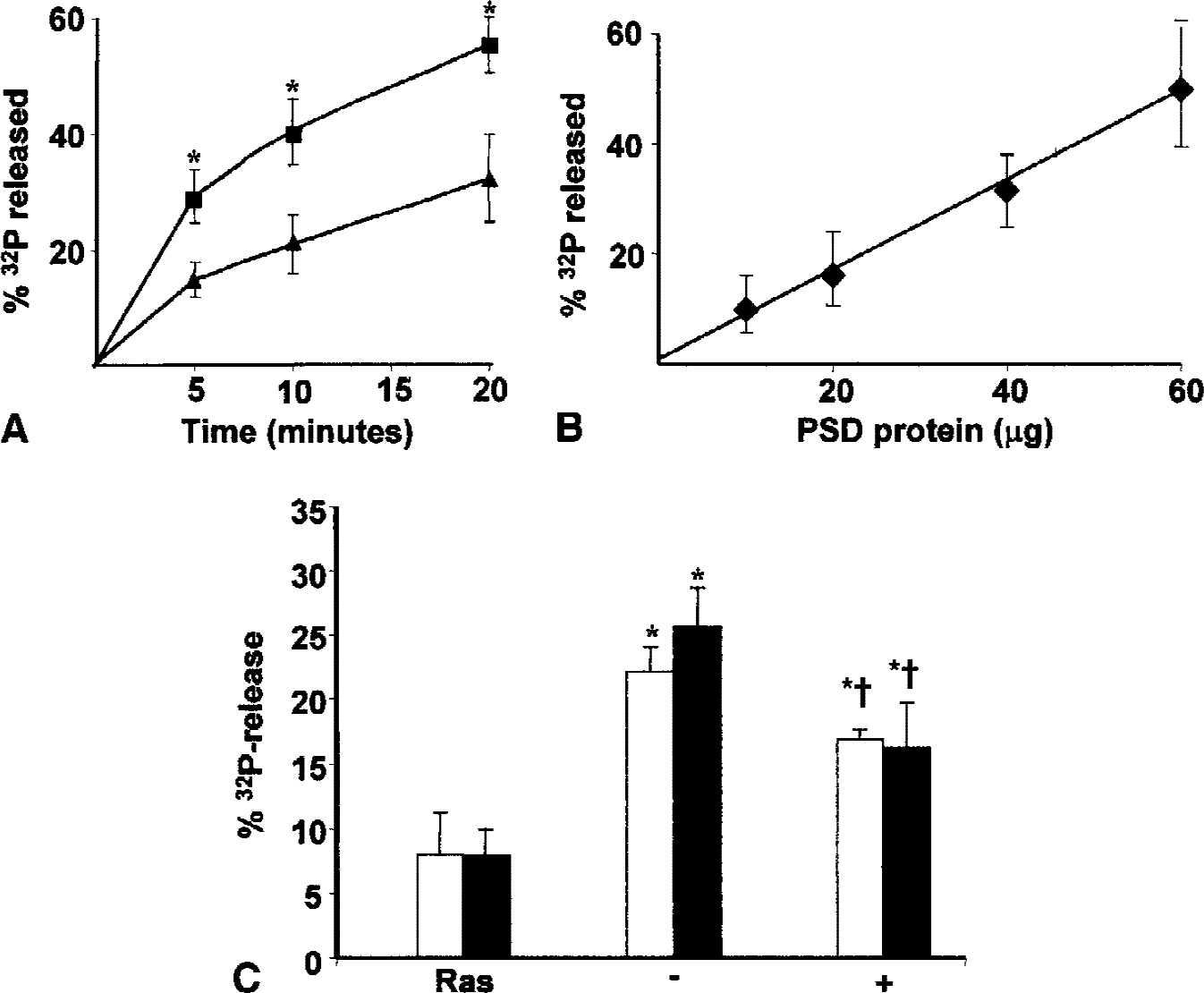
Effect of ischemia on postsynaptic density (PSD) RasGAP activity.
SynGAP binds to the SH2 domains of Src and Fyn
Tyrosine phosphorylation of SynGAP suggested that it may bind to SH2 domains and that this interaction may be altered as a result of the ischemia-induced increase in tyrosine phosphorylation. Because RasGAP interacts with the SH2 domain of Src (Schlesinger et al., 1999), the authors initially investigated the ability of SynGAP to bind to the SH2 domains of Src and Fyn. Postsynaptic densities prepared from naive animals were solubilized with SDS and the soluble proteins were reacted with the SH2 domains of Src and Fyn as described in Materials and Methods. Basally phosphorylated SynGAP bound to both SH2 domains (Fig. 5A). This interaction was prevented by prior treatment of PSDs with protein tyrosine phosphatase and by inclusion of the high affinity Src SH2-binding peptide, EPQpYEEIPIA (Liu et al., 1993), but not its nonphosphorylated analogue, in the reaction mixture (Fig. 5B). Incubation of PSDs with exogenous Src increased the phosphorylation of SynGAP on tyrosine residues and enhanced the binding of SynGAP to the SH2 domain of Src (Fig. 5C and 5D). In no case was binding to the GST moiety alone observed. These results demonstrated that binding of SynGAP to the Src SH2 domain was dependent on tyrosine phosphorylation of SynGAP, and further that binding required the presence of a free phosphotyrosine binding site in the SH2 domain.
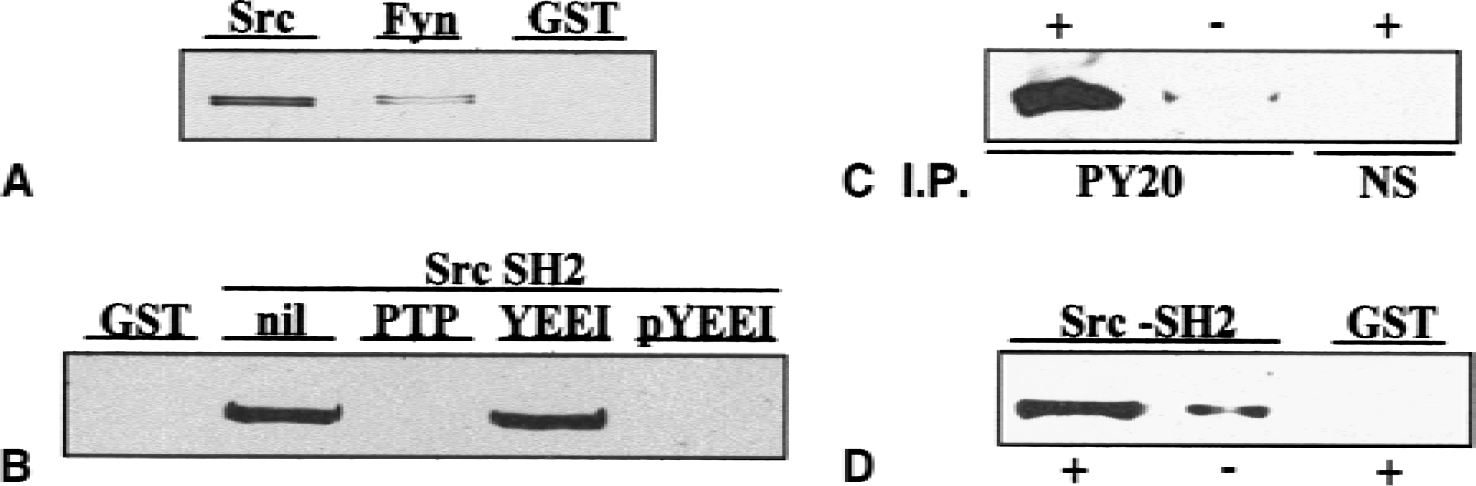
SynGAP binds to the SH2 domain of Src and Fyn.
The authors next determined if the ischemia-induced increase in tyrosine phosphorylation of SynGAP enhanced its ability to bind to the Src SH2 domain. The results presented in Fig. 6 show that after ischemia SynGAP binding to the Src SH2 domain increased by approximately 80%. The binding reaction with SynGAP from ischemic tissue was inhibited by the Src-binding peptide, consistent with its mediation by phosphotyrosine residues.
Effect of ischemia on the binding of SynGAP to the Src SH2 domain.
Transient global ischemia alters the association between SynGAP and PSD95
SynGAP is located at the PSD and binds to the PDZ domains of PSD-95 (Chen et al., 1998; Kim et al., 1998). The authors previous results have indicated that protein interactions involving PSD-95 are altered as a result of an ischemic challenge (Takagi et al., 2000). To determine if the association between SynGAP and PSD-95 was also affected by ischemia, coimmunoprecipitation experiments with membrane proteins from sham and ischemic samples were performed. Deoxycholate soluble proteins (prepared as in Materials and Methods) were immunoprecipitated with antibodies directed against PSD-95 and the resulting immunoprecipitates were analyzed for the presence of SynGAP and PSD-95. Less PSD-95 was present in the immunoprecipitate after ischemia, consistent with an ischemia-induced decrease in solubility of PSD-95 in DOC (Takagi et al., 2000). SynGAP coprecipitated with PSD-95 from both sham and ischemic samples, but the amount that coprecipitated was reduced after ischemia (Fig. 7). This result indicates that the stoichiometry of binding between SynGAP and PSD-95, at least as it occurs in the DOC-soluble fraction, is altered as a result of the ischemic challenge. When the initial immunoprecipitation was performed with SynGAP antibody, the authors did not detect PSD-95 in the immunoprecipitate, presumably because the SynGAP antibody used recognizes the C-terminal sequence of SynGAP, which is involved in binding to PSD-95 (Kim et al., 1998) and may be unavailable for immunoreaction in the complex.
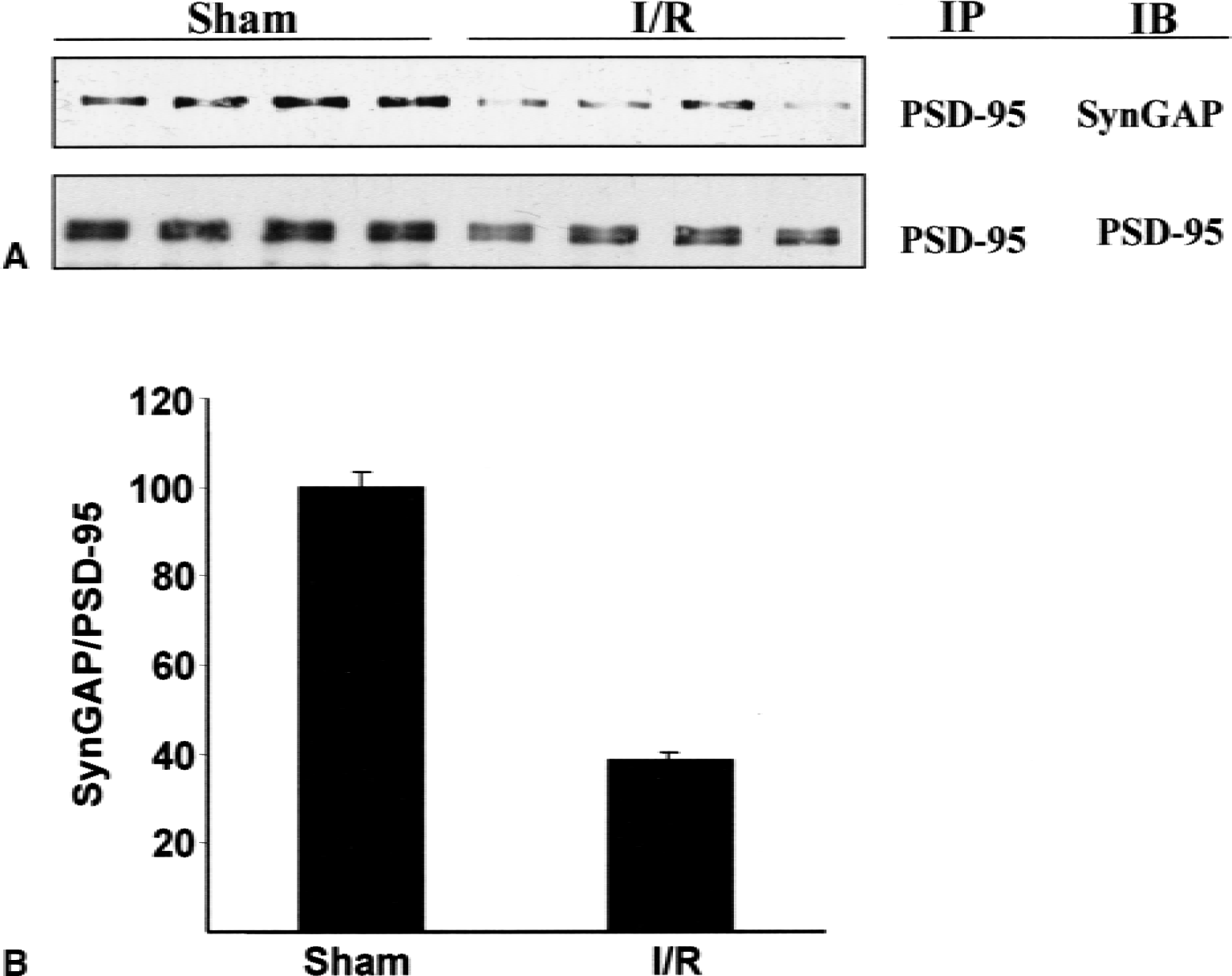
Coimmunoprecipitation of PSD-95 and SynGAP.
DISCUSSION
Postsynaptic densities consist of a complex array of signaling and anchoring proteins that are involved in regulation of synaptic function and in the transduction of synaptic signals to the postsynaptic cell (Husi et al., 2000; Kennedy, 1998; Ziff, 1997). Modulation of the composition or properties of the PSD may be expected to impact on the linkage between the synapse and signaling cascades to which it is connected. An ischemic challenge initiates a series of biochemical and molecular changes that result ultimately in the death of sensitive neurons. Because of its unique juxtaposition between receptors and downstream signaling events, it is likely that the PSD is critically involved in the early stages of ischemia-induced neuropathology. In general accord with this suggestion, ischemia has been found to result in a number of changes in the morphology and composition of PSDs (Cheung et al., 2000; Hu et al., 1998; Martone et al., 1999, 2000; Takagi et al., 2000). The authors have previously reported that ischemia induces an increase in the association of several tyrosine kinases with the PSD and a marked increase in the tyrosine phosphorylation of a number of PSD-associated proteins, including the NMDA receptor subunits NR2A and NR2B (Cheung et al., 2000). The results of the current study extend these observations and identify the synaptic-specific Ras-GTPase activating protein, SynGAP, as a substrate for ischemia-enhanced tyrosine phosphorylation.
The C-terminal domain of SynGAP contains several potential phosphorylation sites for CaMK II and tyrosine kinases. Whereas phosphorylation by the former enzyme inhibits PSD RasGAP activity (Chen et al., 1998; and current results), the authors were unable to detect an effect of ischemia-induced tyrosine phosphorylation on the activity of PSD RasGAP, either before or after phosphorylation in the presence of calcium and calmodulin. Although the simplest interpretation of this result is that tyrosine phosphorylation does not alter SynGAP activity, it is possible that any direct effects of tyrosine phosphorylation on SynGAP might have been masked within the context of the PSD, for example, by the presence of other GAPs in the PSD that reduce the contribution of SynGAP to the total PSD RasGAP activity, or by other ischemia-induced changes in the association of SynGAP with the PSD (see below) that might influence SynGAP activity. Analysis of the effects of tyrosine phosphorylation on the activity of purified SynGAP will be required to resolve this question. Ischemia results in the translocation of CaMK II to the PSD, and at least some of the translocated enzyme appears to be in the activated state (Shamloo et al., 2000). The absence of an effect of ischemia on the activity of PSD Ras GAP, however, suggests that ischemia does not result in phosphorylation of SynGAP by CaMK II.
The authors also investigated the effect of tyrosine phosphorylation of SynGAP on its interaction with the SH2 domains of Src and Fyn. Basally phosphorylated SynGAP bound to the SH2 domains of both Src and Fyn in a tyrosine phosphorylation-dependent manner. Binding to the SH2 domain of Src was increased approximately twofold after ischemia. Although both Src and SynGAP are components of the NMDA receptor complex (Husi et al., 2000; Yu et al., 1997), the authors have been unable to demonstrate an association between SynGAP and Src by coimmunoprecipitation experiments using DOC extracts of forebrain homogenates. This may be because of instability of the Src-SynGAP association under the detergent extraction conditions used, or because of insolubility of the complex. Similar to the current findings, previous studies have found that RasGAP binds to the SH2 domain of Src in a tyrosine phosphorylation-dependent fashion (Brott et al., 1991a, b ; Schlesinger et al., 1999). It was suggested that this interaction might serve a negative modulatory function, limiting the accessibility of Src to potential substrates (Schlesinger et al., 1999). Within the current context, the ischemia-induced increase in tyrosine phosphorylation of SynGAP could result in the transfer of Src between components of the NMDA receptor complex, for example, from NMDA receptor NR2 subunits (Takagi et al., 1999) to SynGAP, with a concomitant change in phosphorylation pattern. Alternatively, the binding of Src to SynGAP might contribute to the recruitment of Src to the PSD in the postischemic period (Cheung et al., 2000).
The decreased association between SynGAP and PSD-95 in DOC extracts of ischemic samples extends the authors' earlier report of ischemia-induced changes in protein interactions involving PSD-95 (Takagi et al., 2000), and is generally consistent with findings indicating that ischemia leads to remodeling of the postsynaptic density (Hu et al., 1998; Martone et al., 1999, 2000; Takagi et al., 2000). In addition to its function as an anchoring protein involved in the structural organization of the PSD, PSD-95 plays a critical role in the transmission of signals from the NMDA receptor to downstream signaling pathways (Migaud et al., 1998; Sattler et al., 1999). Changes in the interaction between SynGAP and PSD-95 may, accordingly, modulate the linkage between the NMDA receptor and the MAP kinase signaling cascade in the postischemic brain. Recently, a novel role for H-Ras in down-regulating tyrosine phosphorylation of the NMDA receptor was proposed (Manabe et al., 2000), suggesting a possible regulatory loop in which ischemia-induced changes in SynGAP functionality may feedback through Ras and regulate the activity of the NMDA receptor.
The molecular events that are responsible for initiating the chain of events that lead to neuronal cell death after an ischemic event remain to be defined. The results of the current study support the hypothesis that the rapid, ischemia-induced increases in tyrosine phosphorylation of PSD proteins play a critical role in these changes.
Footnotes
Acknowledgments:
The authors thank N. Bissoon, H. H. Cheung, and R. Moussa for helpful discussion throughout the course of this work.