
Abstract
Cerebral ischemia causes the presynaptic release of tissue-type plasminogen activator (tPA). The postsynaptic density (PSD) is a postsynaptic structure that provides a matrix where signaling transduction of excitatory synapses takes place. The postsynaptic density protein-95 (PSD-95) is the most abundant scaffolding protein in the postsynaptic density (PSD), where it modulates the postsynaptic response to the presynaptic release of glutamate by regulating the anchoring of glutamate receptors to the PSD. We found that tPA induces the local translation of PSD-95 mRNA and the subsequent recruitment of PSD-95 protein to the PSD, via plasminogen-independent activation of TrkB receptors. Our data show that PSD-95 is removed from the PSD during the early stages of cerebral ischemia, and that this effect is abrogated by either the release of neuronal tPA, or intravenous administration of recombinant tPA (rtPA). We report that the effect of tPA on PSD-95 is associated with inhibition of the phosphorylation and recruitment of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors to the PSD, known to amplify the effect of the excitotoxic injury, and that this is followed by TrkB-mediated protection of dendritic spines from the harmful effects of the hypoxic insult. These data reveal that tPA is a synaptic protector in the ischemic brain.
Keywords
Introduction
Approximately 30% of the energy in the central nervous system (CNS) is spent maintaining synaptic activity. Thus, even a brief interruption of the blood supply to the brain that does not cause cell death has a direct effect on the synapse. 1 In line with these observations, it has been calculated that one minute of cerebral ischemia is sufficient to destroy 1.9 million neurons and 14 billion synapses. 2 The translational importance of these observations is underscored by the fact that synaptic loss is associated with impairment in neurological function following an acute ischemic stroke. 3
Synapses are assembled by opposed pre- and postsynaptic specializations separated by a synaptic cleft. The active zone (AZ) is an electron lucid area of the presynaptic membrane where vesicle exocytosis takes place and neurotransmitters are released. 4 It has been shown that the presynaptic release of neurotransmitters is impaired during the earlier phases of the ischemic injury, 5 and that this is followed at later stages by loss of synaptic boutons and presynaptic projections. 6 The postsynaptic density (PSD) is an electrodense structure localized on the plasma membrane of the postsynaptic terminal of excitatory synapses, in close apposition to the AZ. It anchors signaling molecules and receptors to the postsynaptic membrane, 7 thus providing a matrix where signaling transduction of excitatory synapses and information storage take place. Cerebral ischemia has a profound effect on the PSD. However, despite a plethora of reports describing morphological and functional abnormalities in the PSD at different time-points after the onset of cerebral ischemia, 8 the effects of the very early stages of the ischemic injury on this structure have not been well characterized.
Membrane-associated guanylate kinases (MAGUKs) are a family of scaffolding proteins that play a pivotal role in synaptic transmission. 9 The PSD protein-95 is a member of the MAGUKs family, and the most abundant scaffolding protein in the PSD. 10 PSD-95 interacts directly or indirectly with a large number of proteins in the PSD, including the cytoskeleton, NMDA receptors (NMDARs), AMPA receptors (AMPARs), and adhesion molecules, 11 thus acting as a molecular organizer and regulator of synaptic function. The expression of PSD-95 varies according to the level of synaptic activity, 12 and its abundance is associated with the development and maturation of dendritic spines. 13 In line with these observations, the expression of PSD-95 decreases after the onset of cerebral ischemia, and this effect is followed by synaptic failure, loss of dendritic spines and neurological impairment. 14
Tissue-type plasminogen activator (tPA) is a serine proteinase that in the intravascular space catalyzes the conversion of plasminogen into plasmin. Based on its fibrinolytic properties, the intravenous administration of recombinant tPA (rtPA) is the standard of care for the treatment of acute ischemic stroke patients.15,16 However, in the brain, tPA has many other roles besides plasmin generation. 17 Hence, the release of neuronal tPA during synaptic activity 18 mediates the development of synaptic plasticity,19–21 and its secretion in response to a hypoxic or ischemic injury22–24 activates cell signaling pathways that promote cell survival and synaptic detection and adaptation to metabolic stress.23,25–27
The work presented here indicates that tPA induces the local translation of PSD-95 mRNA and the subsequent recruitment of PSD-95 protein to the PSD via plasmin-independent activation of TrkB receptors. Our in vivo studies with an animal model of cerebral ischemia show that the expression and recruitment of PSD-95 to the PSD decrease during the early stages of an ischemic injury, and in line with our in vitro observations, that this effect is prevented by either the release of neuronal tPA or treatment with rtPA, by a mechanism that does not require plasmin generation. Furthermore, our data indicate that the effect of tPA on PSD-95 in the ischemic brain does not contribute to the amplification of the excitotoxic injury because tPA also prevents the phosphorylation and subsequent insertion of GluR1-containing AMPA receptors into the PSD. Finally, we report that the effect of tPA on the PSD leads to preservation of the integrity of dendritic spines that have suffered an acute hypoxic injury. In summary, our work indicates that the release of neuronal tPA or treatment with rtPA protects the synapse in the ischemic brain.
Materials and methods
Animals and reagents
Experiments were approved by and conducted and reported following guidelines and regulations of the Institutional Animal Care & Use Committee of Emory University (IACUC), and ARRIVE (Animal Research: Reporting In Vivo Experiments). We used 8–12-weeks-old male male mice on a C57BL/6J background, wild-type (Wt), or genetically deficient on plasminogen (Plg−/−), or with a 20-fold increase in tPA expression in neurons (T4 mice). 28 Recombinant murine tPA (rtPA) and proteolytically inactive tPA were acquired from Molecular Innovations (Novi, MI). Other reagents were the RNeasy mini kit (Qiagen; Germantown, MD), high capacity cDNA reverse transcription kit and phalloidin (ThermoFisher Scientific; Grand Island, NY), PCR primers for PSD-95 (IDT; Coralville, Iowa), antibodies against synaptophysin (SYP) and syntaxin I (STX; EMD Millipore; Billerica, MA), β-actin (Sigma-Aldrich; St. Louis, MO), and the PSD protein-95, bassoon, and GluR1 phosphorylated at S831 (Abcam; Cambridge, MA); Cresyl violet, cycloheximide, triphenyltetrazolium chloride (TTC) staining, and the Trk-B inhibitor ANA-12 (Sigma-Aldrich; St. Louis, MO), and propidium iodide and the Hoechst stain (Invitrogen; Carlsbad, CA).
Animal model of cerebral ischemia
To induce sublethal ischemia, the carotid arteries of Wt, T4 and Plg−/− mice were exposed under anesthesia with isofluorane and either clipped during 5 min (bilateral common carotid artery occlusion (BCCAO)), or kept patent (sham operation), as described elsewhere. 23 To produce lethal ischemia, Wt and Plg−/− mice were subjected to transient occlusion of the middle cerebral artery (tMCAO) as described, 27 with a silicone-coated nylon monofilament (6–0, Ethicon; Issy Les Moulineaux, France) introduced during 30 min through the ECA and advanced to the origin of the MCA. Five minutes after BCCAO or 30 min after tMCAO, the suture was removed and animals were treated with 0.9 mg/kg of rtPA or a comparable volume of saline solution (SS) intravenously infused over 60 min with a micro infusion pump. Twenty-four hours after tMCAO, the volume of the ischemic lesion was measured in TTC-stained sections as described. 29 In both models (BCCAO and tMCAO), cerebral perfusion (CP) was monitored with a laser Doppler (Perimed Inc., North Royalton, OH) over either the forebrain at bregma: 0.02 mm and lateral: 2 mm 30 (for the BCCAO model), or the distribution of the MCA (for the tMCAO model), and only animals with a > 80% decrease in CP after occlusion and complete recovery after suture removal were included in this study.
Neuronal cultures
Cerebral cortical neurons were cultured from E16 to 18 Wt mice as described elsewhere. 23 Briefly, the cerebral cortex was dissected, transferred into Hanks' balanced salt solution containing 100 units/ml penicillin, 100 μg/ml streptomycin and 10 mM HEPES, and incubated in trypsin containing 0.02% DNase at 37 ℃ for 15 min. Tissue was triturated and the supernatant was re-suspended in GS21-supplemented neurobasal medium containing 2 mM l-glutamine, and plated onto 0.1 mg/ml poly-l-lysine-coated wells. Experiments were performed at DIV-16.
Determination of neuronal death
The brains of Wt mice (n = 4) were harvested following 5 min of BCCAO, cut onto 20 µm slices and stained with 0.1% Cresyl violet following manufacturer's instructions. Pictures from the cerebral cortex were taken at bregma: + 1.2, + 0.5, 0.0, −0.5 and −2.0 mm 30 with a 40 × lens, and the number of dead cells per field was visually identified and counted with the ImageJ analysis system. DIV 14 Wt neurons were exposed to 55 min of oxygen and glucose deprivation (OGD) in a HypOxystation H35 chamber (HypOxygen; Frederick, MD) followed by recovery under physiological conditions in the presence of 5 nM of tPA or a comparable volume of vehicle (control). Twenty-four hours later, the uptake of propidium iodide was quantified in Hoechst-positive cells following manufacturer's instructions. Pictures were taken with a 40 × lens and quantifications were performed with the Cellsens Dimension software (Olympus). Values were expressed as percentage of live cells in the control group.
Isolation of PSD extracts and synaptic fractions
Wt cerebral cortical neurons were treated as described below. The forebrain of T4 mice and their Wt littermate controls were extracted after 5 min of BCCAO and treatment with either SS or rtPA. Cells and tissue were homogenized and centrifuged at 2000g for 5 min, and the supernatant (S1) was centrifuged at 32,000g for 10 min. To prepare PSD-enriched fractions, the pellet (P2) containing synaptoneurosomes was centrifuged at 275,000g for 1 h, and the pellets (P3) were centrifuged again at 275,000g for 1 h. To isolate synaptic fractions, synaptoneurosomes were osmotically lysed in 2 ml of 5 mM Tris pH 7.4 and their membranes centrifuged at 295,000 × g for 1 h. Membrane pellets were resuspended, layered on top of a 0.3/1.2 M linear sucrose gradient and centrifuged at 93,000 × g for 2 h. The gradient was then fractionated into 14 × 160 µl fractions and pelleted at 625,000 × g for 1.5 h. Supernatants were discarded and pellets were dissolved in 50 µl of 0.5% SDS.
Polymerase chain reaction
Neurons were incubated during 5 min with 5 nM of murine tPA. Total RNA was isolated with the RNeasy Mini kit and equal amounts of total RNA were used for cDNA synthesis with high-capacity cDNA reverse transcription kits. PSD-95 primers were 5′-TCTGTGCGAGAGGTAGCAGA-3′ (forward), and 5′-AAGCACTCCGTGAACTCCTG-3′ (reverse) with an annealing temperature of 60 ℃. As internal control, we used the ribosomal protein l27 with primers 5′-TCGAGATGGGCAAGTTCATGAAACC-3′ (forward) and 5′-GACCAAAACAAAATATACCTAAAAC-3′ (reverse).
Immunoblotting
To study the effect of tPA on PSD-95 expression, Wt cerebral cortical neurons were incubated 0–15 min with 5 nM of proteolytically active or inactive tPA, alone or in the presence of 350 µM of cycloheximide; and the forebrains of Wt and Plg−/− mice were harvested after 5 min of BCCAO and treatment with rtPA or SS. To study the synaptic recruitment of PSD-95 and GluR1-containing AMPA, PSD fractions were prepared as described from Wt neurons incubated 0–5 min with 5 nM of tPA, and from the forebrain of Wt mice harvested after 5 min of BCCAO and treatment with SS or rtPA. To study the effect of tPA on synaptic integrity, synaptic fractions were prepared from the forebrain of Wt and T4 mice harvested after 5 min of BCCAO and treatment with SS or rtPA. Cells and brains were homogenized and 2.5 μg of proteins were loaded per sample, separated by 4–20% linear gradient polyacrylamide gel, transferred to a PVDF membrane and incubated with antibodies against PSD-95 (1:1000), pGluR1 (1:1500), SYP (1:1000), STX (1:5000) and β-actin (1:5000). Densitometry analysis was performed in each band using the UN-SCAN-IT gel software. Each experiment was repeated at least –three to four times as indicated in the figure legend.
Immunochemistry
To study the effect of tPA on PSD-95 expression, Wt neurons were incubated during 5 min with 5 nM of tPA. To study the effect of tPA on dendritic spines, neurons were incubated during 5 min under normoxic or OGD conditions with 5 nM of tPA, alone or in combination with 30 µM of ANA-12. Cells were fixed, washed in TBS, and incubated overnight on a solution containing anti-PSD-95 antibodies (1:1000). Pictures were taken with an Olympus microscope BX51 with a 40 × lens. Quantification and determination of the size of PSD-95-positive puncta were performed manually with ImageJ. Heat maps were made with Photoshop from those pictures used for quantifications. Each observation was repeated in cells from three different cultures. The number and length of dendritic protrusions were determined with ImageJ in pictures taken with a 60 × lens. Each observation was repeated with cells from three different cultures. To study the synaptic effect of treatment with rtPA following the induction of lethal ischemia, Wt mice underwent 30 min of tMCAO followed by intravenous treatment with 0.9 mg/kg of rtPA or a comparable volume of SS. Twenty-four hours later, brains were harvested, fixed, cut onto 20 μm slices, incubated during 2 h in 0.5% triton/TBS and kept overnight in the presence of antibodies against PSD-95 (1:100) and bassoon (1:1000), followed next day by the addition of secondary anti-mouse Alexa Fluor 488 (1:500) and anti-rabbit Alexa 594 (1:500) antibodies for 1 h. Images were taken with a 60 × lens at bregma: 0.02 mm, lateral: 2 mm and ventral: 1 mm 30 using a Fluoview FV10i confocal laser-scanning microscope (Olympus). The number of PSD-95/bassoon-positive puncta was quantified with the plugin puncta analyzer of ImageJ.
Statistical analysis
Statistical analysis was performed with two-tailed t test and one-way ANOVA, with Holm–Sidak corrections. p-values of < 0.05 were deemed as significant.
Results
Effect of tPA on PSD-95 expression
In earlier studies, we found that tPA increases the thickness of the PSD.
31
Because PSD-95 is the most abundant PSD scaffolding protein,
10
here we used immunoblotting and PCR to investigate its expression in Wt cerebral cortical neurons incubated 0–15 min with 5 nM of tPA. We found that tPA increases the expression of PSD-95 protein (Figure 1(a) and (b)), by a mechanism that does not require plasmin generation (Figure 1(c) and (d)). In contrast, although we found PSD-95 mRNA in our preparations, we failed to detect an increase in its abundance following tPA treatment (data not shown). Because neuronal activity induces the local translation of PSD-95 mRNA,
32
we then performed similar observations in neurons treated with tPA in the presence of cycloheximide. Our data show that the effect of tPA on PSD-95 expression is abrogated by inhibition of protein synthesis (Figure 1(e) and (f)). Together, these data indicate that tPA induces the local translation of PSD-95 mRNA in cerebral cortical neurons.
tPA induces the expression of PSD-95 in cerebral cortical neurons. (a) and (b) Representative Western blot analysis (a) and quantification of the mean intensity of the band (b) of PSD-95 expression in Wt cerebral cortical neurons incubated 0–15 min with 5 nM of tPA. n = 4 observations per time-point, lines denote SEM. (c) and (d) Representative Western blot analysis (c) and quantification of the mean intensity of the band (d) of PSD-95 expression in Wt cerebral cortical neurons incubated 5 min with 5 nM of proteolytically inactive tPA (itPA) or vehicle (control). n = 4 observations per time-point; lines denote SEM. (e) and (f) Representative Western blot analysis (e) and quantification of the mean intensity of the band (f) of PSD-95 expression in Wt cerebral cortical neurons incubated 5 min with 5 nM of tPA alone or in the presence of 350 µM of cycloheximide. Lines denote SEM. n = 4 observations per experimental condition.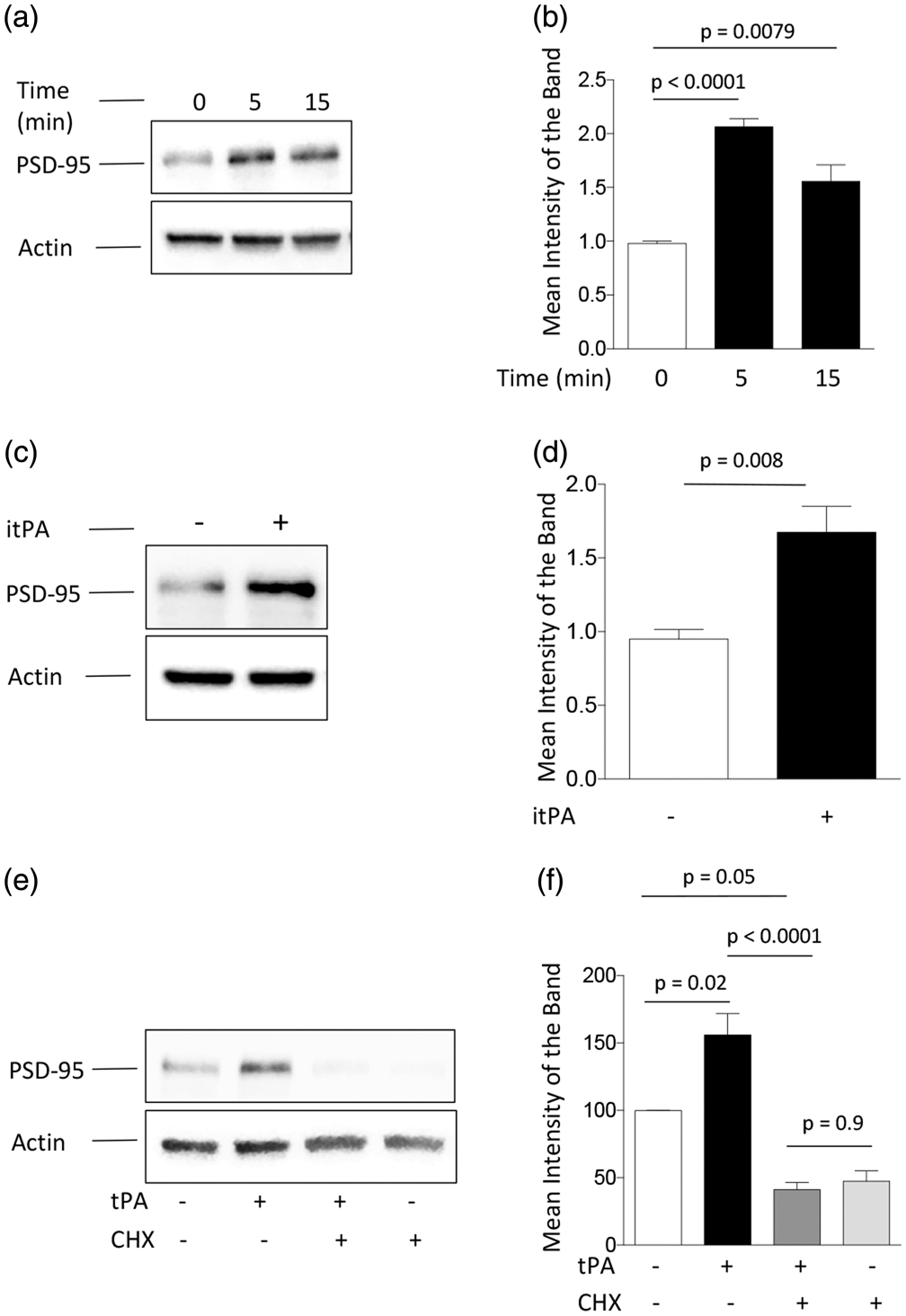
tPA induces TrkB-mediated recruitment of PSD-95 to the PSD
To further characterize these observations, we quantified the number of PSD-95-positive puncta in dendrites from Wt neurons incubated during 5 min with 5 nM of tPA or a comparable volume of vehicle (control). We found that tPA does not increase the number of PSD-95-positive puncta (data not shown) but instead that it enlarges their size and intensity of staining (Figure 2(a) panels a to d and Figure 2(b); n = 55 neurons examined per condition; one-way ANOVA). These data suggest that tPA induces the recruitment of PSD-95 to the PSD. This hypothesis was corroborated by our observation of an increase in the abundance of PSD-95 in PSD extracts prepared from Wt cerebral cortical neurons following 5 min of incubation with 5 nM of tPA (Figure 2(c) and (d)). Because TrkB signaling mediates the synaptic recruitment of PSD-95,33,34 we then measured the intensity of staining of PSD-95-positive puncta in Wt neurons treated with tPA in combination with 30 μM of the TrkB inhibitor ANA-12. Furthermore, we studied PSD-95 expression in PSD extracts prepared from cerebral cortical neurons treated with tPA, alone or in the presence of ANA-12. Our data indicate that TrkB antagonism abrogates tPA-induced recruitment of PSD-95 to the PSD (Figure 2(a) and (b) and (e) and (f); n = 55 neurons examined per group in (a) and (b), and four observations per condition in (e) and (f); one-way ANOVA).
tPA induces TrkB-mediated recruitment of PSD-95 to the post-synaptic density. (a) Panels a, c, e and g correspond to representative micrographs (40 × lens) of PSD-95 expression (green) in dendrites from Wt cerebral cortical neurons treated during 5 min with vehicle (control; a), or 5 nM of tPA, alone (c) or in the presence of 30 μM of the TrkB antagonist ANA-12 (e), or with ANA-12 alone (g). Panels b, d, f and h are heat maps of PSD-95 expression from the corresponding micrographs in panels a, c, e, & g. (b) Mean intensity of PSD-95 immunoreactivity per pixel in dendrites of Wt cerebral cortical neurons exposed to the experimental conditions described in (a). n = 55 neurons per experimental group from three different cultures. (c) to (f) Representative Western blot analysis (c) and (e) and quantification of the mean intensity of the band (d) and (f)) of PSD-95 expression in PSD extracts prepared from Wt cerebral cortical neurons incubated during 5 min with 5 nM of tPA, alone (c) and (d)) or in the presence of ANA-12 (e) and (f)). n = 4 observations per experimental group.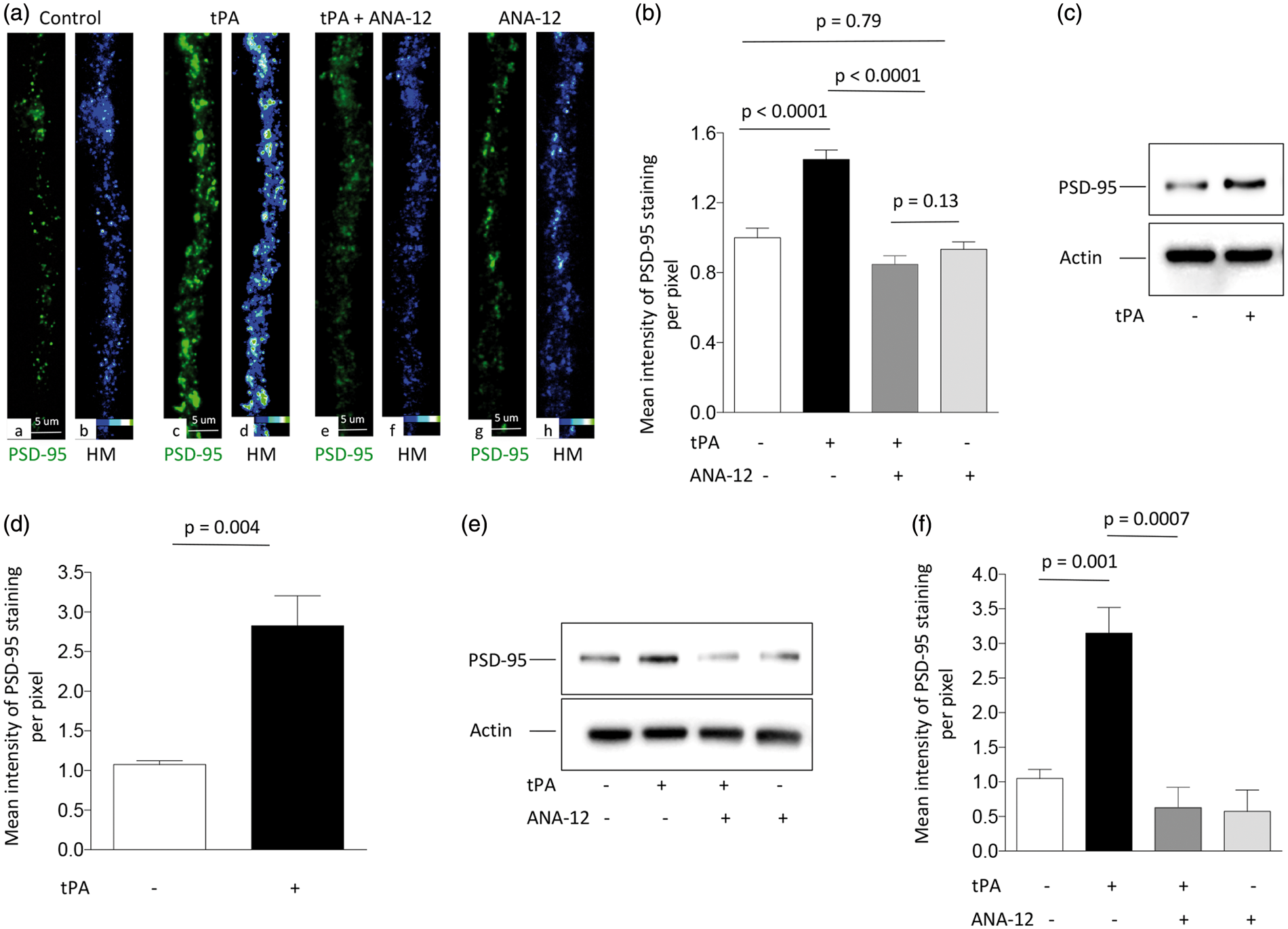
The release of neuronal tPA protects the PSD from the harmful effects of cerebral ischemia
To study the in vivo significance of our findings, we decided to investigate the effect of a brief episode of cerebral ischemia, that does not produce cell death, on the integrity of the pre- and postsynaptic compartments. First, we looked for cell death in Cresyl violet-stained sections from the forebrain of Wt mice subjected to 5 min of BCCAO. We found that 5 min of BCCAO do not induce neuronal death (n = 4; data not shown). Then, synaptoneurosomes prepared from the forebrain of Wt mice exposed to 5 min of sham-operation or BCCAO were fractioned and layered on top of a 0.3/1.2 M linear sucrose gradient as described in the ‘Materials and methods’ section, and immunoblotted with antibodies against synaptophysin (SYP; detects synaptic vesicles), syntaxin I (STX; delineates the presynaptic membrane), and PSD-95. As previously described,35,36 in these preparations, intact synapses are found in those fractions immunoreactive to all three markers: SYP, STX and PSD-95. We found that the mean number of synaptic fractions containing intact synapses decreases from 4.72 ± 0.35 in sham-operated animals to 2.72 ± 0.42 after BCCAO (n = 5 observations per condition; p = 0.007, t-test Figure 3(a) and (b)). Importantly, although we noted a slight decline in the immunoreactivity to SYP and STX after BCCAO, that difference did not reach statistical significance when compared to sham-operated animals (Figure 3(c) and (d)). In contrast, we detected a statistically significant decrease in PSD-95 expression following BCCAO (Figure 3(e); p = 0.003, t-test).
The release of neuronal tPA protects the synapse in the ischemic brain. (a–e) Synaptic fractions were isolated from the forebrain of Wt mice following 5 min of sham-operation (control) or bilateral common carotid artery occlusion (BCCAO), and immunoblotted with antibodies against synaptophysin (SYP), syntaxin I (STX) and PSD-95. (b) Mean number of synaptic fractions containing intact synapses per experimental group. (c–d) Mean intensity of the band for SYP (C), STX (D), and PSD-95 (E) expression in the experimental groups described in (a). Lines denote SEM. Each blot was repeated five times. (f–j) Representative Western blot analysis (f) for SYP, STX and PSD-95 expression in synaptic fractions isolated from the forebrain of T4 mice following 5 min of sham-operation (control) or BCCAO. (g) Mean number of fractions containing intact synapses in both experimental groups. (h–j) Mean intensity of the band for PSD-95 (H), SYP (I), and STX (J) expression per experimental group. Lines denote SEM. Each blot was repeated five times.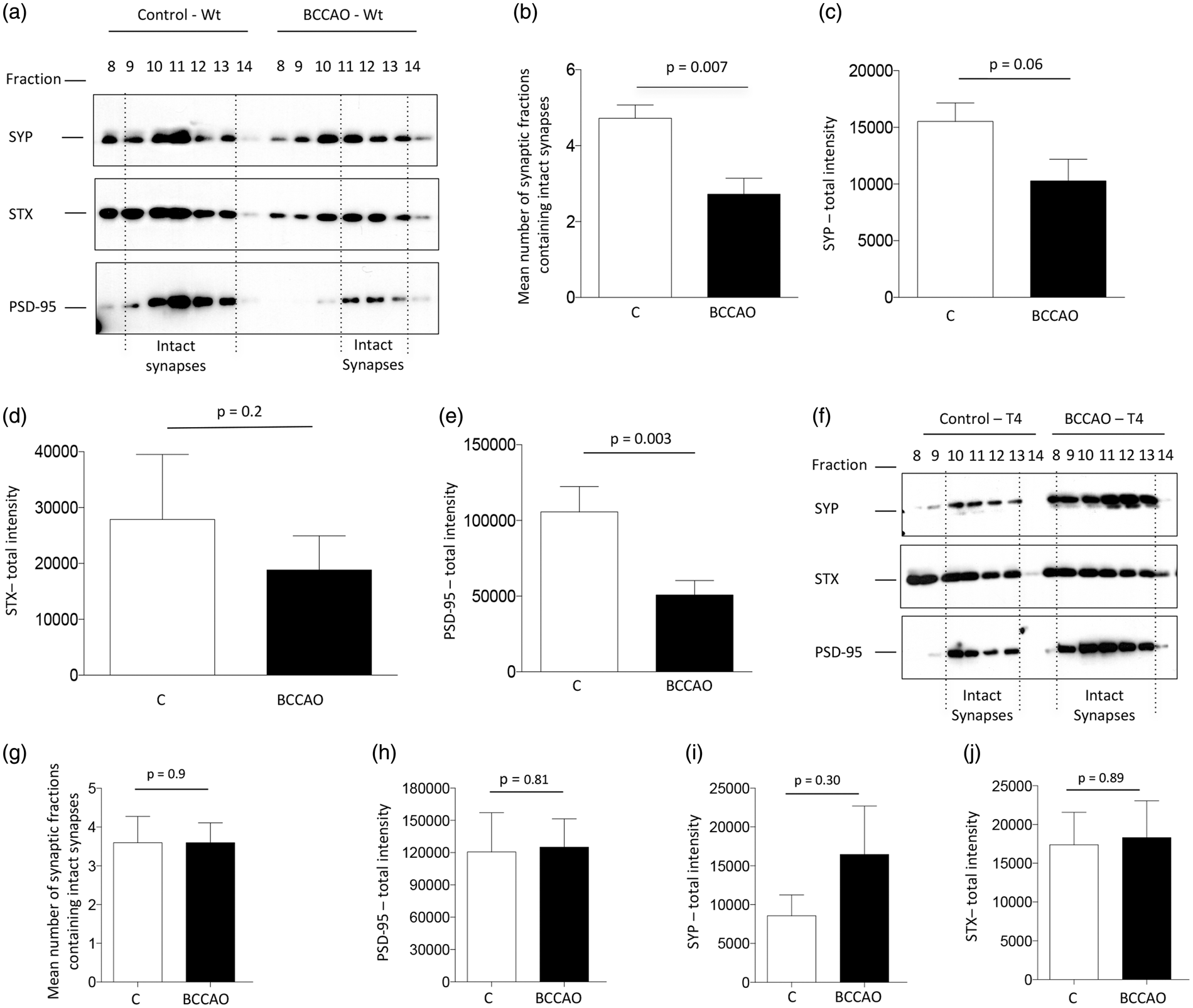
Because a short episode of cerebral ischemia induces the release of neuronal tPA without causing cell death,22,23 we then performed similar observations in T4 mice, with a 20-fold increase in the expression of neuronal tPA. 28 Our data show that in contrast with Wt animals, the number of synaptic fractions containing intact synapses does not decrease in T4 mice following BCCAO (n = 5; Figure 3(f) and (g)). More importantly, and in contrast with our findings with Wt mice, we not only failed to detect a decrease in PSD-95 expression (Figure 3(h)), but instead we found an increase in its abundance in some animals, that however did not reach statistical significance (Figure 3(i) and (j)). These data indicate that the release of neuronal tPA prevents the harmful effects of the ischemic injury on PSD-95 expression and synaptic integrity.
Treatment with rtPA protects the synapse in the ischemic brain
Because following its intravenous administration rtPA crosses through the blood–brain barrier and reaches the ischemic tissue,37–39 we then decided to investigate whether the intravenous administration of rtPA, at doses used to treat acute ischemic stroke patients, has an effect on the expression of PSD-95 in the ischemic tissue. First, we studied the expression of PSD-95 in brain extracts from Wt and Plg−/− mice following 5 min of BCCAO or sham-operation and treatment with either 0.9 mg/kg/IV of rtPA or a comparable volume of SS. Our data indicate that cerebral ischemia causes a rapid decrease in the expression of PSD-95 and that this effect is abrogated by treatment with rtPA (Figure 4(a) and (b)), via a mechanism that does not require plasmin generation (Figure 4(c) and (d)). To determine whether treatment with rtPA also has an effect on the recruitment of PSD-95 to the PSD in vivo, we performed similar observations with PSD extracts prepared from the ischemic tissue of Wt mice subjected to 5 min of either sham operation or BCCAO, followed by intravenous treatment with SS or rtPA. We found that the expression of PSD-95 in the PSD decreases after BCCAO, and that this effect is prevented by treatment with rtPA (Figure 4(e) and (f)). To further characterize these observations, we studied the expression of SYP, STX and PSD-95 in synaptic fractions isolated from Wt mice following 5 min of sham operation, or BCCAO and treatment with rtPA. Our results show that in contrast with untreated Wt mice (Figure 3(a) and (b)), the number of fractions containing intact synapses and the synaptic expression of PSD-95, SYP and STX remain unchanged in Wt animals treated with rtPA after BCCAO (Figure 4(g) to (k); n = 5 observations per experimental group; t-test). These data indicate that treatment with rtPA protects the synapse from the harmful effects of the ischemic injury.
Effect of treatment with rtPA on PSD-95 expression in the ischemic brain. (a–d) Representative immunoblottings (a) and (c) and quantification of the mean intensity of the band (b) and (d) of PSD-95 expression in brain extracts from Wt (a) and (b) and Plg−/− (c) and (d) mice following 5 min of sham-operation or BCCAO, and intravenous treatment with either PBS or 0.9 mg/kg of rtPA. n = 5 observations per condition. Lines depict SEM. (e–f) Representative Western blot analysis of PSD-95 expression in PSD extracts prepared from the forebrain of Wt mice following 5 min of sham operation or BCCAO, and intravenous treatment with either PBS or rtPA. Lines denote SEM. n = 4 observations per experimental group. (g–k) Representative immunoblotting (g) of SYP, STX and PSD-95 expression in synaptic fractions isolated from the forebrain of Wt mice following 5 min of sham-operation (control) or BCCAO, and treatment with rtPA. (h) corresponds to the mean number of fractions containing intact synapses. (i–k) Mean intensity of the band for PSD-95 (I), SYP (J), and STX (K) expression per experimental condition. Lines denote SEM. Each blot was repeated five times.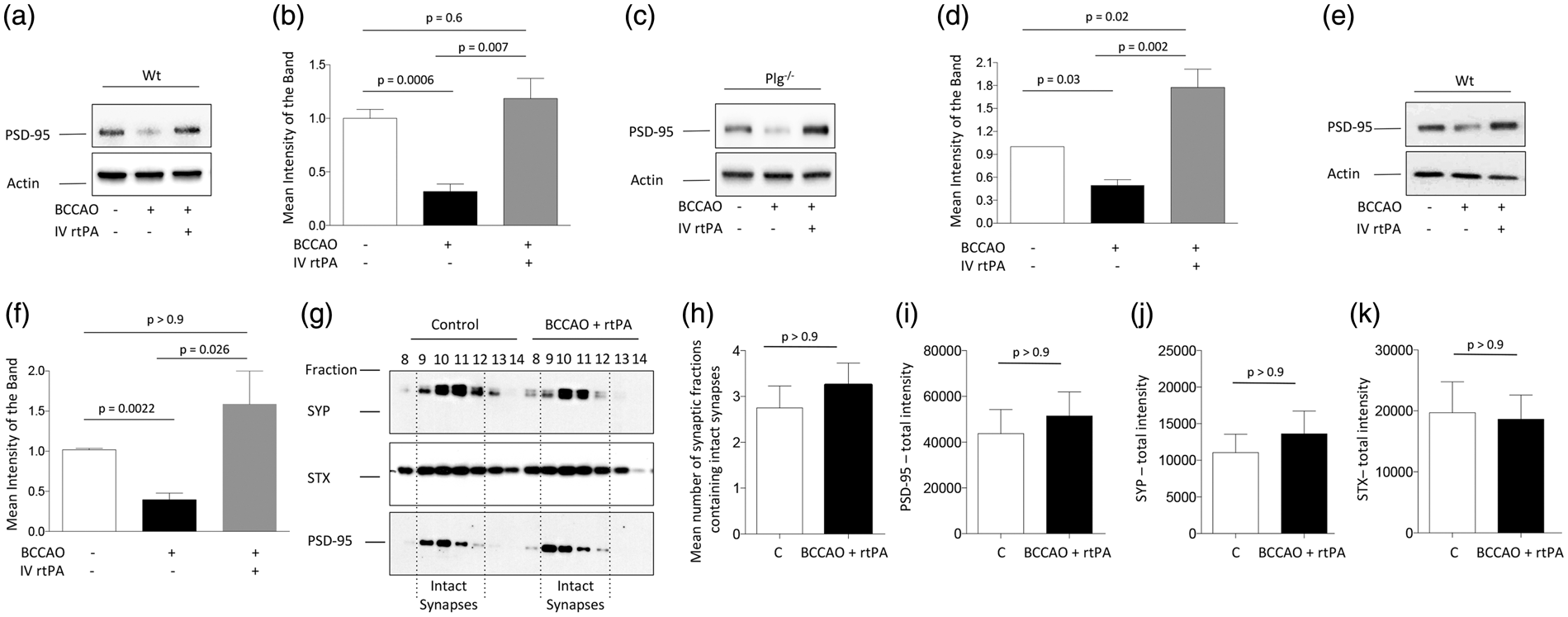
TPA has a protective effect on dendritic spines
It has been recognized that the synaptic recruitment of PSD-95 leads to the formation of dendritic spines.
40
Because our data indicate that tPA induces the expression and synaptic recruitment of PSD-95 via TrkB activation (Figure 2), we then decided to quantify the number of dendritic protrusions in Wt neurons treated during 5 min with 5 nM of tPA, alone or in the presence of the TrkB inhibitor ANA-12. Our data show that the number of dendritic protrusions/50 µm increases from 10.7 ± 0.5 in cells incubated with vehicle (control) to 16.53 ± 0.4 following tPA-treatment (n = 30 cells examined per experimental group; p < 0.0001, one-way ANOVA), and that this effect is abrogated by TrkB antagonism (10.93 ± 0.5; p < 0.0001 compared to cells incubated with tPA without ANA-12; one-way ANOVA; Figure 5(a) and (b)).
Treatment with rtPA protects the post-synaptic terminal via TrkB receptor activation. (a) and (b) Representative micrographs (a) and mean number of dendritic protrusions/50 µm (b) from Wt neurons incubated during 5 min with either PBS (a), or 5 nM of tPA, alone (b) or in combination with 30 μM of ANA-12 (c). n = 30 neurons per condition from three different cultures. Lines denote SEM. Arrows in a – c denote dendritic spines and filopodia. (c) and (d) Representative micrographs (c) and mean number of dendritic protrusions/50 µm (d) in Wt neurons maintained under normoxic conditions (a), or exposed to oxygen and glucose deprivation (OGD) conditions, alone (b) or in the presence of 5 nM of tPA and either PBS (c), or ANA-12 (d). Arrows in (c) (panels a and c) denote dendritic filopodia. n = 50 neurons per condition from three different cultures. (e) and (f) Mean length of dendritic protrusions (e) and cumulative frequency of spine length (f) in Wt neurons exposed to OGD conditions and treatment with rtPA and ANA-12 as described in (c) and (d). n = 120 neurons per experimental condition.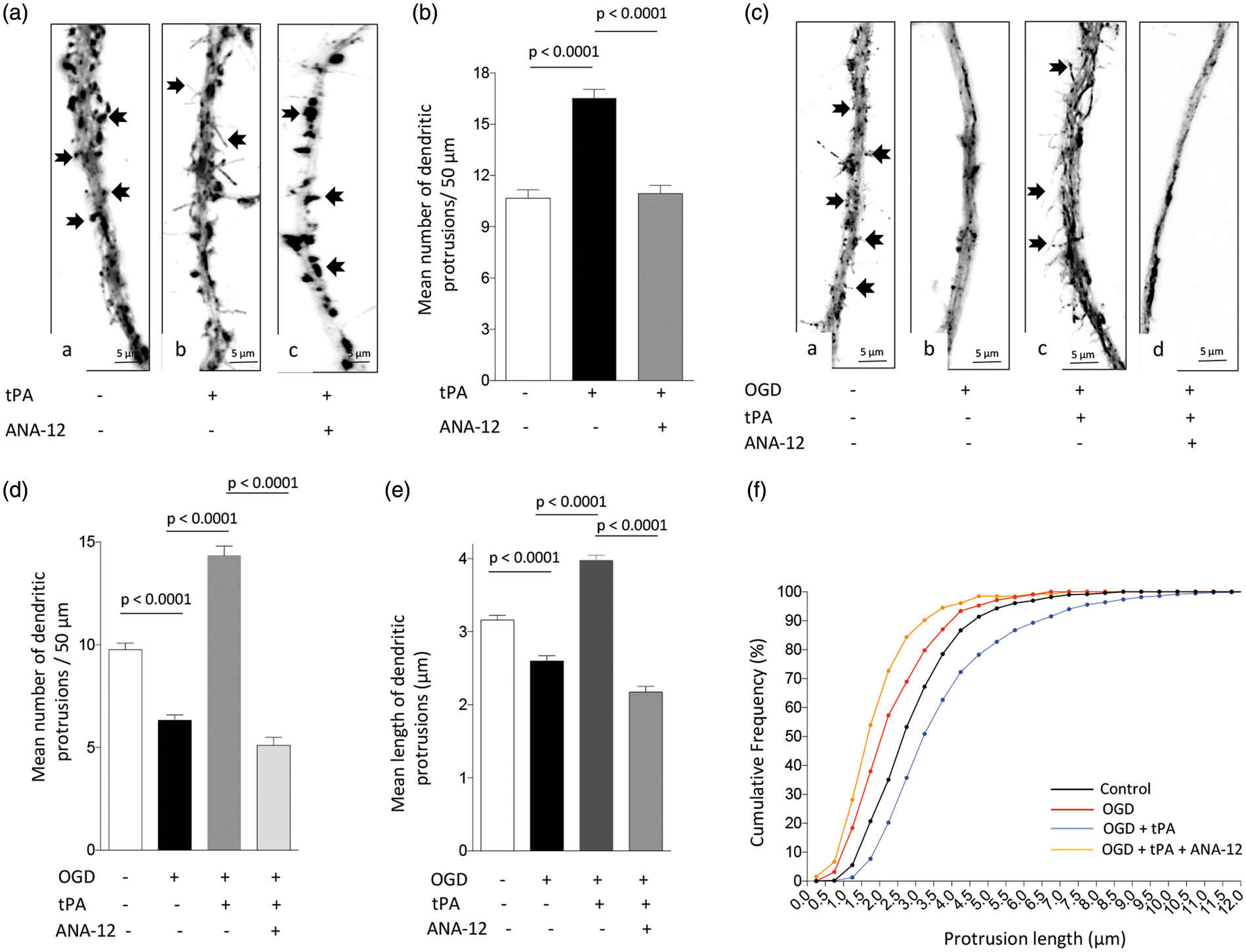
Because hypoxia causes a rapid decrease in dendritic spine density, 41 we then decided to investigate whether tPA has an effect on dendritic spines from neurons exposed to a hypoxic injury. We quantified the number of dendritic protrusions in neurons exposed to 5 min of OGD, in the presence of vehicle (control), or 5 nM of tPA, or a combination of 5 nM of tPA and 30 μM of ANA-12. We found that the number of dendritic protrusions/50 µm decreases from 9.7 ± 0.31 in neurons maintained under normoxic conditions to 6.32 ± 0.26 in neurons exposed to OGD (n = 50 neurons per experimental group, p < 0.0001; one-way ANOVA). Remarkably, in the presence of tPA, the number of protrusions increased to 14.34 ± 0.5 (Figure 5(c) and (d); p < 0.0001 compared to untreated cells, one-way ANOVA), and this effect was abrogated by TrkB antagonism (5.1 ± 0.4; p < 0.0001 compared to neurons treated with tPA alone, one-way ANOVA). Furthermore, a closer examination of these protrusions revealed that OGD not only decreases their number but also their length from 3.1 ± 0.06 µm to 2.3 ± 0.07 µm (n = 120 neurons examined per group; p < 0.0001, one-way ANOVA). Remarkably, treatment with tPA not only increased their number but also their length to 3.97 ± 0.08 µm (n = 120 neurons; p < 0.0001 compared to untreated cells, one-way ANOVA), and this effect was also blocked by TrkB antagonism (2.17 ± 0.06 µm; p < 0.0001 compared to cells treated with tPA alone, one-way ANOVA; Figure 5(e) and (f)).
Effect of treatment with rtPA on the synaptic recruitment of AMPA receptors
It is known that phosphorylation of GluR1 subunits of the AMPA receptor at S831 (pGluR1) and their subsequent PSD-95-mediated recruitment to the PSD
11
are pivotal events for the development of an excitotoxic response to the presynaptic release of glutamate.42,43 These observations are in line with in vivo experimental work indicating that treatment with PSD-95 inhibitors ameliorates the excitotoxic injury in the ischemic brain.
44
Thus, based on these considerations, it is plausible to postulate that by inducting the expression and synaptic recruitment of PSD-95, rtPA may have a harmful effect in the ischemic brain. To investigate this possibility, we decided to study the expression of pGluR1 in PSD extracts prepared from Wt mice after 5 min of sham operation or BCCAO followed by treatment with rtPA or SS. Our results show that cerebral ischemia induces the phosphorylation and synaptic recruitment of pGluR1, and that this effect is abrogated by treatment with rtPA (Figure 6(a) and (b)). These data indicate that the effect of rtPA on the expression of PSD-95 in the PSD does not potentiate the excitotoxic response to the presynaptic release of glutamate, but instead contributes to block it by preventing the phosphorylation and synaptic insertion of GluR1-containing AMPA receptors.
Effect of treatment with rtPA on the phosphorylation and synaptic recruitment of GluR1-containing AMPA receptors. (a) and (b) Representative Western blot analysis (a) and quantification of the mean intensity of the band (b) of the expression of GluR1 subunits phosphorylated at S831 (pGluR1) in post-synaptic density extracts prepared from the forebrain of Wt mice following 5 min of sham-operation or BCCAO and treatment with saline solution or rtPA. Lines in (b) denote SEM. n = 4 per experimental condition.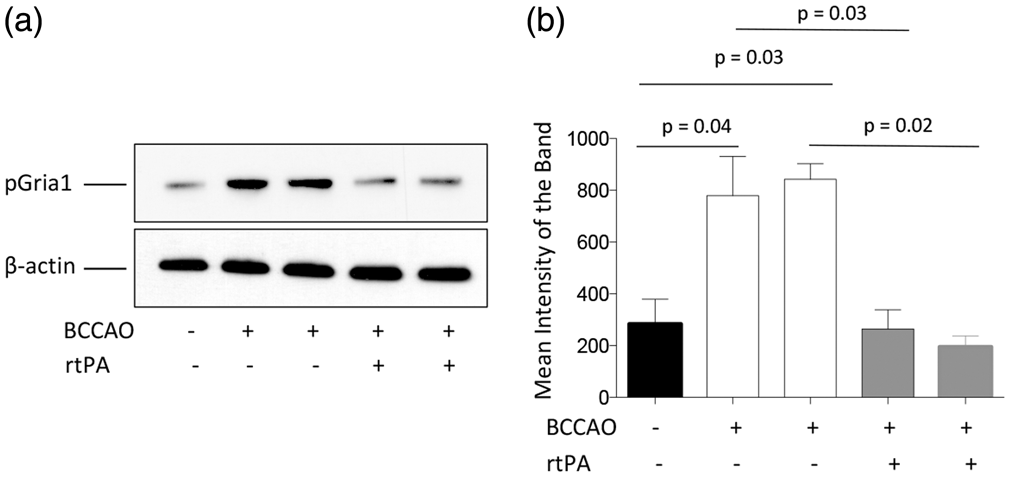
Treatment with rtPA protects the synapse and promotes neuronal survival in the ischemic brain
To determine the synaptic effect of treatment with rtPA following the induction of lethal ischemia, we quantified the number of intact synaptic contacts in the ischemic brain and in a comparable area of the contralateral non-ischemic hemisphere of Wt mice 24 h after tMCAO and intravenous treatment with 0.9 mg/kg of rtPA or a comparable volume of PBS. Our data indicate that the number of synaptic contacts in PBS-treated animals decreases from 313.8 ± 39.83/2100 μm2 in the non-ischemic hemisphere to 92 ± 16.9 in the ischemic tissue (p = 0.02), and that this effect is significantly attenuated by treatment with rtPA (362.3 ± 42.4 synaptic contacts/2100 μm2 in the non-ischemic hemisphere and 237.9 ± 30.60 in the ischemic tissue; p < 0.0001 when the number of synaptic contacts in the ischemic tissue of PBS-treated mice is compared with the number of synaptic contacts in the ischemic area of animals treated with rtPA; Figure 7(a) and (b)). Because irreversible synaptic loss is followed by neuronal death,
1
we then quantified cell survival in Wt cerebral cortical neurons 24 h after 55 min of exposure to OGD conditions and recovery in the presence of either 5 nM of tPA or a comparable volume of vehicle (control). We found that OGD causes a 50.43 ± 4.89% decrease in cell survival (p < 0.0001), and that this effect is significantly attenuated by treatment with tPA (15.36 ± 4.0% decrease in neuronal survival; p = 0.002 when cell survival in vehicle(control)- and rtPA-treated neurons are compared; Figure 7(c)). To study the in vivo relevance of these findings, we measured the volume of the ischemic lesion in Wt and Plg−/− mice 24 h after tMCAO and intravenous treatment with 0.9 mg/kg of rtPA or a comparable volume of PBS. We found that treatment with rtPA decreases the volume of the ischemic lesion in both Wt (from 73 ± 3.46 mm3 to 42.7 ± 5.46 mm3; p = 0.0002) and Plg−/− mice (from 85.50 ± 3.4 mm3 to 55.50 ± 7.1 mm3; p = 0.001; Figure 7(d)).
Treatment with rtPA protects the synapse and promotes neuronal survival in the ischemic brain. (a) Representative confocal microscopy pictures (60 × lens followed by 4.6 × electronic magnification) taken at bregma: 0.02 mm, lateral: 2 mm and ventral: 1 mm in the ischemic (ipsilateral) and non-ischemic (contralateral) hemisphere of Wt mice 24 h after tMCAO followed by the intravenous administration of 0.9 mg/kg of rtPA or a comparable volume of PBS. Green: bassoon; red: PSD-95. (b) Mean number of intact synapses denoted by the presence of bassoon and PSD-95-positive puncta in the coordinates described in A, 24 h after tMCAO and intravenous treatment with 0.9 mg/kg of rtPA or a comparable volume of PBS. n = 3 animals and 25 brain cuts examined per experimental group. Lines denote SEM. (c) Mean cell survival in Wt cerebral cortical neurons examined under physiological conditions (n = 2745 cells), or following 55 min of OGD and 24 h of recovery in the presence of either 5 nM of tPA (n = 2168 cells), or a comparable volume of vehicle (control; n = 1905 cells). Lines denote SEM. (d) Mean volume of the ischemic lesion in Wt and Plg−/− mice 24 h after tMCAO and treatment with 0.9 mg/kg/IV of rtPA or a comparable volume of vehicle (control). n = 10 per experimental condition. Lines denote SEM.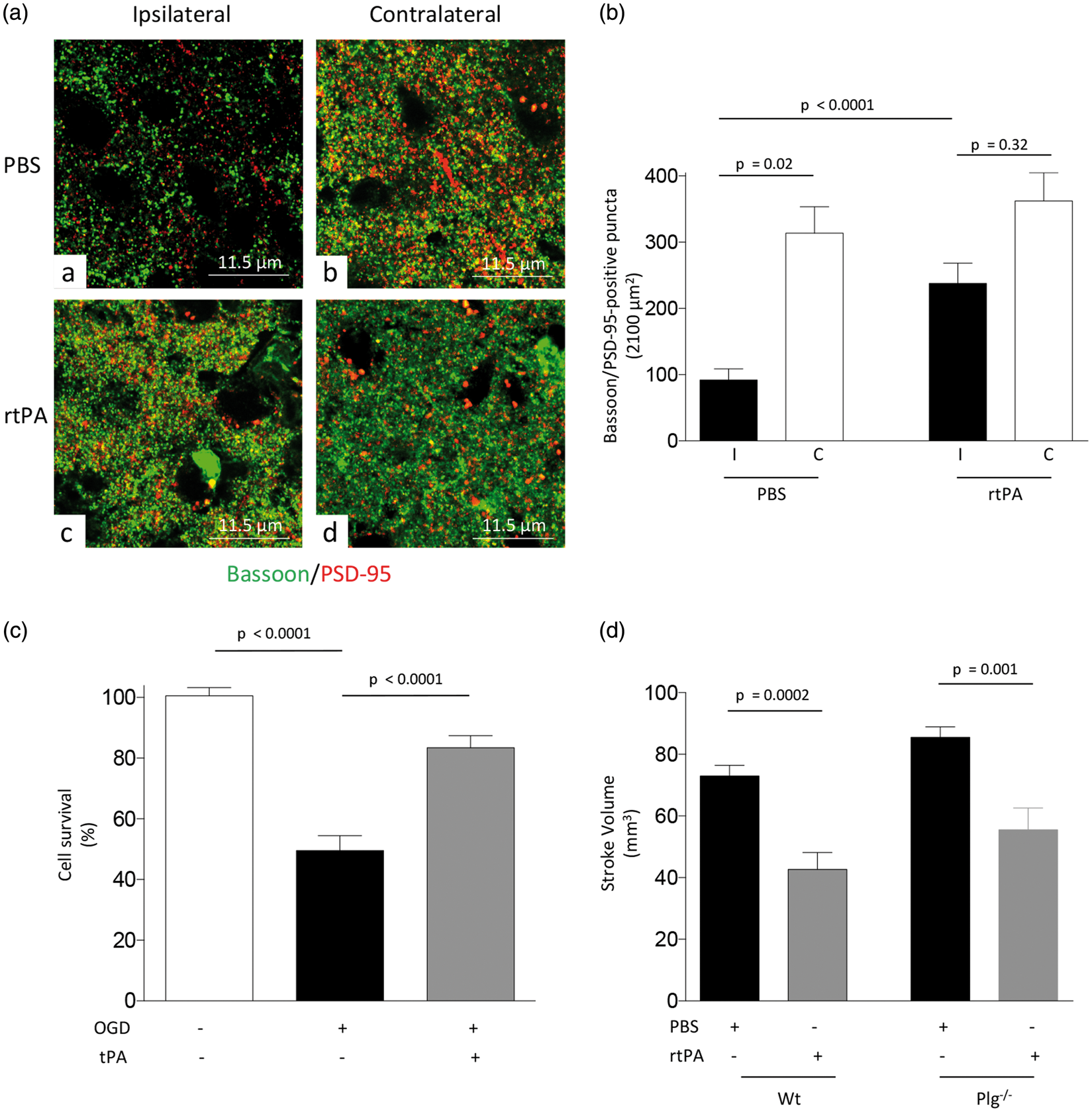
Discussion
Cerebral ischemia has a rapid and profound effect on the structure and function of the synapse that is associated with impairment in neurological function.1,45 However, although this has been widely recognized and numerous studies have described presynaptic and postsynaptic abnormalities at different time-points after the onset of the ischemic injury,1,46 few have investigated the synaptic effects of the early stages of the ischemic insult when cell death has not yet taken place. Our work indicates that 5 min of cerebral ischemia do not induce cell death, but have a significant impact on synaptic structure. More specifically, our fractioning studies show that following 5 min of cerebral ischemia, the number of synaptic fractions containing intact synapses in the ischemic tissue decreases, and that while the presynaptic terminal seems to be more resistant, the PSD is more vulnerable to the ischemic insult. The relevance of these findings is underscored by the fact that since the PSD regulates the strength of excitatory neurotransmission, 47 its early damage during an ischemic injury may determine the degree of functional impairment after an ischemic stroke. Thus, the development of a therapeutic strategy to preserve the integrity of the PSD in the ischemic brain may have a positive impact on synaptic activity and neurological outcome in acute ischemic stroke patients.
A potential problem with the model of sub-lethal ischemia used here is that 5 min of BCCAO are not enough to activate cell death pathways, and thus results obtained with this paradigm may not unveil a potential synaptic effect of tPA in the brain that has suffered a lethal ischemic injury. To address this issue, we quantified the number of synaptic contacts in the ischemic area of Wt mice 24 h after the induction of tMCAO and intravenous treatment with rtPA or PBS. Our data indicate that treatment with rtPA preserves the integrity of synaptic contacts and promotes neuronal survival in the ischemic brain via a mechanism that does not require the conversion of plasminogen into plasmin. Furthermore, our results indicate that the synaptic effect of tPA is not due to the induction of preconditioning. Instead, in line with our earlier observations that tPA has a direct effect on the structure and function of the synapse,36,48 our data suggest that tPA protects the synapse from the harmful effects of an ischemic injury. We also acknowledege that the use of healthy two-month-old males may limit the translation of our results to females and older animals, as well as those with comorbidities. We believe that these are very important aspects that deserve investigation in future studies.
It has long been recognized that cerebral ischemia induces the rapid release of neuronal tPA,23,24 and earlier studies with mice genetically deficient on tPA in endothelial cells, glia and neurons suggested that this release has a neurotoxic effect in the ischemic brain. 24 However, subsequent experimental work with mice overexpressing tPA only in neurons yielded opposite results, indicating the possibility that the release of neuronal tPA may instead have a protective effect in the ischemic brain,23,25–27 by the synaptic detection and adaptation to metabolic stress.26,27 These data are in line with the main finding of the present study, that tPA protects the synapse in the ischemic brain. Importantly, because following its intravenous administration rtPA reaches the ischemic tissue,37–39 then it is plausible to postulate that besides its thrombolytic properties that require the generation of plasmin, rtPA also has a protective effect on the ischemic synapse that is independent of the conversion of plasminogen into plasmin. This hypothesis is supported by our observation that treatment with rtPA protects the synapse and decreases the volume of the ischemic lesion in Wt and Plg−/− mice.
PSD-95 is a member of the MAGUK family of synaptic proteins and the most abundant scaffold protein in the postsynaptic terminal. 49 Owning to its modular structure assembled by three conserved PDZ domains (PSD-95, Drosophila disc large tumor suppressor (Dlg1) and zonula occludens-1) and one Src homology 3-guanylate kinase (SH3-GK) module 50 that integrates the different macromolecular components of the PSD, PSD-95 is a structural organizer of the PSD. 51 Synaptic activity increases the expression of PSD-9512 and in agreement with this observation, the depression of synaptic activity produced by cerebral ischemia causes a profound decrease in PSD-95 expression. 14 Our data indicate that this is a very early event that is not due to ischemic neuronal death. Indeed, our results show that 5 min of cerebral ischemia that do not induce cell death are enough to decrease the expression of PSD-95 in the PSD to almost undetectable levels. Remarkably, this decrease in the abundance of PSD-95 is not mirrored by similar changes in the presynaptic terminal, as the expression of SYP and STX remain intact, indicating a selective vulnerability of PSD-95 during the early stages of the ischemic injury that is protected by tPA.
Synaptic activity increases the transcriptional activity of the PSD-95 promoter. 12 However, the expression of PSD-95 is also regulated at the translational and posttranslational level. 52 Indeed, most of the PSD-95 mRNA is not translated into protein due to the existence of several regulatory mechanisms including miRNAs such as miR-125a, 53 and the controlled degradation of PSD-95 mRNA. 52 We found that in cerebral cortical neurons, tPA induces the expression of PSD-95 protein but not PSD-95 mRNA, and that this effect is abrogated by cycloheximide. These results indicate that tPA promotes the local translation of PSD-95 mRNA into PSD-95 protein. However, additional work is required to determine whether this effect is mediated by antagonism of specific miRNAs or inhibition of PSD-95 mRNA degradation.
PSD-95 interacts with NMDARs 54 and promotes the synaptic insertion of AMPARs by stabilizing the AMPAR/stargazing complex in the PSD. 49 In agreement with these observations, overexpression of PSD-95 promotes the clustering of AMPARs and enhances excitatory neurotransmission. 13 Because several lines of experimental evidence indicate that GluR1-containing AMPA receptors potentiate NMDA and non-NMDA receptor-mediated excitotoxicity, 55 it is conceivable to postulate that tPA may contribute to the development of the excitotoxic injury by inducing the expression and synaptic recruitment of PSD-95, which is known to promote the recruitment of GluR1-containing AMPA receptors to the PSD. Our results indicate that that is not the case, and that tPA not only induces the expression and recruitment of PSD-95 to the PSD, but also inhibits the phosphorylation and synaptic recruitment of GluR1-containing AMPA receptors to the ischemic synapse. More importantly, these data suggest that tPA not only protects the integrity of the PSD but also regulates its composition to prevent the development of an excitotoxic injury.
It is possible that the disruption of synaptic activity during the acute stages of an ischemic injury is a self-protective mechanism to keep neurons alive in an environment depleted of nutrients. With this in mind, it is conceivable to postulate that tPA may block this mechanism by preserving synaptic activity during the ischemic insult, thereby enhancing neuronal vulnerability to the ischemic injury. We believe that our in vivo data showing synaptic protection in mice treated with rtPA after the induction of a lethal ischemic injury, and our in vitro results showing cell survival in neurons treated with tPA after exposure to OGD, indicate that that is not the case, and instead that by preserving synaptic integrity during the ischemic injury, treatment with rtPA also has a neuroprotective effect.
Activation of TrkB receptors is critical for the development and maturation of excitatory synapses 56 and for the recruitment of PSD-95 to the PSD.33,34,57 A link between tPA and TrkB receptors is provided by several lines of experimental evidence indicating that tPA-generated plasmin cleaves proBDNF into mature BDNF, the natural TrkB ligand. 58 In agreement with the existence of a functional interaction between tPA and TrkB receptors, our data indicate that TrkB antagonism abrogates the effect of tPA on PSD-95 recruitment to the PSD. However, our finding that this effect does not require plasmin generation suggests the existence of a non-proteolytic interaction between tPA and either TrkB or BNDF.
In summary, the work presented here indicates that either the release of neuronal tPA or treatment with rtPA induces the expression and recruitment of PSD-95 to the PSD via plasmin-independent TrkB activation. We show that this leads to TrkB-mediated filopodia formation and protection of dendritic spines from the deleterious effects of the ischemic injury. Our data discloses a new role for tPA as synaptic protector in the ischemic brain, and indicate that treatment with rtPA extends the window for neuronal recovery after an acute ischemic injury.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by National Institutes of Health Grants NS-091201 (to MY) and NS-079331 (to MY), and VA MERIT Award IO1BX003441 (to MY).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
Valerie Jeanneret: performed research, Juan Pablo Ospina: performed research, Luis Guillermo Manrique: performed research, Paola Merino: performed research; Enrique Torre: performed research, Fang Wu: performed research, Laura Gutierrez: performed research, Lihong Cheng: performed research, Ariel Diaz: performed research, Manuel Yepes: designed experiments, analyzed data and wrote the paper.