
Abstract
The role of the L-arginine-nitric oxide (NO) system, the role of the endogenous morphine-like substances (endorphins), and the possible interaction between these two systems in the modulation of regional cerebral and spinal CO2 responsiveness was investigated in anesthetized, ventilated, normotensive, normoxic cats. Regional cerebral blood flow was measured with radiolabeled microspheres in hypocapnic, normocapnic, and hypercapnic conditions in nine individual cerebral and spinal cord regions. General opiate receptor blockade by 1 mg/kg naloxone intravenously alone or NO synthase blockade by 3 mg/kg Nω-nitro-L-arginine-methyl ester (L-NAME) intravenously alone caused no changes in regional CO2 responsiveness. Combined administration of these two blocking agents in the very same doses, however, resulted in a strong potentiation, with a statistically significant reduction of the CO2 responsiveness observed. Separation of the blood flow response to hypercapnia and hypocapnia indicates that this reduction occurs only during hypercapnia. Specific μ and δ opiate receptors were blocked by 0.5 mg kg−1 IV β-funaltrexamine and 0.4 mg kg−1 IV naltrindole, respectively. The role of specific μ and δ opiate receptors in the NO–opiate interaction was found to be negligible because neither μ nor δ receptor blockade along with simultaneous NO blockade were able to decrease CO2 responsiveness. The current findings suggest a previously unknown interaction between the endothelium-derived relaxing factor/nitric oxide (EDRF/NO) system and the endogenous opiate system in the cerebrovascular bed during hypercapnic stimulation, with the phenomenon not mediated by μ or δ opiate receptors.
There is significant data suggesting that both the L-arginine/nitric oxide system and the endogenous opiate system take part in cerebrovascular regulation. Both are involved in physiologic control of the cardiovascular system (Holaday, 1983; Furchgott and Zawadzki, 1980), both have an influence on the cerebrovascular bed (Kapadia and deLanerolle, 1984; Bredt and Snyder, 1992), and both interact with other neural, hormonal, and local (Vanhoutte et al., 1986; Olson et al., 1998; Sandor, 1999) regulatory mechanisms. In addition, both systems have a certain commonality in their targets of action at the level of the cellular membrane. Increased intracellular Ca2+ level is of basic importance in activating calcium-dependent nitric oxide synthase (NOS) in the vascular endothelium (Marletta, 1993); however, nitric oxide release causes a decrease in the intracellular Ca2+ of the vascular smooth muscle resulting in vasodilation. On the other hand, opiate-induced inhibition of neurotransmitter release is possibly the consequence of an inhibition of Ca2+ influx into the intracellular space of the presynaptic axonal terminals (Laphcak et al., 1989).
In spite of these experimental findings and the obvious possibilities for an interaction between endorphins and endothelium-derived vasoactive substances, little data exist addressing the possible interaction between these two systems in the regulation of cerebral blood flow (Armstead et al., 1995, 1997). The authors' previous studies (Kovach et al., 1992) demonstrated an important interaction of the two systems in the cerebrovascular bed: acetylcholine-induced, endothelium-dependent relaxation of the cat middle cerebral artery was significantly reduced by hemorrhage, with this effect being completely blocked by general opiate receptor blockade with naloxone.
The current study was undertaken to investigate directly the interaction between endogenous morphine-like peptides and endothelium-derived vasoactive substances in the regulation of cerebral blood flow. Because it is likely that an endogenous opioid mechanism is mobilized only with an alteration or stress of the steady state (Faden and Holaday, 1979), the authors investigated the role of endorphin–endothelium interactions in the cerebrovascular bed by examining blood flow response to changes in arterial PCO2 (CO2 responsiveness). Investigating this interaction may not only result in new data about physiologic control of the cerebral circulation, but also may result in new information that could be important in treating pathologic conditions, such as in hypertension, stroke, diabetes, and hemorrhagic shock, in which both of these systems are known to be activated.
MATERIALS AND METHODS
Animal preparation
Thirty-seven male cats (weighing 2.0 to 3.8 kg) were used in the current study. The animals were anesthetized with an initial dose of 50 mg/kg chloralose (1,2-O-[2,2,2-Trichloroethylidene]-α-D-glucofuranose, Sigma, St. Louis, MO, U.S.A.) and 300 mg/kg urethane (urethane ethyl carbamate; Sigma) injected intraperitoneally. Anesthesia was maintained with successive intravenous (IV) injections of these anesthetics in compliance with the recommendation of the American Heart Association Ethics Committee and the Institutional Animal Care and Use Committee of the University of Pennsylvania. Animals were artificially ventilated by a respirator through an endotracheal cannula with room air, supplemented with oxygen. The core temperature of the animals was kept constant at 37°C with a rectal thermometer (YSI, Model 73a, Yellow Springs, OH, U.S.A.) and a servo-controlled heating lamp. Catheters were inserted into the right brachial and femoral arteries (for blood gas determination and for reference radioactive sampling), into the left brachial and femoral arteries (for continuous blood pressure monitoring on a polygraph (Grass, Model 7E, Quincy, MA, U.S.A.), into the left femoral vein (for drug injection), and into the left ventricle of the heart (for radiolabeled microsphere administration). Heparin (500 U/kg; Elkins-Sinn, Cherry Hill, NJ, U.S.A.) was administered intravenously.
Measurement of regional cerebral and spinal cord blood flow
Regional cerebral blood flow (rCBF) and regional spinal cord blood flow (rSBF) were determined simultaneously with radiolabeled microspheres using the reference sample method (Marcus et al., 1976). Microspheres (15 ± 1.5 μm in diameter; DuPont, NEN-TRAC, Wilmington, DE, U.S.A.) were suspended in 0.9% saline with 0.01% Tween-80 and were injected into the left ventricle of the heart in a volume of 0.3 to 0.5 mL, with an activity of 40 μCi. Vials containing the microspheres were vigorously shaken with an electric shaker for at least 30 minutes before the injections to ensure even dispersion of the microspheres. For each measurement of flow, 1.6 × 106 microspheres (57Co, 85Sr, 46Sc) were injected over a 10-second period, an amount which allowed at least 1000 microspheres to be delivered to 1 g of cerebral tissue. The injection was followed by a 10-second flush of 2.0 mL saline. Reference blood samples were collected through a femoral and a brachial artery using a Harvard withdrawal syringe pump at 1.0 mL/min, beginning 30 seconds before the injection and continuing for 2 minutes after the saline flush. The microsphere injection did not alter arterial blood pressure or heart rate. At the end of the experiments, the cats received an overdose of 150 mg/kg pentobarbital intravenously (Abbott Laboratories, Chicago, IL, U.S.A.), and the brain was infused through the carotid artery with heparinized saline followed by 10% formalin. After removing the brain from the skull, the following discrete areas of the brain were cut and placed in 5-mL vials: thalamus, hypothalamus (medio-basal area, including the median eminence), white matter (corpus callosum), fronto-parietal cortex (mid. suprasylvian and mid. ectosylvian gyri), cerebellum (vermis), pons-medulla oblongata (NTS region), and cervical, thoracic, and lumbar spinal cord (coronal sections, gray and white matter together). Tissue samples were weighed and counted in a gamma scintillation spectrometer (Packard, Model MINAXI g, Auto-gamma 5000 series, Downers Grove, IL, U.S.A.). The energy windows used for 57Co, 85Sr, and 46Sc were 80 to 165 keV, 464 to 564 keV, and 700 to 1400 keV, respectively. Backscatter of lower energy emission from isotopes with higher energy windows was subtracted to obtain a corrected count value. Cerebral blood flow was calculated from the following equation: rCBF = C b × RBF/Cr where rCBF is regional cerebral blood flow in mL g−1min−1, Cb is counts per gram of brain tissue, RBF is reference arterial blood sample withdrawal rate in mL/min, and Cr is total counts in the reference arterial blood sample.
Blood gas analysis
Arterial PaCO2, PaO2, and pH were determined from femoral arterial blood samples immediately after the samples were obtained, using a blood gas analyzer (ABL-30; Radiometer, Copenhagen, Denmark). Arterial PaO2 was kept in a normoxic range (>90 mm Hg) throughout each experiment to avoid the interference of an hypoxic component. Arterial pH was set between 7.21 and 7.30 before drug administration by adjusting the respiratory rate and by bicarbonate administration. End-expiratory CO2 was continuously monitored using an end-tidal CO2 analyzer (Beckman, LB-2, Fullerton, CA, U.S.A.), and was adjusted to 36 to 38 mm Hg before drug administration.
Calculation of regional CO2 responsiveness
Regional cerebral CO2 responsiveness was calculated by measuring the local rCBF and SBF values of nine distinct cerebral and spinal cord regions at normal, decreased, and increased arterial PaCO2 values. The authors examined separately the blood flow responses to hypocapnia and hypercapnia. Hypocapnia was obtained by increasing the rate of the respiration and hypercapnia was produced by 5% CO2 gas inhalation. The rCBF and rSBF measurements at each PaCO2 level were performed after a period of at least 10 minutes at the desired PaCO2 level (continuously monitored with the end-tidal CO2 recording) to ensure that after each PaCO2 change a new steady state was attained and that there was an approximate period of 25 to 30 minutes between each consecutive PaCO2 level alteration. The authors determined the hypocapnic and hypercapnic blood flow reactivity by calculating the mL g−1min−1 change in blood flow per mm Hg PaCO2 change in each discrete brain and spinal cord region.
Experimental groups
Experiments were performed in six groups. Group 1: normotensive, saline-treated animals (n= 6); group 2: normotensive, naloxone-treated animals (n = 6); group 3: normotensive, Nω-nitro-L-arginine-methyl ester (L-NAME)-treated animals (n = 7); group 4: normotensive, L-NAME + naloxone-treated animals (n = 6); group 5: normotensive, L-NAME + β funaltrexamine-treated animals (n = 6); group 6: normotensive, L-NAME + naltrindole-treated animals (n = 6). Regional CBF was determined in each cat during hypocapnia (PaCO2 = 25 to 29 mm Hg), normocapnia (PaCO2 = 36 to 38 mm Hg), and hypercapnia (PaCO2 = 50 to 60 mm Hg). The sequence of the three different PCO2 levels during these studies was randomized.
Drugs
Blockade of the L-arginine-nitric oxide pathway was performed with 3 mg/kg L-NAME administered intravenously, dissolved in 1 mL/kg saline. General opiate receptor blockade was achieved with 1 mg/kg naloxone intravenously, whereas specific blockade of the μ opiate receptors was performed by 0.5 mg kg−1 β funaltrexamine intravenously, and blockade of the opiate δ receptors by 0.4 mg kg−1 naltrindole intravenously. All drugs were obtained from Sigma.
Statistical analysis
Regional CO2 responsiveness of the cerebral and spinal vessels was calculated in each animal as outlined above. Individual hypocapnic and hypercapnic blood flow reactivities were compared across all six experimental groups using a 3-way analysis of variance (ANOVA) with repeated measures (factor 1 = treatment, factor 2 = hypo and hypercapnic reaction as a repeated measure, factor 3 = region). This analysis indicated that the blood flow response to hypocapnia was different from the response to hypercapnia (see Results); therefore, the authors performed a 2-way ANOVA (without repeated measures) examining the hypocapnic and hypercapnic responses separately, and used a Bonferonni t-test to correct for multiple comparisons. The authors also applied the one-factor ANOVA to examine the drug effects regionally (factor = treatment). Fischer's PLSD test was used to examine differences in the physiologic parameters. P ≤ 0.05 was considered significant in all cases.
RESULTS
Effect of general opiate receptor blockade and NOS inhibition on regional cerebral and spinal cord CO2 responsiveness
The CO2 -induced flow increases in the naloxone-treated cats were not statistically different from that of the saline-treated animals (Figs. 1 to 3, Table 1). Small doses (3 mg/kg) of L-NAME also had no effect on regional cerebrovascular CO2 responsiveness in any of the cerebral and spinal cord regions examined, except in the cerebellum (Bonferonni t-test) (Figs. 1 to 3, Table 1). However, simultaneous inhibition of NOS by L-NAME and blockade of all opiate receptor subtypes by naloxone resulted in an almost complete abolition of the vascular CO2 responsiveness. Based on t-test analysis, the blood flow response to CO2 was decreased in all brain and spinal cord regions examined (unpaired t-test; data not shown). After correction for multiple comparisons using the conservative Bonferonni t-test, however, these differences were only statistically significant in the medulla, cortex, thalamus, and cerebellum (Figs. 1 to 3, Table 1). In addition, this interaction of the endothelium-derived relaxing factor/nitric oxide (NO/EDRF) and endogenous opiate system was seen only during hypercapnia. No significant change in the blood flow responsiveness to hypocapnia was observed after simultaneous NOS and endogenous opiate receptor blockade (Table 1). Results obtained with combined opiate receptor blockade and NOS blockade indicate a strong interaction between the endogenous opiate system and the L-arginine-nitric oxide system in the regulation of regional hypercapnic cerebrovascular reaction.
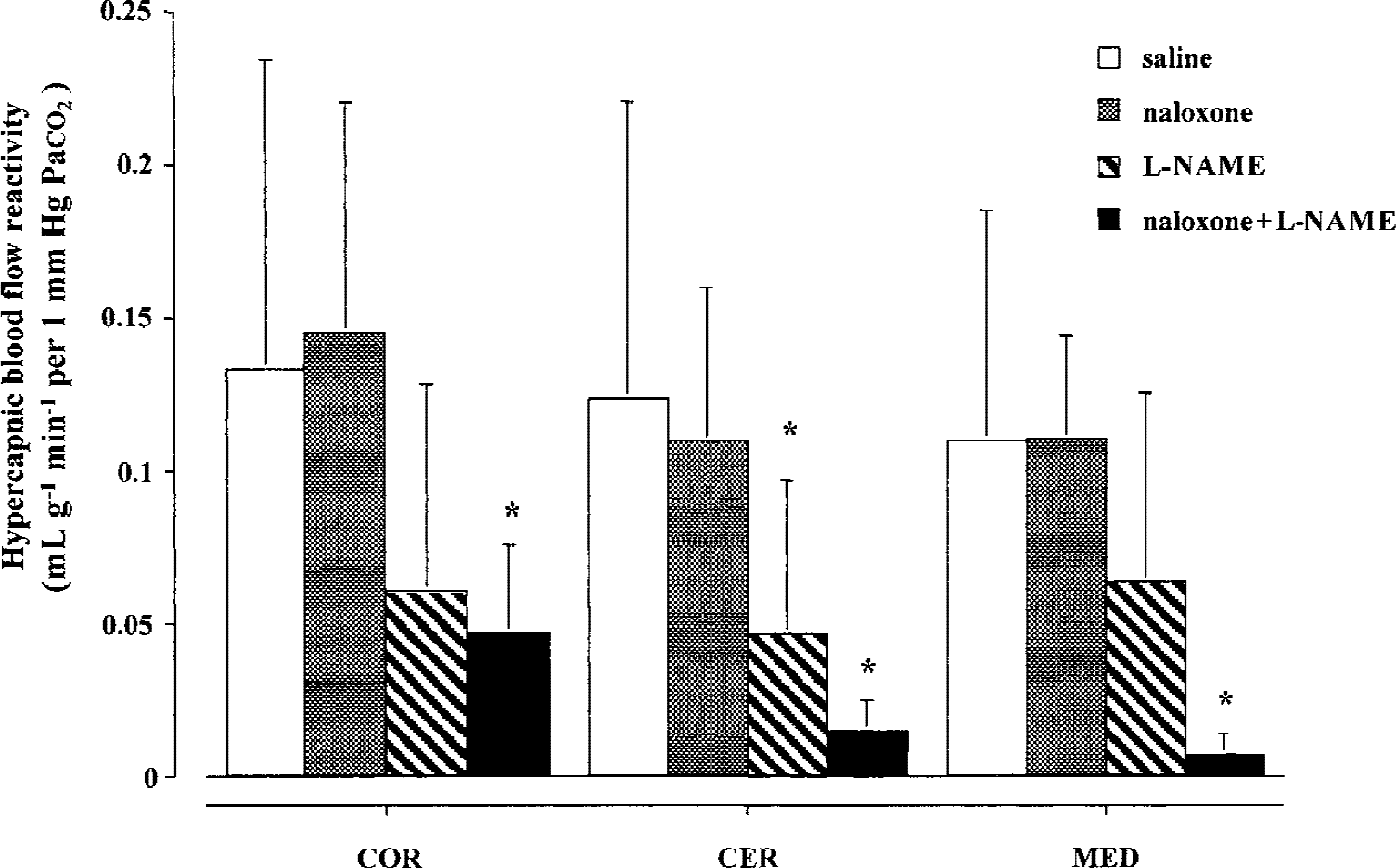
Regional hypercapnic cerebrovascular CO2 sensitivity of the cerebral cortex (COR), cerebellum (CER), and pons-medulla oblongata (MED) in saline-treated (n = 6), naloxone-treated (n = 6), L-NAME–treated (n = 7), and naloxone + L-NAME–treated (n = 6) cats. Bars represent mean ± SD. * P < 0.05 compared with the corresponding saline control CO2 sensitivity (Bonferonni t-test).
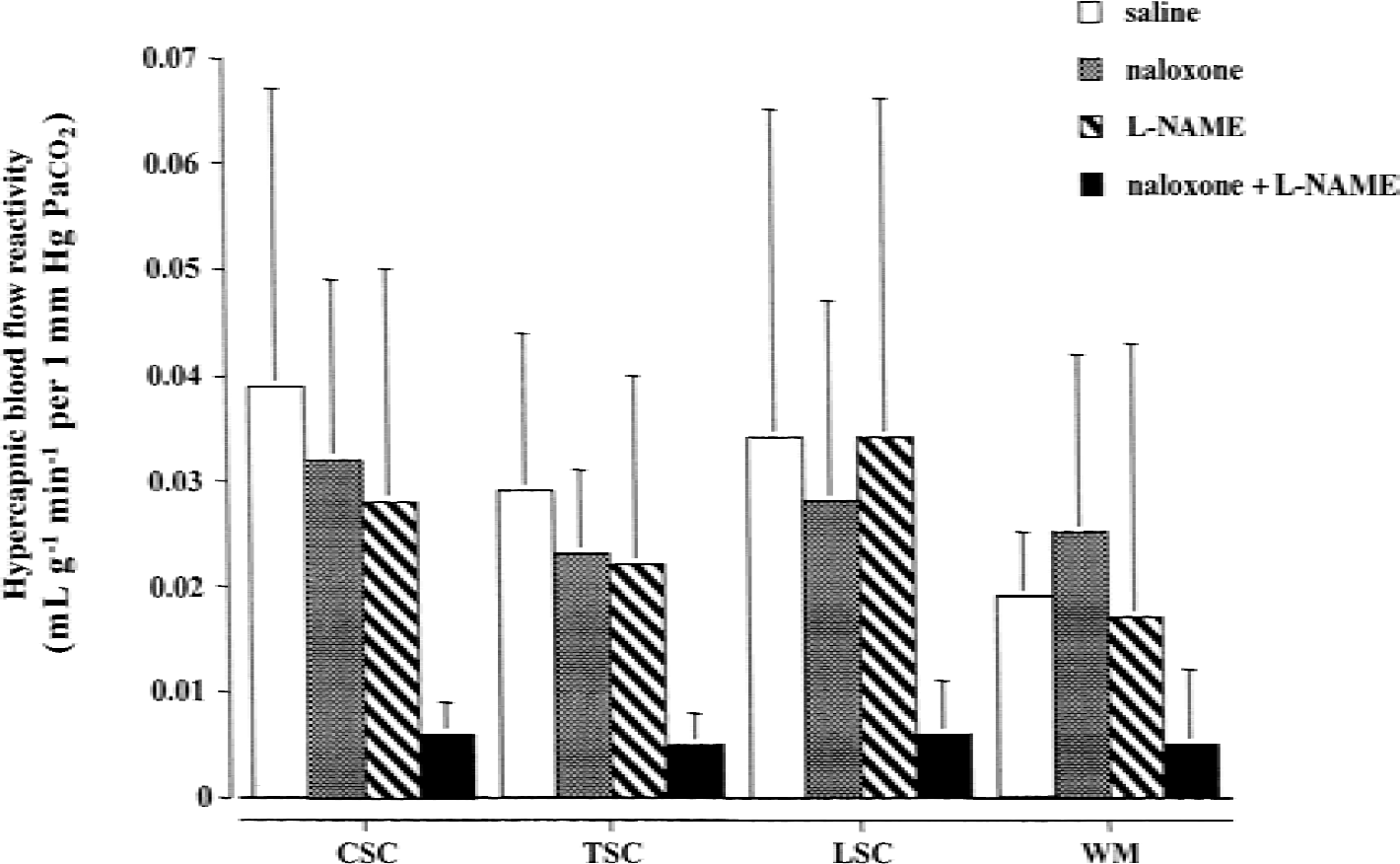
Regional hypercapnic cerebrovascular CO2 sensitivity of the white matter (WM), cervical (CSC), thoracic (TSC), and lumbar spinal cord (LSC) in saline-treated (n = 6), naloxone-treated (n = 6), L-NAME–treated (n = 7), and naloxone + L-NAME–treated (n = 6) cats. Bars represent mean ± SD.
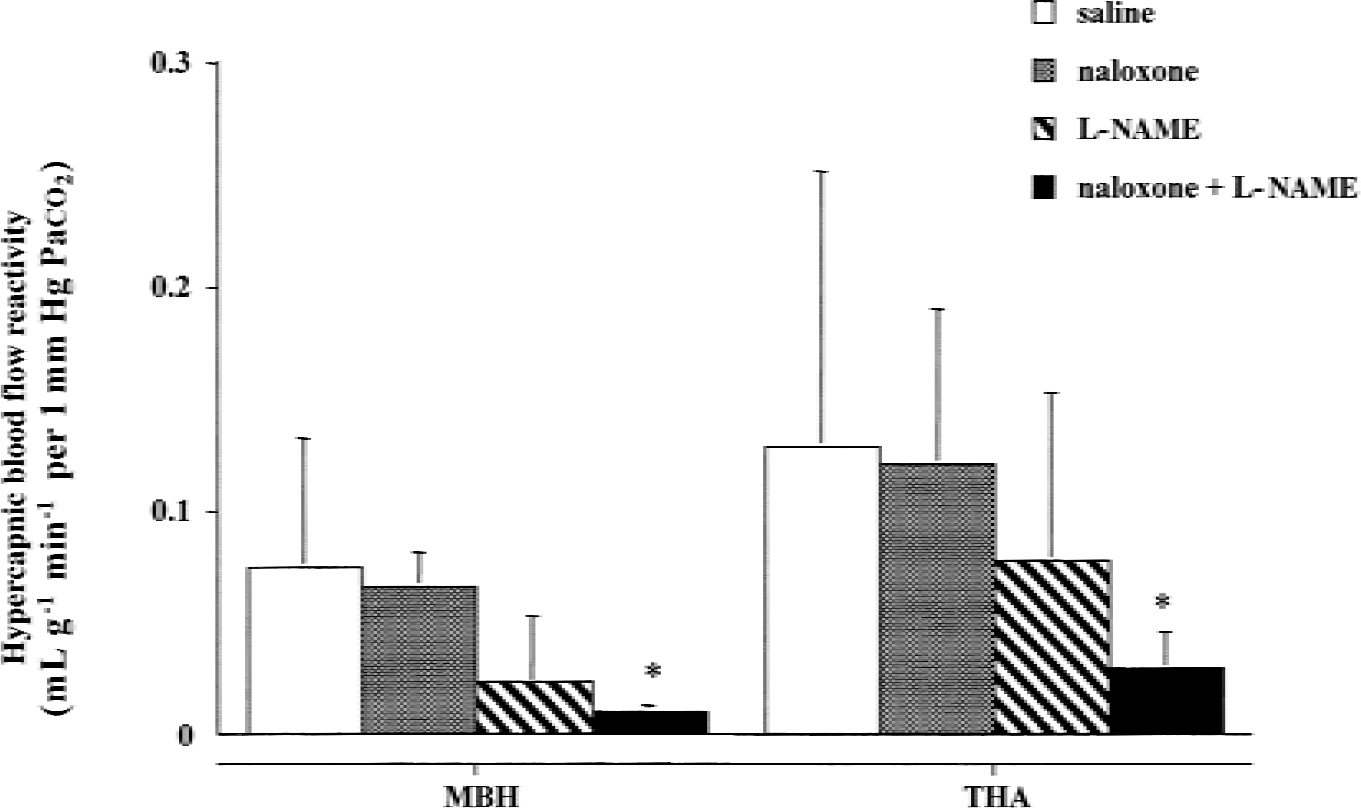
Regional hypercapnic cerebrovascular CO2 sensitivity of the hypothalamus (MBH) and of the thalamus (THA) in saline-treated (n = 6), naloxone-treated (n = 6), L-NAME–treated (n = 7), and naloxone + L-NAME–treated (n = 6) cats. Bars represent mean ± SD. * P < 0.05 compared with the corresponding saline control CO2 sensitivity (Bonferonni t-test).
Cerebrovascular CO2 reaction
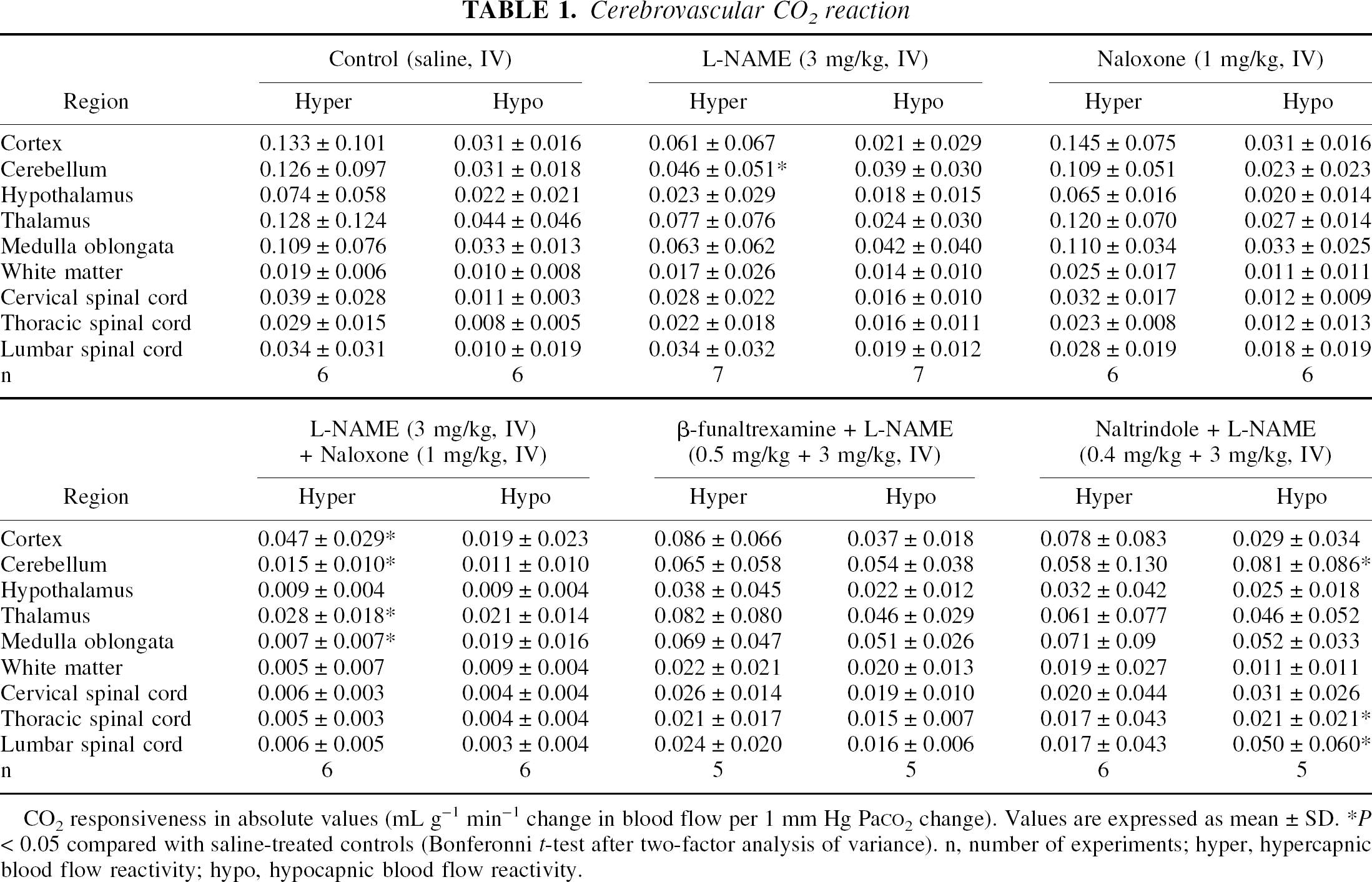
CO2 responsiveness in absolute values (mL g−1 min−1 change in blood flow per 1 mm Hg PaCO2 change). Values are expressed as mean ± SD.
P < 0.05 compared with saline-treated controls (Bonferonni t-test after two-factor analysis of variance). n, number of experiments; hyper, hypercapnic blood flow reactivity; hypo, hypocapnic blood flow reactivity.
Effect of general opiate receptor blockade and NOS inhibition on steady-state arterial pressure, rCBF, and rSBF
Naloxone treatment caused no statistically significant alteration of systemic mean arterial blood pressure (MABP). L-NAME administration resulted in a small rise in MABP (129 ± 39 mm Hg) that was not statistically significant from the MABP values observed in the saline-treated control group (123 ± 24 mm Hg), whereas naloxone and L-NAME coadministration caused a significant increase in MABP (157 ± 19 mm Hg;P < 0.05) (Table 2).
Physiologic parameters
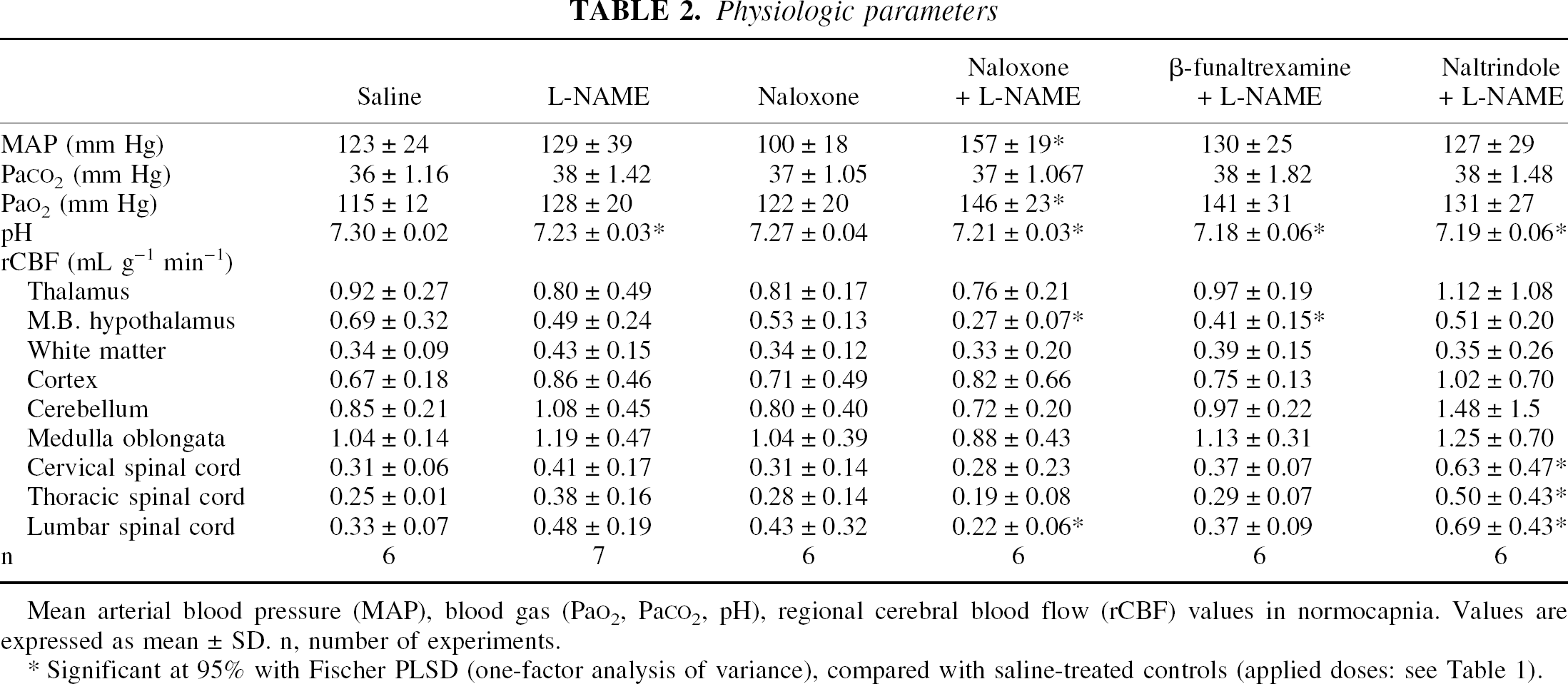
Mean arterial blood pressure (MAP), blood gas (PaO2, PaCO2, pH), regional cerebral blood flow (rCBF) values in normocapnia. Values are expressed as mean ± SD. n, number of experiments.
Significant at 95% with Fischer PLSD (one-factor analysis of variance), compared with saline-treated controls (applied doses: see Table 1).
Basal flow values of the cerebral and spinal regions showed no statistically significant alterations after naloxone-induced general opiate receptor blockade or after L-NAME–induced NOS blockade. Coadministration of naloxone and L-NAME resulted in no statistically significant basal flow alterations in any region except in the hypothalamus where steady-state flow values decreased significantly compared with the saline-treated controls (Table 2).
Effect of simultaneously induced specific opiate receptor blockade and NOS inhibition on regional cerebral and spinal cord CO2 responsiveness
To examine which opiate receptor subtype is responsible for the previously described interaction between L-arginine-NO system and endogenous opiate system in respect of regional cerebrovascular CO2 responsiveness, the authors applied different specific opiate receptor blockers coadministered with L-NAME. Simultaneous IV administration of the μ receptor blocker β-funaltrexamine (0.5 mg kg−1, IV) and the NOS blocker L-NAME (3 mg kg−1) resulted in no statistically significant decrease of CO2 sensitivity during hypercapnia compared with the saline-treated controls in any of the brain regions investigated (Table 1). The simultaneous administration of the δ receptor blocker naltrindole (0.4 mg kg−1, IV) and the NOS blocker L-NAME (3 mg kg−1) also did not produce any statistically significant decrease in CO2 responsiveness during hypercapnia compared with the saline-treated controls (Table 1). During hypocapnia, however, the blood flow responsiveness significantly increased in the cerebellum, thoracic-, and lumbar spinal cord area after simultaneous naltrindole and L-NAME treatment (Bonferonni t-test.). The marked increase in CO2 sensitivity during hypocapnia in the subgroup receiving the delta receptor blockade is possibly the direct consequence of the marked increase in the basal normocapnic flow values in these brain areas.
Effect of simultaneously induced specific opiate receptor blockade and NOS inhibition on steady-state arterial pressure, rCBF, and rSBF
Isolated μ-, δ opiate receptor blocker, and L-NAME coadministration caused no significant change in blood pressure (Table 2). Coadministration of β-funaltrexamine and L-NAME did not produce any changes in basal blood flow except in the hypothalamus where steady-state flow values decreased significantly compared with the saline-treated controls. Coadministration of naltrindole and L-NAME caused significant increases in basal flow only in the cervical, thoracic, and lumbar spinal cord area with no further blood flow changes found in any other brain region examined (Table 2).
DISCUSSION
The current study shows a strong interaction between the endogenous opiate system and the L-arginine–NO system in regulating the cerebrovascular system after alterations in PaCO2. The coadministration of the L-arginine analog L-NAME and the general opiate receptor blocker naloxone resulted in an almost total abolishment of the CO2 responsiveness during hypercapnia. However, the very same doses of these two drugs given alone caused few significant changes in the CO2 responsiveness of the brain and spinal cord regions examined.
Recognition of the multiple regulatory roles of neuropeptides and the discovery of the endogenous morphine-like substances, “endorphins” (Hughes et al., 1975), as well as the demonstration of the many functions of the vascular endothelium and discovery of its highly active chemical products, EDRF and endothelium-derived constricting factor (EDCF) (Furchgott and Zawadski, 1980; Vanhoutte et al., 1986), has completely changed the authors' views about vascular regulation.
Based upon the results of the current study, the authors propose an interaction between the endogenous opioid peptides and NO in the mediation of cerebral and spinal blood flow CO2 responsiveness. In analyzing the main actions of these two regulatory systems it is seen that both participate in the physiologic control of the cardiovascular system, both have an influence on the cerebrovascular bed, both interact with other neural, hormonal, and local regulatory mechanisms, and both show similarities in their mechanism of action at the level of the cellular membranes.
Endorphins are present in the neurons and axon terminals of the central nervous system and in cerebrospinal fluid. In the brainstem, it has been reported that endorphin and enkephalin-like immunoreactive dendrites and capillary basement membranes are in close proximity (Kapadia and deLanerolle, 1984). Opiate receptors are present in the cerebral microvessels (Peroutka et al., 1980), and the anatomic distribution of opiate containing neurons and opiate receptors has been studied extensively. Endogenous opioids modulate the effects of cathecholamines, acethylcholine, dopamine, substance P, oxytocin, vasopressin, TSH, LHRH and LH, PRL and GRH, and ACTH (Olson et al., 1998).
It is likely that opiates affect cerebral vessels through an increased potassium conductance of the smooth muscle cells (Harder and Madden, 1984). Since the late 1970's a large amount of data has accumulated on the cerebrovascular effects of the endogenous opioid peptides in vitro and in vivo, as well as on the cerebrovascular effects of general or selective opiate receptor blockade (Benyo and Wahl, 1996). The question of a possible protective effect of general or specific opiate receptor blockade on cerebral ischemia is still a matter for discussion.
The effect of endogenous opioid peptides on cerebral CO2 sensitivity is somewhat contradictory. In the current study, the authors did not find any statistically significant change of CO2 responsiveness in nine different cerebral and spinal cord regions in normotension after the administration of naloxone, an opiate receptor antagonist. However, the authors previously found that in severe hemorrhagic hypotension in cats, the very same treatment resulted in a dramatic reduction of the cerebrovascular CO2 responsiveness (Sandor et al., 2001).
Endothelium-derived vasoactive substances are released basely and after stimulation of the N-methyl- d -aspartate and glutamate receptors in the central nervous system (Bredt and Snyder, 1992). Endothelium-dependent vascular responses may be induced by vasopressin and oxytocin, angiotensin II, bradykinin, histamine, Substance P, VIP, and CGRP. A review of these interactions was given by Vanhoutte et al. (1986). Endothelium-derived relaxing factors play a significant role in cerebral blood flow regulation and cerebrovascular CO2 sensitivity, both of which may depend on an intact vascular endothelium. In addition, these endothelium-derived substances may play a role in the cerebrovascular bed in hypertension, hypoxia and ischemia, and subarachnoid hemorrhage. The majority of data supports the hypothesis that NO participates in the cerebrovascular vasodilation elicited by hypercapnia (Iadecola et al., 1994). Previous work from the authors' laboratory showed that the selective blockade of NOS produced dose-dependent abolition of the regional cerebrovascular CO2 responsiveness (Sándor et al., 1994). In the current study, the authors described the effect of a small dose (3 mg/kg) of L-NAME that produced a significant reduction in CO2 response in only 1 of 9 cerebral regions (cerebellum). However, it is unlikely that NO is the only vasoactive agent involved in the CBF response to hypercapnia because NOS inhibitors attenuate the vasodilation only within a certain range of PCO2 levels and because even in the most susceptible PCO2 range, a residual response is always present even after high dose NOS inhibition (Iadecola and Zhang, 1994).
In spite of the experimental findings described above, and the obvious possibilities for an interaction between endorphins and endothelium-derived vasoactive substances, little data exist dealing with the possible interaction between these two systems in the regulation of cerebral blood flow. The authors previously demonstrated an important interaction of the two systems in the cerebrovascular bed: acetylcholine-induced, endothelium-dependent relaxation of the cat middle cerebral artery was significantly reduced by hemorrhage, but this effect was completely blocked by general opiate receptor blockade with naloxone (Kovach et al., 1992). In the current study, the authors have shown a strong interaction between these two systems in the responsiveness of the cerebral vessels to alterations in CO2. The data show that neither μ- normgr;-nor δ-opiate receptors participate in the synergistic interaction between the general opiate blocking agent naloxone and the NOS blocker L-NAME in reducing cerebrovascular CO2 responsiveness. Based on the current data, the authors propose the specific strong interaction between the effector mechanisms of opiate receptor activation and that of the L-arginine-NO pathway in producing hypercapnic cerebral vasodilation.
It is widely accepted that the cerebral arterial dilatation in response to increases in blood or tissue PCO2 is mediated by extracellular acidification (Puscas et al., 2000), although the means by which H+ dilates cerebral vessels remains unclear. The effect of protons on the smooth muscle seems to be predominantly extracellular which then may lead to hyperpolarization (Dietrich and Dacey, 1994) through opening of potassium channels. The postacidification decrease in calcium influx will be because of hyperpolarization (Aaljaker and Poston, 1996), as well as the closing of the L-type Ca2+ channels (West et al., 1992). The low intracellular Ca2+ levels may contribute to the vasodilation. The vasodilator effect of acidosis depends on the effect of protons on the vascular smooth muscle. However, the magnitude of this effect is enhanced by the presence of endogenous substances such as NO and the prostanoids, although the importance of these substances depends on the species, age, and the vascular bed. A number of studies have demonstrated that blockade of the brain NOS activity by systemic or topical administration of arginine analogues is capable of attenuating or abolishing the cerebrovascular response to hypercapnia (Iadecola et al., 1994). Results in NOS mutant mice (Ma et al., 1996), and with direct cerebrocortical NO concentration measurements in rats (Harada et al., 1997), suggest that NO derived from neuronal and not endothelial NOS is involved in hypercapnic cerebral blood flow regulation. However, NO is not the sole determinant of hypercapnic vasodilation in the cerebral circulation, and there are NO-dependent and NO-independent components in the cerebrovasodilation elicited by hypercapnia (Iadecola and Zhang, 1994). Numerous data (Parfenova and Leffler, 1996) are consistent with the hypothesis that endothelial prostacyclin synthesis induced by hypercapnia participates in the dilation of smooth muscle. A permissive role of endothelial prostaglandin synthesis represents an important signal for adjacent smooth muscle in response to hypercapnic stimuli especially in newborns (Leffler et al., 1995; Kövecs et al., 2001), compared with neuronal NO synthesis, which represents another important permissive signal but mainly in adults (Iadecola and Zhang, 1996).
Thus, it seems that both components are present in adults and newborns, but that their relative roles may be different. Along with NO and the prostanoids, other vasoactive systems also may be involved in the mediation of cerebrovascular CO2 responsiveness. While the role of adenosine is still controversial, the pivotal role of the K ATP channels in the regulation of CO2 responsiveness is well grounded (Faraci et al., 1994). Another important finding is that the adenosine triphosphate (ATP)-sensitive K+ channel may have an arginine site that influences its function (Kontos and Wei, 1996).
Taken together, several mechanisms appear to be involved in the CO2 -induced response of the cerebral circulation, with numerous interactions between these mechanisms: (1) Bari et al. (1996) data are consistent with the hypothesis of interaction between ATP-sensitive K+ channels and nitric oxide, (2) acidosis induced by hypercapnia may increase NOS activity (Niwa et al., 1993), (3) rapid decline of intracellular pH induced by hypercapnia plays a central role in mediating prostanoid synthesis in the microvascular endothelial cells (Hsu et al., 1993), (4) NO-and prostaglandin-induced carbonic anhydrase activity inhibition is involved in the vasodilation produced by hypercapnia due to subsequent pH changes (Puscas et al., 2000). A previously undetermined fifth interaction may exist, because the current data indicate a strong interaction between the endogenous opiate system and the L-arginine-NO system in the cerebrovascular responses to CO2.
Although a strong interaction between endogenous opiates, K+ channels, prostanoids, and nitric oxide in hypoxic cerebral artery dilatation has already been described (Armstead et al., 1995, 1997), the current data suggest for the first time a similar interaction between these systems in hypercapnic cerebro-vasodilatation.
Based on the current data, the authors propose the existence of several parallel functioning pathways to the complete hypercapnic flow response and a complex interaction among these pathways and factors in hypercapnic vasodilation.