
Abstract
Two hours of transient focal brain ischemia causes acute neuronal death in the striatal core region and a somewhat more delayed type of neuronal death in neocortex. The objective of the current study was to investigate protein aggregation and neuronal death after focal brain ischemia in rats. Brain ischemia was induced by 2 hours of middle cerebral artery occlusion. Protein aggregation was analyzed by electron microscopy, laser-scanning confocal microscopy, and Western blotting. Two hours of focal brain ischemia induced protein aggregation in ischemic neocortical neurons at 1 hour of reperfusion, and protein aggregation persisted until neuronal death at 24 hours of reperfusion. Protein aggregates were found in the neuronal soma, dendrites, and axons, and they were associated with intracellular membranous structures during the postischemic phase. High-resolution confocal microscopy showed that clumped protein aggregates surrounding nuclei and along dendrites were formed after brain ischemia. On Western blots, ubiquitinated proteins (ubi-proteins) were dramatically increased in neocortical tissues in the postischemic phase. The ubi-proteins were Triton-insoluble, indicating that they might be irreversibly aggregated. The formation of ubi-protein aggregates after ischemia correlated well with the observed decrease in free ubiquitin and neuronal death. The authors concluded that proteins are severely damaged and aggregated in neurons after focal ischemia. The authors propose that protein damage or aggregation may contribute to ischemic neuronal death.
Keywords
Proteins must be correctly folded to be functional. Nonfolded (newly synthesized or denatured) or misfolded proteins exposing their hydrophobic segments remain abnormal and are prone to aggregate. To avoid aggregation, abnormal proteins are either protected by molecular chaperones or are quickly degraded by the ubiquitin/proteasome system (Cohen, 1999). Protein ubiquitination is a series of adenosine triphosphate–dependent reactions to form isopeptidyl bonds ligating ubiquitin to the exposed hydrophobic segments of abnormal proteins. Ubiquitination tags abnormal proteins for degradation rather than chaperonelike protection. Under pathologic conditions, when abnormal proteins in cells are too numerous to be protected or degraded, they will aggregate (Cohen, 1999). Neuronal accumulation of abnormal protein aggregates has been commonly found in all neurodegenerative diseases, although the causes of protein aggregation vary among the diseases (Cole and Timiras, 1987; Kakizuka, 1998; Koo et al., 1999). All protein aggregates commonly contain ubiquitin immunoreactivity, suggesting that ubiquitinated proteins (ubi-proteins) are components of protein aggregates (Alves-Rodrigues et al., 1998). Protein aggregation also occurs in neurons after acute brain injury such as transient forebrain ischemia (Hu et al., 2000).
Two hours of focal brain ischemia induced by middle cerebral artery occlusion (MCAO) produces acute massive neuronal death in the striatal core region and a slow type of neuronal death in the neocortical penumbra area. It is generally believed that the slower type of cell death can be salvaged by intervening in the death mechanisms (Siesjö, 1992). In the authors' previous study, they demonstrated by ethanolic phosphotungstic acid (EPTA) electron microscopy that protein aggregation persistently occurs in CA1 neurons after transient global forebrain ischemia. Ethanolic phosphotungstic acid preferentially stains protein aggregates formed after transient cerebral ischemia, and the protein aggregates can be double-stained with ubiquitin immunogold as demonstrated by immunoelectron microscopy (Hu et al., 2000). In this study, the authors investigated whether protein aggregation also occurs in ischemic neurons after focal ischemia. The authors show that proteins become irreversibly aggregated in ischemic neurons before their death after focal ischemia. The authors hypothesize that protein damage or protein aggregation may contribute to the slower type of neuronal degeneration after focal brain ischemia.
MATERIALS AND METHODS
Ischemia model
Male Wistar rats (weighing 260 to 310 g; Charles River, Wilmington, MA, U.S.A.) were fasted overnight with free access to water. All experimental procedures were approved by the Subcommittee on Animal Studies of the Queen's Medical Center and the Animal Care and Use Committee at the University of Miami School of Medicine. All efforts were made to minimize animal suffering and to reduce the number of animals used. Anesthesia was induced by inhalation of 3% halothane in N2O:O2 (70%:30%) and was maintained at 0.7% to 1.5% during surgical procedures. The tail artery was cannulated to monitor blood gases, pH, blood glucose, and blood pressure. Heparin (0.1 mL, 300 U/mL) was administered through the tail artery to avoid formation of thrombosis distal to the occluding filament. A surgical incision was made to expose the right common carotid artery (CCA), internal carotid artery, and external carotid artery. The external carotid artery was ligated proximal to the origin of any branches, such as the occipital artery. The proximal CCA then was ligated and temporarily closed proximal to the carotid bifurcation by a microvascular clip. A small incision was made in the CCA. The occlusion filament was inserted into the internal carotid artery through the CCA 19 to 21mm distal from the bifurcation to occlude the origin of the MCA. The filament was prepared of monofilament fishing line and covered with a distal cylinder of silicon rubber (diameter 0.31 to 0.32 mm). After the MCAO was performed, animals were extubated and allowed to awaken and resume spontaneous breathing. Two hours after induction of ischemia, the filament was withdrawn while animals were breathing halothane in N2O:O2 through a face mask. Temperature was maintained at 37°C ± 0.5 by a heating pad during the operation. After surgery, the animals were cooled in a cold room (4°C) for up to 2 hours of recirculation to avoid hyperthermia. Rectal temperature was kept at 37°C ± 0.5°C by monitoring the animals in every 5 minutes. The neurologic status of each rat was evaluated according to a previously published neurologic grading scale (Bederson et al., 1986). Animals with a neurologic grade of 0, 1, or 2 were excluded from the current study.
Three series of 2-hour MCAO animals were prepared for biochemical analysis, electron microscopy, and confocal microscopy, respectively. They were reanesthetized at either 1, 4, or 24 hours of reperfusion (n = 4 in each experimental group). For biochemical studies, brains were obtained by freezing them in situ with liquid nitrogen while the animals were artificially ventilated (Ponten et al., 1973). Brain tissues were dissected in a −12°C glove box and were cut into ∼3 mm slices in the striatal coronal plane (Fig. 1). Tissue samples weighing approximately 20 mg, representing the dorsolateral caudoputamen (region 1) and the lateral (region 2) and dorsolateral (region 3) neocortical regions of the ipsilateral hemisphere, were collected as shown in Fig. 1. For electron microscopy, brains were perfused with ice-cold 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 mol/L cacodylate buffer, and brain regions (including region 4 of the contralateral hemisphere; Fig. 1) were examined by electron microscopy. For laser-scanning confocal microscopy, brains were perfused with ice-cold 4% paraformaldehyde in phosphate-buffered saline (PBS) and the same brain regions were examined by confocal microscopy. Sham-operated rats were subjected to the same surgical procedures but without induction of brain ischemia.
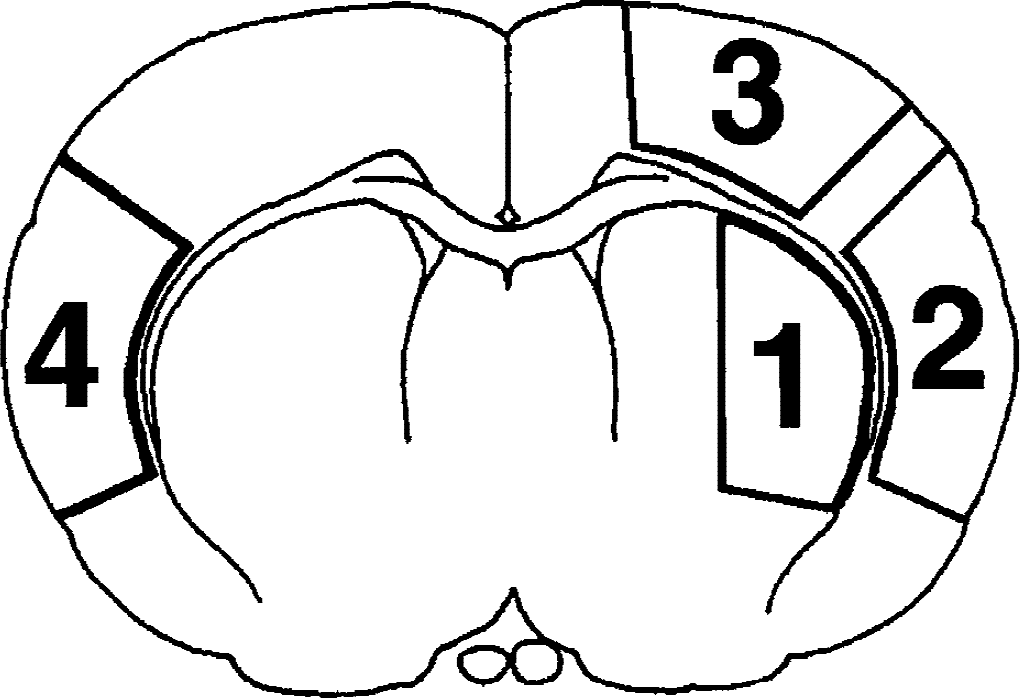
Schematic drawing of the four brain regions analyzed in this study. The coronal section was taken from bregma −0.48 mm. Regions 1 to 3 are in the ischemic hemisphere, and region 4 is on the contralateral side.
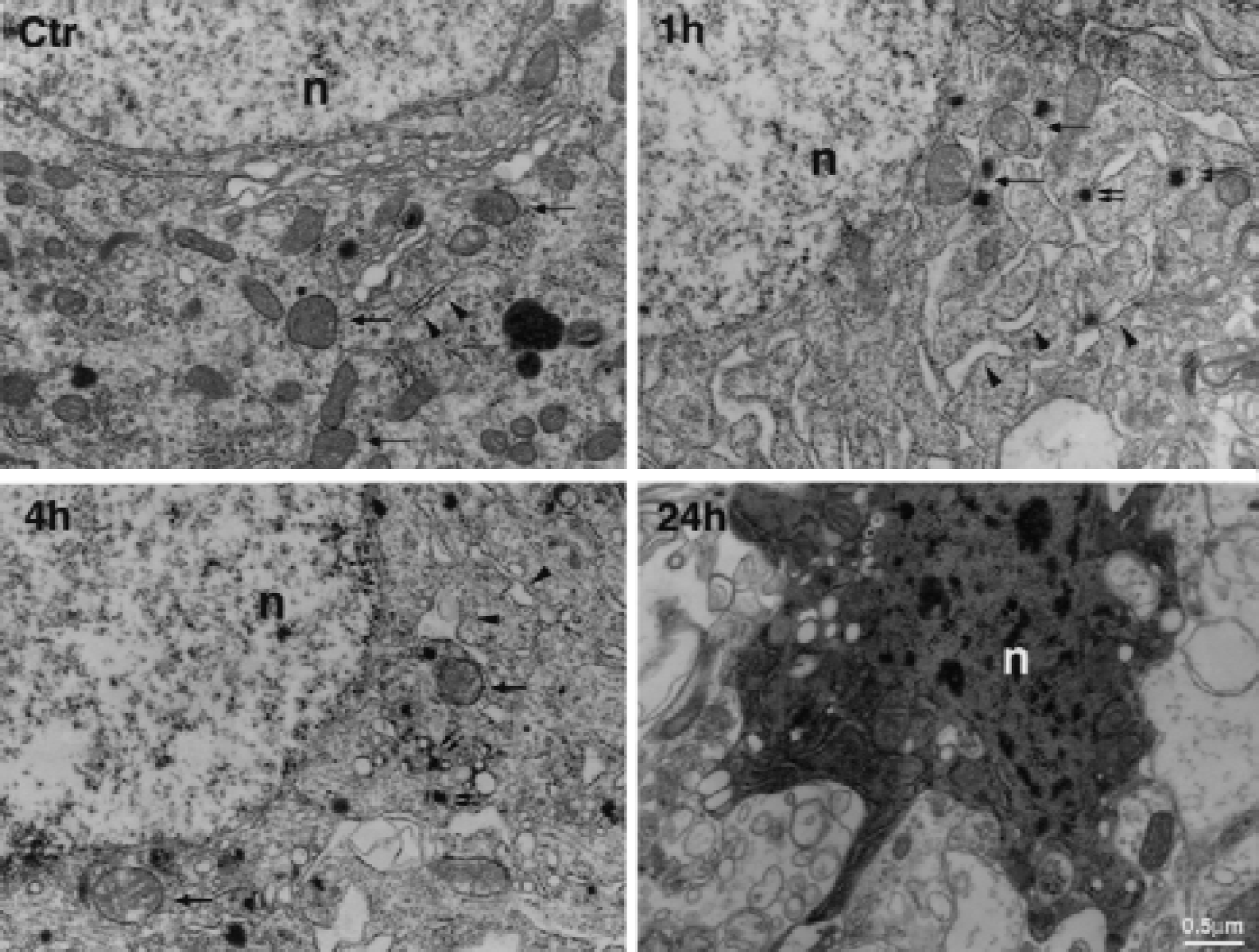
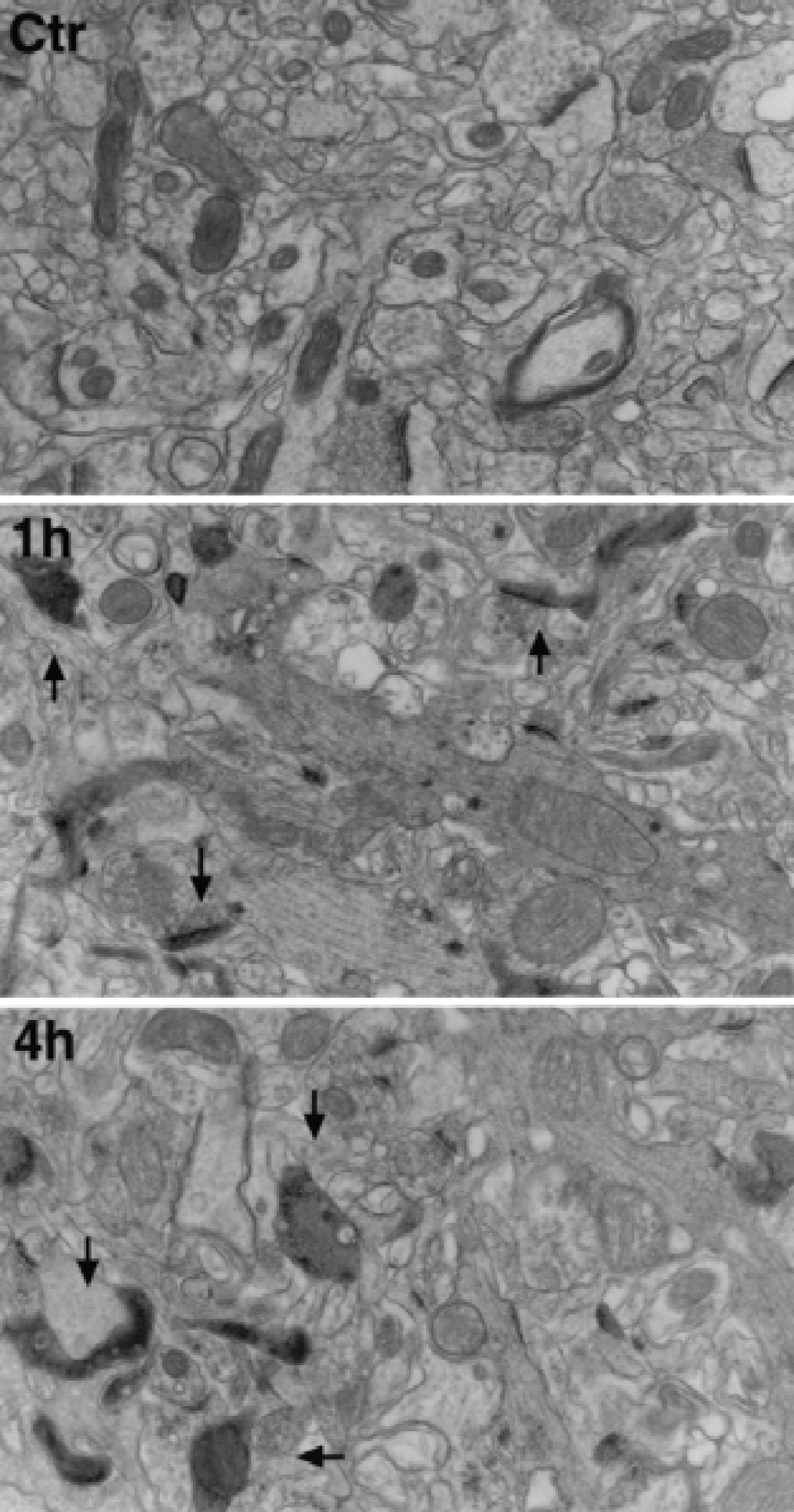
Electron micrographs of neuronal soma stained with osmium-uranium-lead in region 2 of a sham-operated control (Ctr) and at 1, 4, and 24 hours of reperfusion after 2 hours of middle cerebral artery occlusion. Swollen mitochondria (arrows), dilation of rough endoplasmic reticulum (arrowheads), and disaggregation of polyribosomes were observed in postischemic neurons. Some electron-dense deposits (smaller double arrows) also were seen in the cytoplasm during the postischemic phase, but were not visible in control neurons. Neurons were darkly stained after 24 hours of reperfusion. n, nuclei.
Electron microscopy
Electron microscopic studies were performed on brain tissue sections stained either by 1% EPTA or by the conventional osmium-uranyl-lead method as described previously (Martone et al., 1999; Hu et al., 2000). Briefly, coronal brain sections (150-μm-thick) were cut with a vibratome at the striatal level (Fig. 1) and were postfixed for 1 hour with 4% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4). For conventional osmium-uranyl-lead staining, brain sections were postfixed for 2 hours in 1% osmium tetroxide in 0.1 mol/L cacodylate buffer, rinsed in distilled water, and stained with 1% aqueous uranyl acetate overnight. Tissue sections then were dehydrated in an ascending series of ethanols to 100% followed by dry acetone, and were embedded in Durcopan ACM. Ultrathin sections (0.1 μm) were prepared and stained with lead citrate. For EPTA staining, sections were dehydrated in an ascending series of ethanols to 100% and stained for 45 minutes with 1% phosphotungstic acid (PTA) prepared by dissolving 0.1 g PTA in 10 mL of 100% ethanol and adding 4 drops of 95% ethanol. Sections then were embedded in Durcopan ACM. Ultrathin sections (0.1 μm) were prepared and examined with an electron microscope without additional staining.
Laser-scanning confocal microscopy
Double-label fluorescence confocal microscopy was performed on coronal brain sections (50 μm) at the striatal level (Fig. 1) from sham-operated control and animals subjected to 2 hours of MCAO followed by 1, 4, or 24 hours of reperfusion. A monoclonal antibody against ubiquitin (Catalog No. MAB1510, CA, U.S.A.) was used (Morimoto et al., 1996). The brain sections were washed twice in Tris-buffered saline (TBS) for 10 minutes at room temperature and then in TBS containing 0.2% TX100 for 10 minutes. Nonspecific binding sites were blocked in 3% bovine serum albumin in TBS/0.1% TX100 for 1 hour. The monoclonal antiubiquitin primary antibody was diluted in TBS/0.1% TX100 and 3% bovine serum albumin at 1:500. After incubation overnight at 4°C, the sections were washed 3 times for 10 minutes at room temperature in TBS containing 0.1% TX100. Sections then were incubated with a secondary fluorescein-labeled anti-mouse from Amersham (NJ, U.S.A.) diluted 1:200 each and propidium iodide (15 μg/mL) in TBS containing 1% bovine serum albumin and applied for 1 hour at room temperature. Sections were washed several times in TBS, mounted on glass slides, and coverslipped using Gelvatol. The slides were analyzed on a BioRad MRC 1024 laser-scanning confocal microscope (BioRad, CA, U.S.A.). Histologic examination was conducted using vibratome sections (50-μm-thick). The sections were mounted on glass slides, dried, and stained with acid fuchsin and celestin blue. Dead neurons stained with fuchsin were shrunken and acidophilic as examined by light microscopy (Auer et al., 1989).
Subcellular fractionation
A crude pellet fraction and cytosolic fraction were prepared. Brain tissues were homogenized with a Dounce homogenizer (25 strokes) in 10 vol of ice-cold homogenization buffer containing 15 mmol/L Tris base/HCl pH 7.6, 1 mmol/L dithiothreitol (DTT), 0.25 mol/L sucrose, 1 mmol/L MgCl2, 1 μg/mL pepstatin A, 5 μg/mL leupeptin, 2.5 μg/mL aproptonin, 0.5 mmol/L phenylmethylsulfonyl fluoride (PMSF), 2.5 mmol/L EDTA, 1 mmol/L EGTA, 0.25 mol/L Na3VO4, 25 mmol/L NaF, and 2 mmol/L sodium pyrophosphate. The homogenates were centrifuged at 10,000 ×g at 4°C for 10 minutes. The 10,000 ×g supernatants were further centrifuged 200,000 ×g at 4°C for 1 hour to obtain the cytosolic fraction. The 10,000 ×g pellet fractions were sonicated and washed on a shaker with ice-cold homogenization buffer containing 2% TX100 and 150 mmol/L KCl for 1 hour at 4°C, then were centrifuged at 10,000 ×g to obtain a TX100/KCl insoluble pellet fraction. Protein concentration was determined by the Bio-Rad DC protein assay kit.
Western blot analysis
Western blot analysis was performed on 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for intracellular free ubiquitin in the cytosolic fraction, and on 8% SDS-PAGE for the TX100/KCl insoluble pellet fraction, according to the method of Laemmli as described previously (Hu and Wieloch, 1994). Equal amounts of samples containing 15 μg protein in pellets and 40 μg protein in the cytosolic fraction from the control group and experimental groups were applied to each lane in a slab gel of SDS-PAGE. After electrophoresis, proteins were transferred to an Immobilon-P membrane. The respective membranes were incubated overnight at 4°C with primary antibody against ubiquitin from Chemicon (1:2000). Membranes then were incubated with horseradish-peroxidase-conjugated anti-mouse secondary antibody for 1 hour at room temperature. The blots were developed by an enhanced chemiluminescent (ECL) detection method (Amersham).
RESULTS
Histopathology
Neuronal damage in regions 1 to 3 (Fig. 1) after 2 hours of focal ischemia was examined on vibratome sections stained with acid fuchsin and celestin blue. Massive neuronal death was already present at 1 hour of reperfusion in region 1 of the striatum. In regions 2 and 3, a few dead neurons (<10%) were found at 1 and 4 hours of reperfusion. By 24 hours of reperfusion, an infarct was evident in region 2 of neocortex, and selective neuronal death was seen in region 3. No neuronal death was found in the region 4 of the contralateral hemisphere or in sham-operated controls (data not shown).
EPTA stains protein aggregates formed after focal brain ischemia
The authors used the EPTA staining method to characterize protein aggregation after focal ischemia. Rats were subjected to 2 hours MCAO by intraluminal suture, followed by 1, 4, and 24 hours of reperfusion. Brains sections at the striatum level (Fig. 1) were stained with EPTA and by conventional osmium-uranium-lead staining methods, and were examined by electron microscopy. Ultrastructure of control neurons (Fig. 2, Ctr) stained with the conventional method was normal, with normal nucleus (n), mitochondria (arrows), endoplastic reticulum, and its associated polyribosomes (arrowheads). In comparison, many pathologic ultrastructural features were seen in regions 1 to 3 after ischemia. Consistent with their light microscopic appearance, most neurons in region 1 were already dead at 1 hour of reperfusion, as judged by their severe cell membrane damage by electron microscopy (data not shown). In regions 2 and 3 at 1 and 4 hours of postischemic reperfusion, swelling of mitochondria (Fig. 2, 1h and 4h, arrows), dilation of rough endoplastic reticulum (Fig. 2, 1h and 4h, arrowheads), and disaggregation of polyribosomes were observed in ischemic neurons. However, these neurons in regions 2 and 3 still had intact cell membranes and normal nuclear morphology (Fig. 2, 1h and 4h). Dark aggregates also were noted in these neurons of regions 2 and 3 at 1 and 4 hours of reperfusion (Fig. 2, 1h and 4h, smaller double arrows). These dark aggregates often were distributed in association with intracellular vesicles (Fig. 2, 4h). By 24 hours of reperfusion, neurons of region 2 were darkly stained, intracellular membranes were severely destroyed, and neurons were shrunken (Fig. 2, 24h). In region 3 at 24 hours of reperfusion, both normal neurons and dead neurons were present. All neurons in region 4 were normal after ischemia.
On EPTA-stained brain sections, EPTA normally stained the synaptic structure and nucleus in control brains (Fig. 3, Ctr), but also stained intracellular protein aggregates of regions 2 and 3 in neurons at 1 and 4 hours of reperfusion (Fig. 3, 1h and 4h, arrowheads). EPTA-stained protein aggregates were found on intracellular vesicles in the soma and apical dendrites (Fig. 3, 1h and 4h, arrowheads). Consistent with the conventional electron microscopic observations, most striatal neurons of region 1 were dead at 1 hour of reperfusion (data not shown), and most neocortical neurons in region 2 were dead at 24 hours of reperfusion (Fig. 3, 24h). However, some neocortical neurons in region 3 still appeared viable (data not shown).
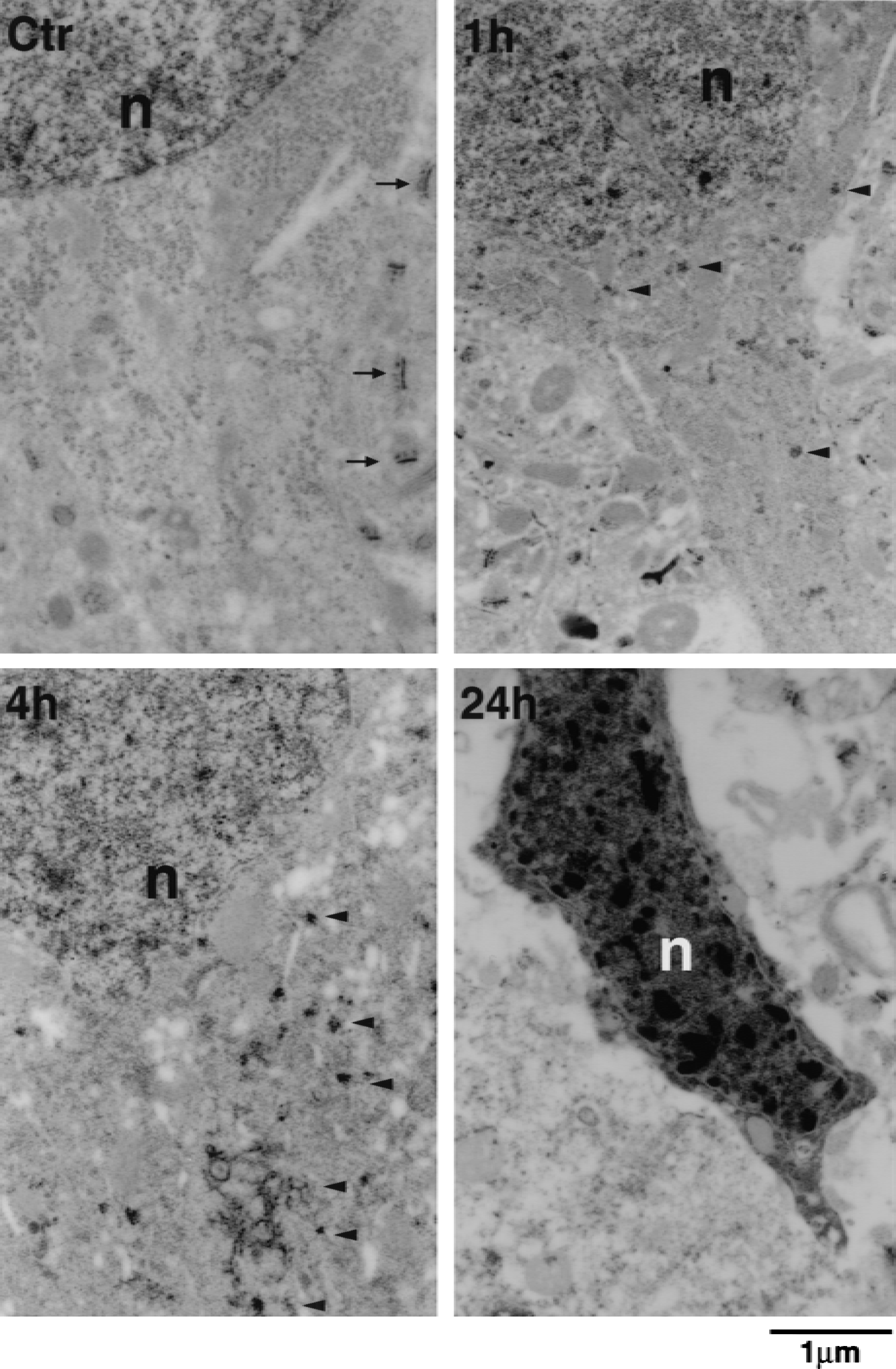
Electron micrographs of neuronal soma stained with EPTA in region 2 of sham-operated rats (Ctr) and at 1, 4, and 24 hours of reperfusion after 2 hours of middle cerebral artery occlusion. EPTA strongly stained synaptic structures (arrows) and nuclei (n), but reacted weakly with other subcellular structures in controls. In the postischemic brain sections, EPTA stained additional intracellular protein aggregates (arrowheads). These aggregates were preferentially distributed in the cell soma, often associated with intracellular vesicles and along the apical dendrites at 1 and 4 hours of reperfusion. Shrunken nuclei with chromatin clumping were observed in most neurons of region 2 at 24 hours of reperfusion.
In the neuropil of neocortex, EPTA normally stained synaptic structures in control and postischemic neurons (Fig. 4A, Ctr, arrows), but EPTA also stained protein aggregates in region 2 (Fig. 4A, 1h, 4h and 24h, arrowheads) and region 3 (data not shown). These protein aggregates were not present in region 4 of the contralateral hemisphere (data not shown) or in sham-operated control brains (Fig. 4A, Ctr). The threadlike EPTA-stained protein aggregates were often seen both in the small dendritic trunk and in thin dendritic spines (Fig. 4A, 1h and 4h, arrowheads). By 24 hours of reperfusion, EPTA-stained aggregates were still present in region 2 (Fig. 4A, 24h), but cellular structures were destroyed (Fig. 2, 24h), and these neurons had lost their ubiquitin immunoreactivity (Fig. 6B, 24h, green). Higher magnification demonstrated that some mitochondria in the dendritic trunk in region 2 were prominently stained with EPTA at both 1 (data not shown) and 4 hours of reperfusion (Fig. 4B, upper). Some axoplasms in regions 1 to 3 were darkly stained with EPTA, and they were often observed after ischemia, particularly after 24 hours of reperfusion (Fig. 4B, lower). Axons were identified by their surrounding myelin sheath (Fig. 4B, lower). Although the myelin sheath was not directly visible in the EPTA-stained material because of lipid extraction without osmication, they were clearly visible in negative relief allowing for easy identification (Fig. 4B, arrows). EPTA did not stain axoplasm in sham-control brain sections. In comparison with EPTA-stained sections, electron-dense material also was present in the neuropil of brain sections stained with osmium-uranium-lead (Fig. 5, 1h and 4h, arrows). The osmium-uranium-lead–stained deposits were superimposed upon other normally stained subcellular structures, and it was usually difficult to determine the relation between them. However, the regional and temporal distribution of the osmium-uranium-lead–stained electron-dense material was similar to that of the protein aggregates stained by EPTA (compare Fig. 4A with 5).
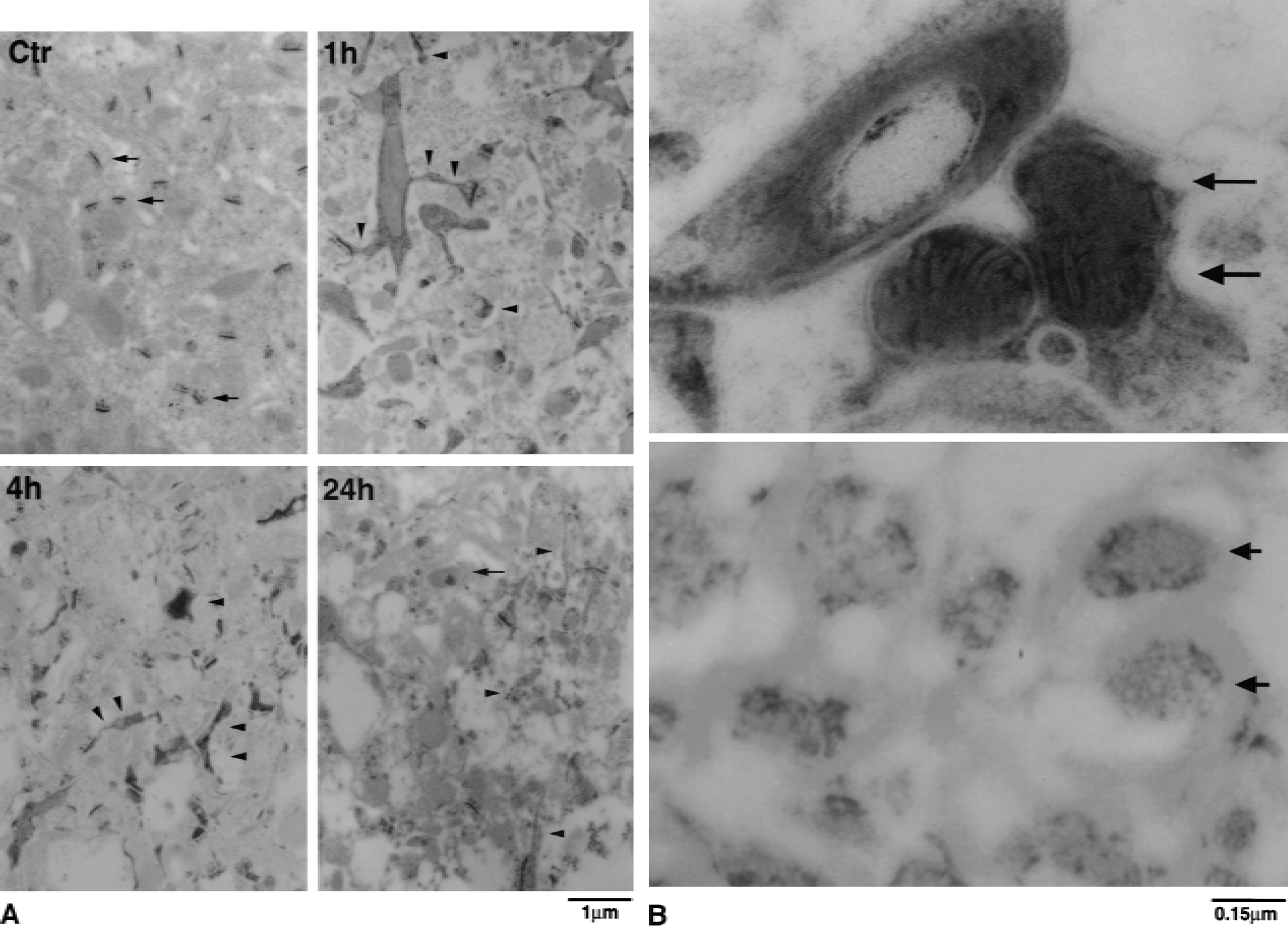
Electron micrographs of the neuropil area stained with osmium-uranium-lead in region 2 of sham-operated controls (Ctr) and at 1 hour and 4 hours of reperfusion after 2 hours of middle cerebral artery occlusion. In postischemic brains, some relatively dark structures (arrows), possibly representing the threadlike structures found in EPTA-stained sections (Fig. 4A), were seen at 1 and 4 hours of reperfusion.
Formation of ubi-protein aggregates
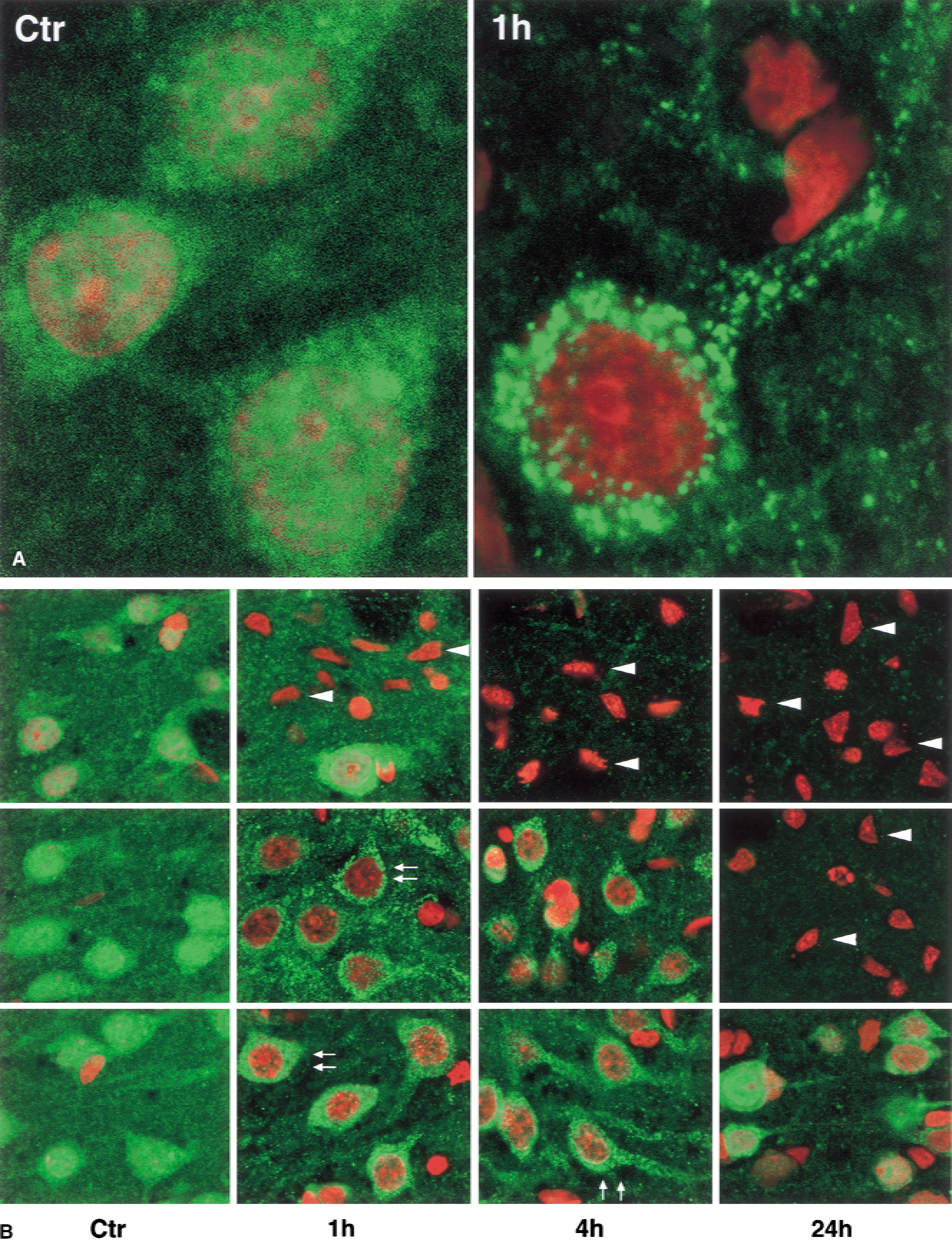
Brain sections from controls and animals with 1 hour of reperfusion after 2 hours of MCAO were double labeled with a monoclonal anti-ubiquitin antibody (green) and propidium iodide (red) (Fig. 6A). Ubiquitin immunolabeling was distributed in the nuclei, cytoplasm, and neuropil area of control neurons (Fig. 6A, sham, green). After ischemia, however, the immunolabeling pattern clearly changed from a relatively even distribution to heterogeneous aggregates (Fig. 6A, 1h, green). These anti-ubiquitin–positive aggregates were scattered around nuclei and were clustered along dendrites (Fig. 6A, 1h, green). The propidium iodide staining in control nuclei (Fig. 6A, Ctr, red) was masked by the ubiquitin staining (Fig. 6A, green).
The regional distribution and progression of ubi-protein aggregation are further shown in Fig. 6B. Ubiquitin immunostaining was changed from a relatively even distribution to clumped aggregates in regions 2 and 3 (Fig. 6B, middle and lower panels, green). As demonstrated below (Fig. 7), the even ubiquitin immunostaining might represent free ubiquitin in control neurons, whereas the clumped aggregates of ubiquitin immunostaining in postischemic neurons might result from depletion of free ubiquitin and ubi-protein aggregation. In the striatal core region, ubiquitin immunostaining had already disappeared in most neuronal bodies at 1 hour of reperfusion, but immunostaining was still present at this time point in the background and in a few large neurons (Fig. 6B, upper panel, 1h, green). This may be because most neurons in this region had died but other cellular components or neuronal connections were still intact and thus retained ubiquitin immunoreactivity. Most neurons in the striatal core region were dead after 1 hour of reperfusion, as judged by their shrunken triangular nuclei that were highly stained by propidium iodide (Fig. 6B, upper panel, 4h and 24h, red). These dead neurons had lost their ubiquitin immunoreactivity (Fig. 6B, upper panel, arrowheads). In region 2 (Fig. 6B, middle panel, green) and region 3 (Fig. 6B, lower panel, green) of the neocortex, the immunostaining pattern changed from an even distribution to clumped aggregates in the cytoplasm and dendrites but disappeared from the nuclei at 1 and 4 hours of reperfusion (Fig. 6B, arrows). By 24 hours of reperfusion, the immunolabeling (green) had disappeared from region 2, where massive neuronal death had occurred (Fig. 6B, middle panel, 24h, arrowheads). However, there were still some surviving neurons in the boarder area between regions 2 and 3. Some neurons in region 3 (penumbra), as well as the remaining neurons in the boarder area between regions 2 and 3, were highly labeled with anti-ubiquitin antibody at 24 hours of reperfusion (Fig. 6B, lower panel, 24h, green).
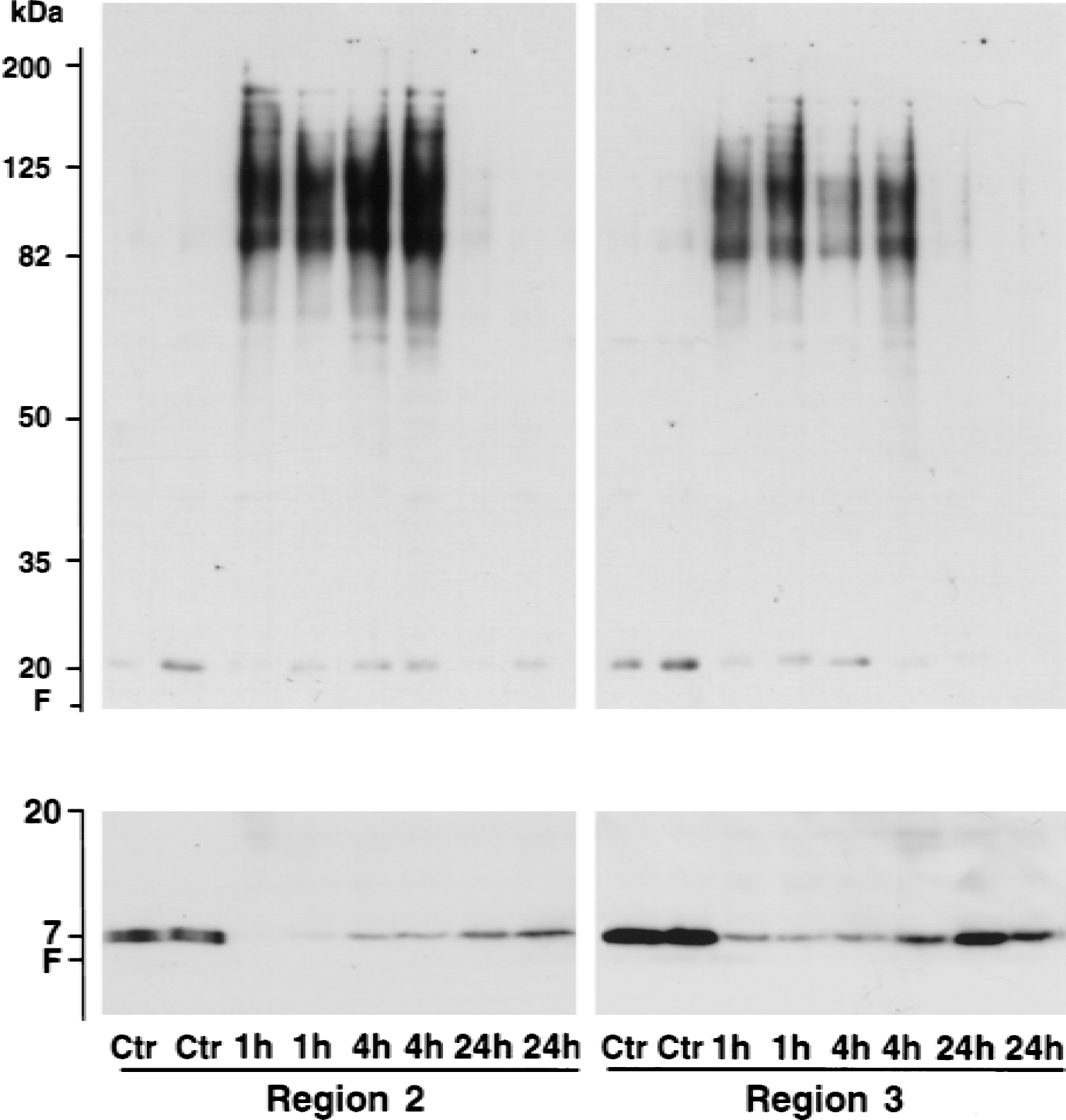
Immunoblots of ubiquitin in the Triton-resistant pellets (top) and cytosolic fraction (bottom). Samples of regions 2 and 3 were from sham control rats (Ctr) and rats subjected to 2 hours of middle cerebral artery occlusion followed by 1, 4, and 24 hours of reperfusion. Each sample was derived from one rat. Two separate samples in each experimental group were run on sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The blots were labeled with an anti-ubiquitin antibody and visualized with the ECL system. Molecular size markers are indicated on the left. F, sample front edge on the gel.
To study protein aggregation further during reperfusion after 2 hours of MCAO, the authors prepared a Triton-resistant pellet fraction and a cytosolic fraction. Ubi-proteins were dramatically increased at 1 and 4 hours of reperfusion in the pellet fractions from both regions 2 and 3 (Fig. 7, upper panels). These changes were more pronounced in region 2 than in region 3 (Fig. 7, upper panels). This might be because the ischemic insult was more severe in region 2 than in region 3. On Western blots, ubi-proteins were only slightly increased in region 1 after ischemia (data not shown), consistent with the early loss of ubiquitin immunoreactivity after cell death shown by confocal microscopy (Fig. 6B, upper panel, green). Concomitantly, intracellular free ubiquitin in the cytosol was markedly decreased at 1 hour of reperfusion, then recovered to various degrees at 4 and 24 hours of reperfusion in both regions 2 and 3 (Fig. 7, lower panels). These results indicated that free ubiquitin in the cytosol was used for the ubiquitination of abnormal proteins in the pellet fraction during the early reperfusion period. This suggests that recovery of free ubiquitin on immunoblots may be because of both de novo ubiquitin synthesis in the remaining neurons and deubiquitination of the ubi-proteins. Free ubiquitin is a heat shock protein that is induced after focal ischemia (Fig. 6B, lower panel, 24h, green; (Dewar et al., 1993, 1994; Noga et al., 1997). Thus, the recovery of free ubiquitin in the brain may be attributable to both newly synthesized free ubiquitin and deubiquitination of ubi-proteins.
DISCUSSION
In this study, the authors have demonstrated that intracellular proteins become severely aggregated and ubiquitinated in ischemic neurons after 2 hours of MCAO. Protein aggregates exist neither in control neurons nor in nonischemic neurons after focal ischemia. Protein aggregates are clearly visible in neocortical neurons stained with EPTA and examined by electron microscopy. They are deposited mainly on membranous structures such as intracellular vesicles in the cell body and apical dendrites. They are also located in axons and, on ultrathin brain sections, form a threadlike pattern in the neurophil during the postischemic phase. The clumped ubi-protein aggregates seen by confocal microscopy have a similar regional and temporal distribution as the EPTA-stained protein aggregates viewed by electron microscopy. Consistent with the results obtained by confocal and electron microscopy, Western blot analysis showed severe accumulation of ubi-proteins during the postischemic phase. Ischemia-induced ubi-proteins are Triton-resistant, suggesting that they may be irreversibly aggregated with each other (Cole and Timiras, 1987; Hayashi et al., 1992a, 1992b; Kazantsev et al., 1999). This study extends the authors' previous study on protein aggregation after transient cerebral ischemia, and provides direct evidence that protein damage and aggregation occur in neurons after focal ischemia. The EPTA-stained intracellular aggregates in living cells are composed of ubi-proteins as demonstrated by antiubiquitin immunogold electron microscopy (Hu et al., 2000). This is also consistent with several previous studies showing signs of protein damage and aggregation after focal and global brain ischemia. Dark material was found in neurons destined to die after transient cerebral ischemia (Kirino et al., 1984; Deshpande et al., 1992). Changes in the immunoreactivities of ubiquitin, tau (Dewar et al., 1993, 1994; Shackelford and Yeh, 1998), and amyloid precursor protein (Stephenson et al., 1992; Yam et al., 1997; Dietrich et al., 1998; Lin et al., 1999; Valeriani et al., 2000) have been observed in regions of neurodegeneration after focal brain ischemia. Transient cerebral ischemia induces depletion of free ubiquitin and marked increases in ubi-proteins (Magnusson and Wieloch, 1989; Morimoto et al., 1996; Hayashi et al., 1992a; Ide et al., 1999).
Distribution of protein aggregates
At the electron microscopic level, the presence of abnormal protein aggregates in ischemic neurons is most clearly demonstrated by the EPTA staining method, consistent with a previous study in a forebrain ischemia model (Hu et al., 2000). ETPA-stained protein aggregates are already formed in regions 2 and 3 at 1-hour postischemia, and are continuously present until 4 hours in region 2, and until 24 hours in region 3 after 2 hours of MCAO. In region 1 of the striatum, necrotic neuronal death occurs promptly after 2 hours of MCAO as judged by severe membrane damage and shrunken nuclei observed by light and electron microscopy. There are some differences in protein aggregation between focal ischemia as shown in the current study and global brain ischemia in the authors' previous article (Hu et al., 2000). First, the timing is different. Protein aggregates appear at 1 to 4 hours of reperfusion and progressively accumulate with longer periods of reperfusion after 15 minutes of global ischemia (Hu et al., 2000). However, protein aggregates are already seen at 1 hour of reperfusion in regions 2 and 3 after 2 hours of focal ischemia, and neurons will die by 24 hours of reperfusion in these brain regions. This may be because the longer ischemic duration in the focal ischemia model leads to more severe protein damage and aggregation, compared with the shorter ischemic period in global ischemia model. Second, the pattern of protein aggregation is different. After 15 minutes of global ischemia, protein aggregation occurs mainly in intracellular vesicles and endoplasmic reticulum at 4 hours of reperfusion, then in mitochondria and dendritic membranes at 24 hours of reperfusion. In comparison, after focal ischemia, besides intracellular vesicles and mitochondria, protein aggregates also form threadlike structures in regions 2 and 3, where neurons die slowly. Higher magnification shows that these threadlike structures are thin dendrites and axons. Because most neuronal dendritic trunks in these areas are not heavily stained with EPTA at 1 hour of reperfusion, these EPTA-stained threadlike dendrites and axons may be derived from the neurons that are already damaged in the other areas. Alternatively, proteins in the EPTA-stained threadlike dendrites and axons may be more susceptible to ischemia so that they aggregate before neurodegeneration. The finding of protein aggregates in axoplasm is interesting because axonal damage caused by ischemia could impact neuronal function in the remote regions. Three-dimensional tomography of the EPTA-stained brain sections may provide more information about where these threadlike structures and axons are derived from.
Consistent with the results obtained by EPTA electron microscopy, ubiquitin immunostaining changes from a relatively even pattern to a heterogeneous distribution after focal ischemia, as shown by confocal microscopy. The punctate ubi-protein immunoreactivity is distributed around nuclei and along dendrites, similar to the pattern of EPTA-stained protein aggregates seen by electron microscopy. The confocal microscopic morphology of neurons with ubi-protein aggregates is virtually normal, relative to control neurons, suggesting that these neurons are still alive and able to retain ubiquitin immunoreactivity. When neurons have degenerated as shown by their shrunken nuclei highly stained with propidium iodide and triangular in shape, they lose their ubiquitin immunoreactivity. This suggests that ubi-protein aggregates are present only in viable neurons. This seems quite reasonable because protein ubiquitination is an adenosine triphosphate–dependent process (Ciechanover et al., 2000); thus, the loss of ubiquitin immunoreactivity after cell death may be because of adenosine triphosphate depletion. Consistent with the confocal data, immunoblots showed that ubi-proteins were markedly increased at 1 and 4 hours of reperfusion, then disappeared by 24 hours of reperfusion, when the neurons were dead in regions 2 and 3. These findings are all consistent with the observation that ubi-protein aggregates are only present in living cells after ischemia. The newly formed ubi-proteins after focal ischemia are Triton-resistant, consistent with the nature of irreversible protein aggregation found in other pathologic conditions (Cole and Timiras 1987; Hayashi et al., 1992b; Kazantsev et al., 1999).
Protein aggregation and neuronal death
The fact that ubi-protein aggregation occurs only in living cells, but most of them are on their way to die, suggests a rationale for the possibility that protein damage and aggregation causes neuronal death after focal ischemia. However, additional studies involving biochemical, genetic, or pharmacologic interventions will further uncover the roles of protein aggregation in neuronal pathology after brain ischemia. It is known that protein aggregation results mostly from the protein damage or accumulation of unfolded proteins produced during and after ischemia. According to the results of this study, it is unlikely that the accumulation of unfolded proteins and protein aggregation play a major role in the acute cell death in the ischemic core region because the neuronal death occurs at an early stage of reperfusion. However, it may be possible that the accumulation of unfolded and damaged proteins in the early stage of reperfusion, which is followed by protein aggregation, contributes to the more delayed neurodegeneration in the incomplete ischemic region after focal ischemia. Consistent with this possibility, it has been shown that the induction of heat shock protein 70 by either viral infection (Wagstaff et al., 1998; Yenari et al., 1998) or transgenic overexpression (Hutter et al., 1996; Plumier et al., 1997; Sharp et al., 1999) before ischemia protects neurons from injury after focal ischemia. Considering that heat shock protein 70 is a molecular chaperone (Fink, 1999), the protection may be mediated by preventing unfolded and damaged proteins from the association with membranous structures or from intermolecular aggregation. Thus, the authors hypothesize that the accumulation of unfolded and damaged abnormal proteins after ischemia exceeds the capacity of the processing machinery such as the chaperone protection or ubiquitin-proteasome degradation, thus they aggregate. Accumulation of unfolded and damaged abnormal proteins, their aggregates, or both, may contribute to the slow or delayed type of neurodegeneration observed after focal ischemia.