
Abstract
Brain pH is precisely regulated, and pH transients associated with activity are rapidly restored under physiological conditions. During ischemia, the brain's ability to buffer pH changes is rapidly depleted. Tissue oxygen deprivation causes a shift from aerobic to anaerobic metabolism and the accumulation of lactic acid and protons. Although the degree of tissue acidosis resulting from ischemia depends on the severity of the ischemia, spreading depolarization (SD) events emerge as central elements to determining ischemic tissue acidosis. A marked decrease in tissue pH during cerebral ischemia may exacerbate neuronal injury, which has become known as acidotoxicity, in analogy to excitotoxicity. The cellular pathways underlying acidotoxicity have recently been described in increasing detail. The molecular structure of acid or base carriers and acidosis-activated ion channels, the precise (dys)homeostatic conditions under which they are activated, and their possible role in severe ischemia have been addressed. The expanded understanding of acidotoxic mechanisms now provides an opportunity to reevaluate the contexts that lead to acidotoxic injury. Here, we review the specific cellular pathways of acidotoxicity and demonstrate that SD plays a central role in activating the molecular machinery leading to acid-induced damage. We propose that SD is a key contributor to acidotoxic injury in cerebral ischemia.
Brief excursus: pH homeostasis and activity-related pH transients in the brain
pH homeostasis and regulation – a prerequisite for proper brain function
The dimensionless “pH” is a term that (approximately) represents the negative decadic logarithm of the proton (or hydrogen ion/H+) concentration in aqueous solutions. In the healthy mammalian brain, extracellular and intracellular pH (pHe/pHi) are around 7.3, which implies a free H+ concentration of about 50 nM in both compartments 1 (Fig. S1). As a result, the Nernst potential for H+ is close to 0 mV, which, due to the negative resting membrane potentials (around −60 mV for neurons and −80 mV for astrocytes), creates a significant electrical gradient that drives H+ into the cells. This requires specific transport mechanisms for the export of protons in order to prevent cellular acidification.1,2 (Fig. S1).
At the same time, brain cells are metabolically highly active and the major pathways for ATP production, glycolysis and mitochondrial respiration, produce acid/base equivalents. In order to maintain their intracellular pH, brain cells must actively secrete acid. Lactic acid, the end product of glycolysis, dissociates into lactate and H+, which are exported across the plasma membrane by monocarboxylate transporters (MCTs) 3 (Fig. S1). Neurons and astrocytes express different MCT isoforms, with neurons expressing MCT2 and astrocytes expressing MCT1/4. A wealth of evidence suggests that lactate produced by astrocytes serves as metabolic substrate for neurons. 4 This concept, however, is also debated as a number of studies have shown that neuronal ATP production and oxidative phosphorylation are not dependent on lactate derived from astrocytes. 5 ,6 Moreover, MCTs, like all secondary-active transporters, have the ability to function in both directions. Depending on the concentrations of its substrates inside and outside the cell, neurons may also release lactate and H+. 7
Moreover, neurons and astrocytes are constantly producing CO2 through mitochondrial oxidative phosphorylation. As a result of the activity of cellular carbonic anhydrases, CO2 is rapidly converted to carbonic acid (H2CO3) and then to H+ and bicarbonate (
pH has a significant impact on most biological processes and reactions, as protons readily bind to proteins and alter their function. Most cellular proteins and peptides are negatively charged because their isoelectric point is around 5, whereas the cytoplasmic pH is around 7.3. Therefore, it is important to counteract major changes in pH.9,10 The optimal pH for many cellular enzymes is around the typical resting pHe/i of 7.1–7.3, and any deviation from this point alters their activity. A prime example is phosphofructokinase, a key enzyme of glycolysis that has an extremely steep pH dependence. Its activity decreases by sixfold with a mere 0.05 pH decrease. 11 Note also that the pH scale is logarithmic, so a seemingly small decrease in pH from 7.2 to 6.9 doubles the free H+ concentration (from 64 to 125 nM). Moreover, when a cell is exposed to a large amount of acid, its buffering capacity may become reduced or even saturated. As a result, any subsequent exposure to H+ will lead to greater pH changes. If brain cells are unable to quickly recover from such acid loads, as in the case of a stroke, the consequences can be catastrophic.
The main acid extruders for neurons and astrocytes are the secondary-active Na+/H+ exchangers of the SLC9 family. The most widely expressed isoform is the Na+/H+ exchanger 1 (NHE1) (SLC9A1)
10
(Fig. S1). In addition, both cell types express
pH regulation involves strong chemical H+-buffering and the transport of acid/base equivalents across plasma membranes. In addition, CO2 produced by the mitochondria can escape from cellular carbonic anhydrases, passively cross membranes and be converted to H+ and
Neuronal activity is accompanied by changes in pH
Due to the high pH buffering capacity of brain cells, one might assume that activity in the healthy brain would not significantly alter pHe and/or pHi. However, it was already described almost 100 years ago that electrical stimulation causes an extracellular acidification in the vertebrate cortex.
19
Since then, studies in various animal models have firmly established that active neurons undergo a reversible decrease in pHi, the amplitude and duration of which depends on the degree and duration of activity.1,2,20 Additionally, magnetic resonance imaging has shown localized acidification accompanying normal brain function in humans.
21
Several cellular mechanisms contribute to activity-induced neuronal acidification, including mitochondrial production of CO2.
22
Moreover, an increase in intracellular Ca2+ and subsequent activation of the PMCA, as well as the efflux of
In contrast to neurons, astrocytes become more alkaline with neuronal stimulation.
25
This is primarily caused by a depolarization-induced activation of inward NBCe1 and the resulting influx of
Disruption of ion homeostasis is a hallmark of the ischemic brain
Brain ischemia causes breakdown of ion homeostasis and membrane potentials
Brain ischemia occurs when blood flow to the brain is reduced or stopped, leading to an inadequate supply of glucose (hypoglycemia/aglycemia) and oxygen (hypoxia/anoxia) to the tissue. Global ischemia, which can occur after cardiac arrest, leads to unconsciousness within less than 10 seconds. 29 Focal ischemia is often caused by thrombosis or embolism blocking an artery supplying a specific area of the brain. 30 Depending on the brain area supplied by the vessel and the duration of occlusion, the resulting ischemic stroke may lead to a range of neurological deficits, from mild to severe. If the ischemia lasts long enough to cause irreversible cell damage, the neurological deficit may be permanent. In the core of a focal ischemic area, cerebral blood flow falls below 15% of normal (<10 ml/100 g/min), while the surrounding ischemic penumbra is defined by a reduction in blood flow to 15–40% (10–35 ml/100 g/min).30,31
Much of our knowledge on brain ischemia and its consequences was gained using different in vivo and ex vivo animal models. Common in vivo models include middle cerebral artery occlusion (MCAO) or photothrombosis in rodents.32,33 Ischemic conditions can also be generated in tissue slices from specific brain regions or in cell cultures. While the latter approaches allow for a superior control of experimental conditions, they inherently lack vascular aspects of brain ischemia. 32 Notably, and as stressed before, 34 the variability in the experimental preparations and conditions used must be carefully and critically considered in the interpretation of the results.
Despite various model systems, it is widely acknowledged that brain ischemia causes drastic shifts in extra- and intracellular ion concentrations, which translate to changes in cellular membrane potentials.29,35 These changes are caused by a rapid decrease in cellular ATP levels, which can drop to less than 15% within a few minutes.17,30 In response to the decreasing ATP/ADP ratio, neuronal ATP-sensitive K+ (KATP) channels open and facilitate K+ efflux (Figure 1). Under hypoxia, K+ efflux also manifests via Ca2+-dependent K+ channels or G protein-regulated K+ channels.36,37 This shift contributes to an initial membrane hyperpolarization, making the neurons less excitable. The malfunction of the neuronal NKA causes a further elevation in the extracellular K+ concentration ([K+]e). This is due to the failing exchange of intracellular Na+ with extracellular K+ by the NKA when ATP is not readily available. Furthermore, failure of the astrocytic NKA causes disrupted clearance of K+. 38 Overall, these processes initiate a gradual increase of [K+]e from 4–5 mM to a ceiling level of 10–12 mM over several minutes after the onset of ischemia, accompanied by a slow depolarization of neurons (Figure 1). As [K+]e progressively approaches 10–12 mM, an abrupt ionic shift occurs, reflected by a sharp increase in [K+]e to 30–60 mM (Figure 1). The sharp rise in [K+]e is coincident with a large, sustained, almost complete collapse of neuronal membrane potentials and electrical silencing.29,39 This event is known as spreading depolarization (SD). A hallmark feature of SD in extracellular electrophysiological recordings is a robust, negative deflection of the local field potential filtered in direct current (DC) mode. This deflection has an amplitude of −15 to −20 mV and appears on adjacent electrodes with a delay that is consistent with the rate of SD propagation, which is typically a few millimeters per minute40 (Figures 1 and 2). In summary, ischemia, which is caused by insufficient blood flow to the brain that leads to a lack of oxygen and nutrients, initiates transmembrane ionic shifts. In turn, SD is thought to occur due to ionic translocations between the intracellular and extracellular compartments. SD is then coupled with a blood flow response that can exacerbate the lack of oxygen and energy substrates. 41
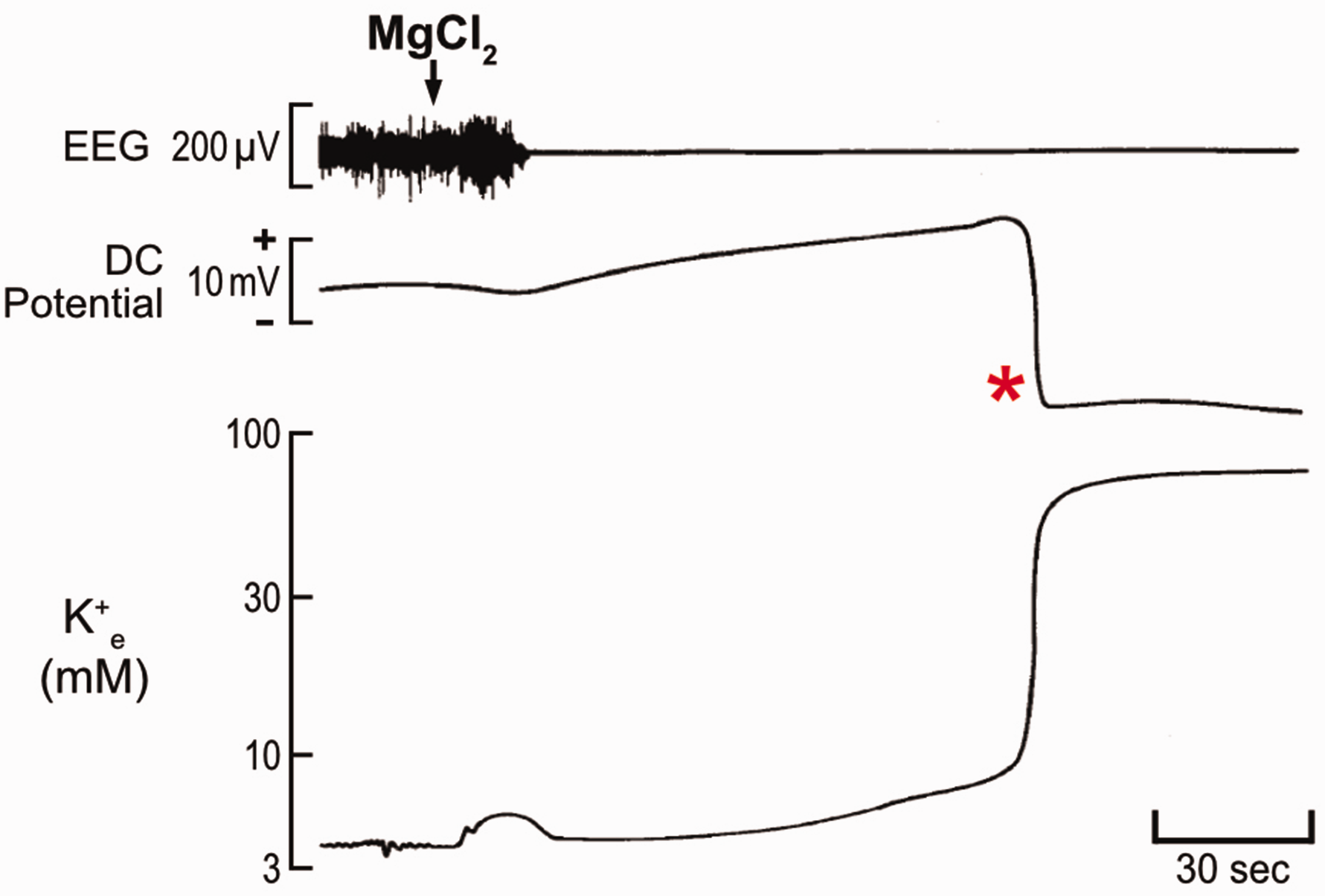
Changes in local field potential and
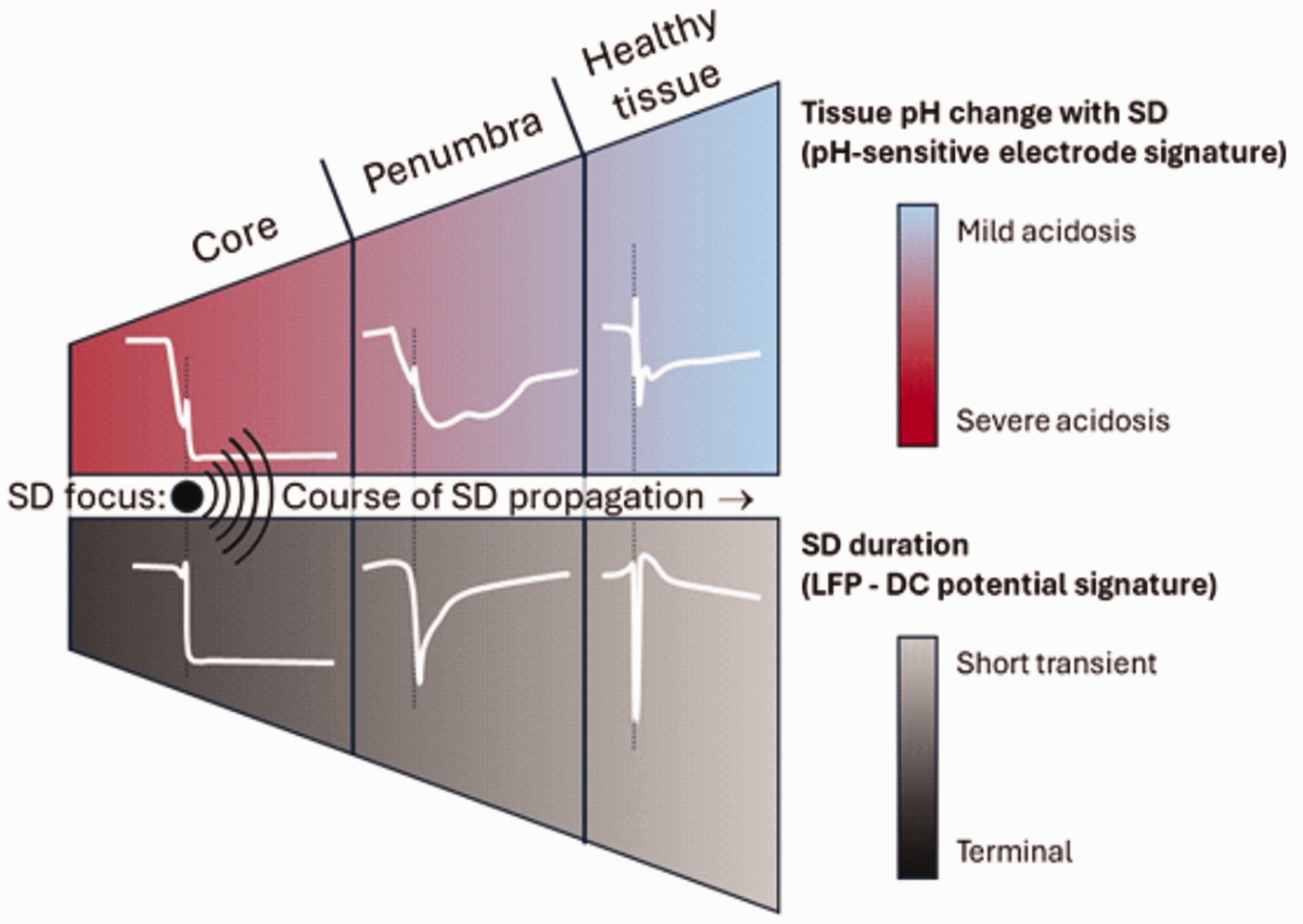
The continuum of spreading depolarization (SD) and extracellular acidification. Irreversible terminal SD is seen in the ischemic core. Transient SD events are observed in the surrounding penumbra and adjacent healthy tissue. The time course and amplitude of changes in extracellular pH essentially reflect this continuum in the different regions.
As mentioned above, marked changes in ion concentrations characterize SD. Extracellular K+ accumulates to 30–60 mM, with a concomitant decrease of neuronal K+ content from about 135 to around 100 mM. Furthermore, there are prominent ionic translocations of Na+ resulting in a decrease in [Na+]e from ∼150 to 60 mM and an increase in neuronal [Na+] from 10 to ∼40 mM. 42 Likewise, [Ca2+]e drops from 1.3 mM to 80 μM, while [Ca2+]i in neurons increases from about 60 nM to as high as 25 μM. SD thus represents the near-complete breakdown of transmembrane ion gradients synchronized in a critical volume of brain tissue (about 1 mm3), and propagates slowly as a wavefront over the gray matter in a self-igniting fashion. 40 Repolarization occurs through the re-establishment of transmembrane ionic gradients in the wake of SD, primarily by NKA, if enough ATP is available. 40
While astrocytes also depolarize in ischemic conditions and with SD, they are generally less susceptible to ischemic damage. This is presumably due to the slower loss of ATP and breakdown of ion gradients in astrocytes compared to neurons. 43 – 48 In addition, astrocytes, but not neurons, possess glycogen stores, indicating that glycolysis and lactate production can continue for a limited time even during complete ischemia to meet their metabolic needs. 49 Lactate production and release by astrocytes may eventually benefit neurons as well. 46 Ongoing glycolysis however also results in continued production of acid equivalents, and the impact of astrocytic glycogenolysis and lactate production on the ischemic brain remains uncertain.17,46,50
The duration of SDs, or the time to repolarization, depends on the metabolic status of the tissue. In energy depleted gray matter, repolarization is delayed, which is caused by the failure of the ATP-dependent NKA. Neuronal NKA thus fails to restore resting neuronal membrane potentials. At the same time, astrocytic NKA fails to support glial K+ clearance. Consequently, the concentrations of [K+]e and [Na+]i remain high, leading to a significant prolongation of SD.51 –53 Severe metabolic crisis and cardiac arrest can result in terminal SD, with no repolarization, indicating irreversible damage (Figure 2). This event was previously known as anoxic depolarization or terminal depolarization and has lately been recognized as a terminal form of SD,53,54 also because of its typical wave-like propagation pattern.55,56
A continuum of SD has been consequently defined, taking into account the specific metabolic status of the tissue involved, the duration of SD, and the potential damage caused53,54 (Figure 2). At one end of the spectrum, persistent, terminal SD is not followed by repolarization. Terminal SD occurs during severe metabolic stress and is associated with irreversible neuronal injury. This type of SD is typical of circulatory arrest or the ischemic core in focal cerebral ischemia. At the other end of the spectrum, short-lasting and innocuous SD propagates in optimally perfused gray matter, such as with migraine aura or in optimally perfused tissue beyond ischemic zones in focal cerebral ischemia. 53 Between these two ends of the spectrum, SD events with postponed repolarization and varying duration spread in ischemic tissue at risk of injury, and carry injurious potential. 53 These stages of the SD continuum can be recognized along the path of a given SD event 53 (Figure 2). In focal ischemia, an SD event is initiated near the ischemic core where it is terminal, but as the very same event propagates through the penumbra to normal tissue, it becomes nearly normoxic with increasing distance from the ischemic region. In other words, the SD spectrum can be appreciated as transition from terminal to normoxic along the path of the same SD.
SDs evolve in a recurrent pattern for at least two weeks after the primary impact in the injured human brain.57,58 Specific features of SD occurrence offer prognostic value for poor outcomes after subarachnoid hemorrhage, malignant hemispheric stroke and traumatic brain injury.59,60 The initial SD in acute experimental ischemia occurs within minutes of vascular occlusion in case tissue perfusion drops below a critical threshold of 20–25% of baseline.61 –63 SDs can also be triggered by hypotension transients or metabolic supply-demand mismatch in tissue at risk of injury. 64 Recurrent SDs propagate at a low rate of millimeters per minute across the ischemic penumbra and their direct injurious potential has been long contemplated.53,65 –67
Ischemia rapidly acidifies brain tissue
In addition to the ionic translocations and membrane potential changes described above, brain ischemia induces intracellular acidosis in neurons and astrocytes.39,44,65,68 –72 This tissue acidification enables non-invasive detection of ischemic areas by pH Magnetic Resonance Imaging in acute stroke in animal models and in stroke patients.73,74 The reported magnitude of pHi changes in animal studies varies depending on the model used.30,46,73 Most studies concluded that the ischemia-induced intracellular acidification was mainly caused by the production of lactate and H+.30,69,75 –77 Furthermore, an increase in partial pressure of CO2 (PCO2) and activation of PMCA in response to Ca2+ influx may contribute to the decrease in pHi.24,69,78
Ischemia activates NHE1, which plays a pivotal role in the export of H+ from neurons and astrocytes into the extracellular space at more acidic pHi.1,79 –84 NHE1 activation has been shown, for example, in hippocampal neurons isolated from rodent brains following brief periods of anoxia.71,85,86 In the healthy brain, acid extrusion by astrocytes is mainly dominated by (inward) NBCe1. In ischemic conditions, however, NHE1 plays a critical role as its contribution increases at lower pHi.68,87 –90 The specific role of NBCe1 in ischemia is still not known in detail.90 –92 A recent study performed in acute mouse brain tissue slices, found that NBCe1 operates in the inward mode during brief chemical ischemia, resulting in reduced astrocytic acidification, increased Na+ influx, and a decline in cellular ATP. 48
Extracellular pH variations caused by brain ischemia or pharmacological inhibition of cellular ATP production follow a temporal progression. A sustained decline in pHe begins within 15–30 seconds and may be preceded by a brief alkaline shift.30,35,44,46,69,79,91,93 –96 Again, in addition to an increase in the PCO2, tissue acidification is mainly attributed to increased cellular glycolysis and production of lactate as opposed to the generation of ATP by mitochondrial metabolism.69,75,95 Consequently, tissue acidosis is markedly exacerbated by hyperglycemia, which can cause pHe to fall below 6.5.35,69,77,78,97,98 The magnitude of extracellular acidosis varies strongly depending on the status of the tissue, but ranges typically between 0.4–0.7 pH units. However, it can be assumed that the wide variation in the degree of tissue acidosis reported in different studies is probably due to the fact that the occurrence of SDs was rarely taken into account, so that when the pH reading coincidentally coincided with or directly followed the occurrence of SDs, more severe acidosis was noted. The following sections highlight the crucial role of SDs in ischemic tissue acidosis.
Spreading depolarization exacerbates tissue acidosis in cerebral ischemia
Extracellular pH variations with spreading depolarization
Vascular occlusion leads to extracellular acidification first with slow progression, which is coincident with the slow extracellular accumulation of K+ (see Fig. S2). If the ischemic challenge is moderate and SD is not detected, tissue pH recovers gradually over a period of 10–15 minutes. 62 However, when perfusion upon vascular occlusion drops sharply below 20–25% and ischemia is severe, SD occurs within a few minutes. The SD onset is accompanied by rapid acidification superimposed on the slow pH reduction.22,39,95 Hypoxia in brain slice preparations causes SD and associated pH variations compatible to what is seen in in vivo.99,100
The SD continuum can be understood at the level of tissue pH, as well. In principle, the duration of the acid shift associated with SD corresponds to the duration of SD. In cases of terminal SD, acidosis becomes irreversible 79 (Figure 3(a)). In malperfused tissue at risk of injury, SD causes substantial and prolonged acidosis with delayed recovery 62 (Figure 3(b)), while in intact tissue with optimally regulated pH, SD is coupled with short, transient acidosis79,101 (Figure 3(c)). These stages form a continuum along the path of an SD. In the core region of a focal ischemic insult, terminal SD is coupled with acidification without recovery. As the SD wave propagates into the penumbra, the acidic shift becomes reversible with increasingly shorter duration as SD travels further away from its site of origin and into optimally perfused tissue (Figures 2 and 3).
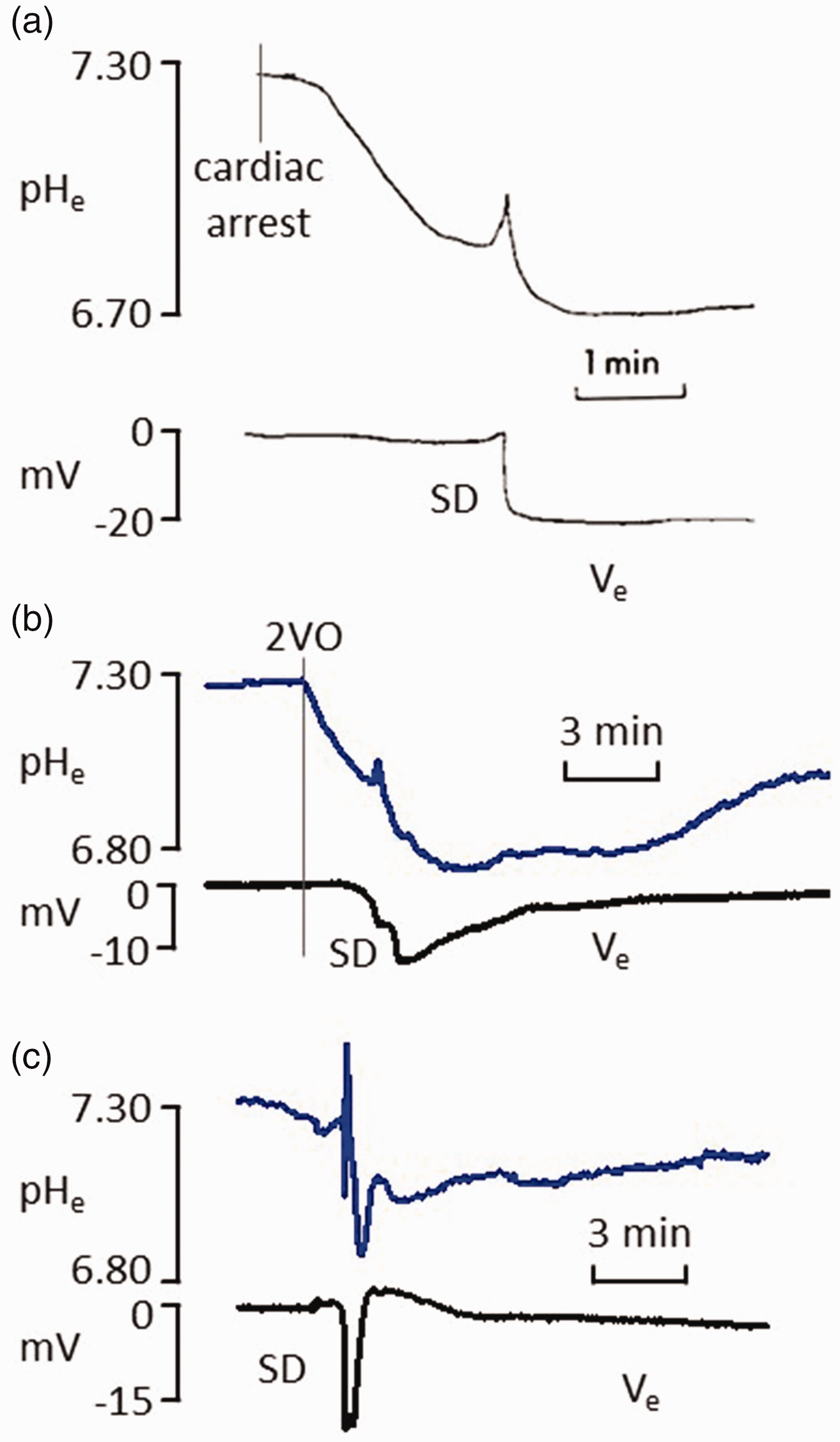
The continuum of extracellular acidification associated with spreading depolarization (SD). (a–c) recordings from the rat cerebral cortex with pH-sensitive microelectrodes (upper traces) and a reference electrode acquiring local field potential filtered in direct current mode (lower traces). (a) pHe gradually decreased after circulatory arrest and settled at its lowest level after terminal SD that arose spontaneously minutes after cardiac arrest. (b) SD occurred spontaneously within minutes after the perfusion drop caused by the bilateral occlusion of the common carotid arteries (2VO). The prolonged SD was followed by a substantial acid load superimposed on the pHe decrease with ischemia onset and (c) SD was triggered experimentally in the optimally perfused rat cerebral cortex. The short-lasting event was coupled with a pHe drop followed by gradual recovery. Traces in (a) are modified and reproduced with permission from Mutch and Hansen, 1984; 79 traces in (b) are modified and reproduced with permission from M Tóth et al., 2020.S57
The complexity of tissue pH variations associated with SD is best understood from experiments in which SD was triggered in the optimally perfused rodent cortex. The depolarization initiates extracellular pH shifts that correspond with the negative DC potential signature of SD.62,79,102 The pHe variation includes an initial brief acidic shift (∼0.1 pH units), a subsequent equally brief alkalotic deflection (∼0.13 pH units) and a concluding prolonged and defining acidosis (∼0.43 pH units). The duration of the acidosis (∼40 s at half amplitude) strongly correlates with the duration of the DC potential deflection62,79,101 (Figure 4(a)). The recovery from tissue acidosis occurs in two phases. The first phase involves an initial steep restitution of pH, along with the repolarization from SD, occurring at a rate of approximately 0.4–0.7 pH units per second. The second phase involves a slow return to physiological tissue pH over a period of 8–10 minutes post-SD62,79 (Figure 4(a)). Extracellular acidosis with SD may build up due to proton export from neurons and astrocytes via the NHE1, as well as the release of lactic acid peaking at 6–8 micromol g−179,103 –105 (Figure 4(b)).
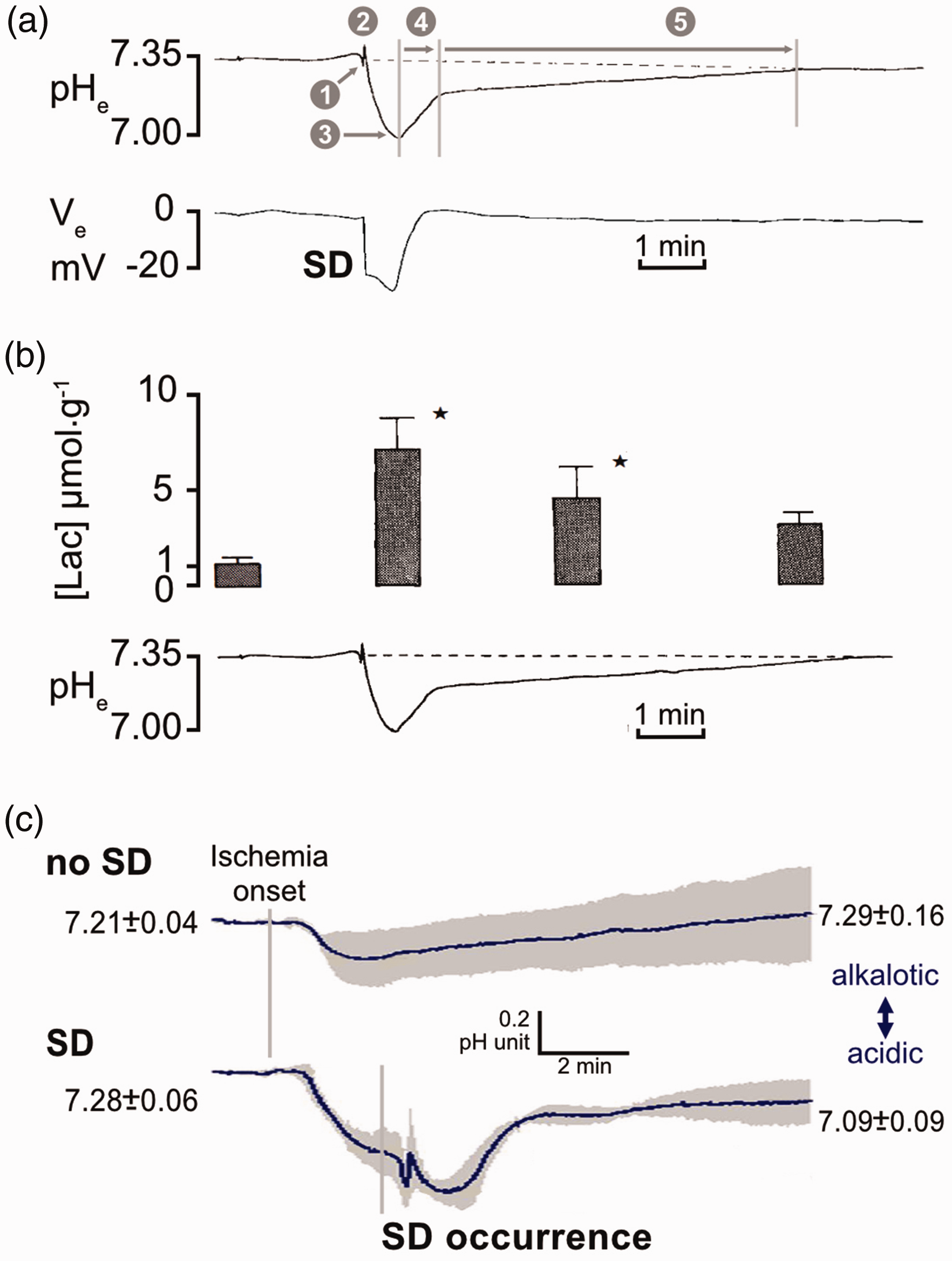
Changes in extracellular pH (pHe) and lactate concentration in the wake of spreading depolarization (SD) in the optimally perfused (a–b) and the ischemic (c) rodent cerebral cortex. (a) Representative traces recorded with microelectrodes to show the subsequent phases of the pHe variation coupled with SD (top) with respect to the direct current (DC) potential signature of SD (bottom); ❶, initial brief acidic shift; ❷, brief alkalotic deflection; ❸, peak of dominant acidosis; ❹, rapid phase of recovery; ❺ slow phase of recovery. (b) Tissue lactate concentration (mean ± stdev; p < 0.05, Student's t-test for paired data) with respect to a representative trace of pHe variations with SD and (c) pHe changes after the onset of global forebrain ischemia (bilateral common carotid artery occlusion) with no SD (top) or aggravated by the spontaneous occurrence of SD (bottom). pH-sensitive microelectrode recordings and pHe values are given as mean ± stdev. (a) and (b) are modified with permission from Mutch and Hansen, 1984; 79 (c) is modified with permission from Menyhárt et al., 2017. 62
At the other end of the SD spectrum, acidification with terminal SD also exhibits reproducible features. An important difference with respect to SD in intact tissue is that the extracellular acidification begins before the occurrence of large, terminal SD-related changes in the extracellular field potential or membrane potentials.22,29,39,95,99 Terminal SD induces an additional rapid, sustained acidification, in some cases again preceded by a brief alkaline shift.22,95,99,106,107 Acidosis with terminal SD is exacerbated by hyperglycemia, which can cause pHe to fall below 6.5.35,69,77,78,108 –110 Finally, a distinctive feature of tissue acidosis with terminal SD is its permanent nature. 79
Experimental data focusing on the ischemic penumbra in the hyperacute phase of cerebral ischemia (0–24 hours after vascular occlusion) revealed tissue pH kinetics similar to those associated with terminal SD, with the only difference in the recovery from acidosis. Gradual tissue acidosis evolved in penumbra-like tissue (perfusion between 20–40%) after vascular occlusion, then the occurrence of spontaneous transient SD within minutes sharply deepened acidosis to a pHe of 6.48 as compared to a pHe of 6.93 in the absence of SD 62 (Figure 4(c)). Recovery from the acidic shift occurred about 90 s later. 62 Furthermore, pHe after SD remained acidic for well over 10 minutes (pHe ∼7.09), whereas pHe at the absence of SD approached physiological values (pHe ∼7.29) over an equivalent time frame 62 (Figure 4(c)).
The mechanism behind acid accumulation during ischemic SDs is not yet fully understood. Nevertheless, extracellular lactate load greatly increased with ischemic SDs compared to the lactate levels measured in ischemic brain tissue prior to SD. 111 Accordingly, SD is believed to cause lactic acidosis in ischemic brain tissue. The kinetics of tissue pH variations with recurrent SDs in the acute phase of stroke (the first two weeks) have not yet been measured. However, microdialysis confirmed a rise in lactate levels with each SD detected between 24 h and 5 days in viable tissue at risk of secondary injury in a patient of malignant hemispheric stroke. 112
Intracellular pH variations with spreading depolarization
Intracellular pH changes with SD in vivo are less well studied than pHe variations. However, fluorescent imaging found that the temporal resolution and the relative amplitude of the SD-coupled acidic shift exhibited a good match between pHi and pHe. 62 Neurons in metabolically intact neocortical brain slice preparations exhibit intracellular acidosis in response to SD induced with KCl, as indicated by a fluorescent pH indicator. 102 Astrocytes, in contrast, showed an increase in pH due to the activation of inward NBCe1 in response to the increase in extracellular K+ induced by SD.25,102,113
The amplitude and duration of the intracellular acidic shift were markedly increased with SD in the ischemic compared to the optimally perfused cortex.
62
Yet, the specific share of neurons and glia cells remains uncertain. The acidification of neurons is widely accepted, while the pHi changes of astrocytes with ischemic SDs remain inconclusive. As described above, there is compelling evidence that astrocytes, like neurons, become more acidic during prolonged ischemia and terminal SD due to a significant loss of ATP. At the same time, the K+-induced depolarization of astrocytes during SDs will – at least initially – activate inward NBCe1, resulting in inward transport of
In conclusion, SD inflicts both extracellular and intracellular acidosis in the ischemic nervous tissue graver than vascular occlusion alone. Furthermore, it is reasonable to assume that recurrent SDs in close succession will lead to delayed recovery in ischemic areas and to gradual decreases in pH after each event, resulting in sustained acidification. Notably, in earlier studies, the degree of tissue acidosis in ischemic brain tissue was usually reported unaware of whether SDs occurred in temporal and spatial proximity to the readings. Therefore, the significance of SD in ischemic acidotoxicity was largely overlooked. The sections below will discuss the cellular mechanisms of acidotoxicity and propose SD as a central event to activate acid-sensing membrane carriers and channels, ultimately leading to acidosis-related injury in cerebral ischemia.
Cellular mechanisms of acidosis-linked ischemic tissue injury
With an increasing diversity of regulated and unregulated cell death types being recognized, an ischemic insult inevitably leads to uncontrolled cation fluxes as the first step towards injury. Intracellular Na+ accumulation draws water into cells, leading to cytotoxic edema, which prompts oncotic cell death. The substantial increase in intracellular Ca2+ concentration activates intracellular signaling cascades that may cause rapid apoptotic or necrotic neural death. Cation translocations are mediated by cation channels and primary and secondary active transport. In ischemia, primary active transport is directly compromised by metabolic failure, and secondary active transport operates less effectively due to the reduced electrochemical gradient of Na+ across the cell membrane. Finally, acidosis, which is typical of cerebral ischemia, quickly activates cation carriers and channels to disrupt ionic homeostasis. 115
The following chapters have compiled experimental evidence from complementary in vivo and ex vivo preparations to illustrate potential cellular mechanisms leading to ischemic acidotoxicity. It must be recognized that these are some of many potential injury pathways in ischemic nervous tissue that may be alternative or ultimately be synergistic. It should also be noted that the following sections are intended to provide mechanistic insight without implying direct clinical translation.
Intracellular acidification and neuronal injury linked to NHE1 and NCX
Although mild-to-moderate acidification has been recognized as neuroprotective, more severe ischemia-related tissue acidosis can cause neuronal injury. The injury is imposed, in part, by the saturation or the overstimulation of the molecular machinery that extrudes intracellular protons. 116 Internal acidification of neurons quickly activates the plasmalemmal electroneutral and voltage-independent NHE1. 117 NHE1 is a secondary active transporter that moves protons out of the cell, energized by the downhill influx of Na+. Protons not only serve as the substrate of the transporter but also act as its allosteric activator. 118 Consequently, NHE1 activity increases rapidly as pHi decreases, reaching a half maximal activation (pH50) at pHi 7.0–6.8 and maximal activation around pHi 6.6. 119 Interestingly, a rising intracellular Ca2+ concentration, which is typical for neurons at risk of ischemic injury, has been reported to activate NHE1 as well, via Ca2+/calmodulin (CaM) binding to NHE1. Ca2+/CaM binding relieves the autoinhibition of the transporter120,121 or augments transport efficacy by NHE1 dimerization. 122
The removal of intracellular acid load by NHE1 aims to restore physiological pHi to support normal cell function. However, pre-clinical models of cardiac or cerebral ischemia/reperfusion suggest that NHE1 may exacerbate injury.83,90 First, it appears likely that NHE1 function saturates during the progression of ischemia, rendering it incapable of alleviating intracellular acidosis (Figure 5(a)). As H+ are exported from cells, the extracellular space becomes increasingly more acidic during ischemic acidosis. The extrusion of additional H+ then can be impeded by the activation of an acid-sensitive extracellular inhibitory site of NHE1 or an increase in the electrochemical gradient of H+. 123 Second, limited ATP availability during ischemia hinders energy-dependent Na+ extrusion by the NKA, significantly reducing the Na+ gradient that powers NHE1. 124 This reduction is particularly relevant during SD, 40 especially when it is prolonged. 51 Of note, [Na+]e may decrease from 150 mM to 60 mM during SD. 40 In the mouse brain, two-photon Na+ imaging detected a transient increase in [Na+]i of more than 20 mM during the passage of an SD in the ischemic penumbra. 124 These findings suggest that NHE1 confronted with a waning driving force must become quickly overwhelmed, leading to a sustained intracellular acid load (Figure 5(a)).
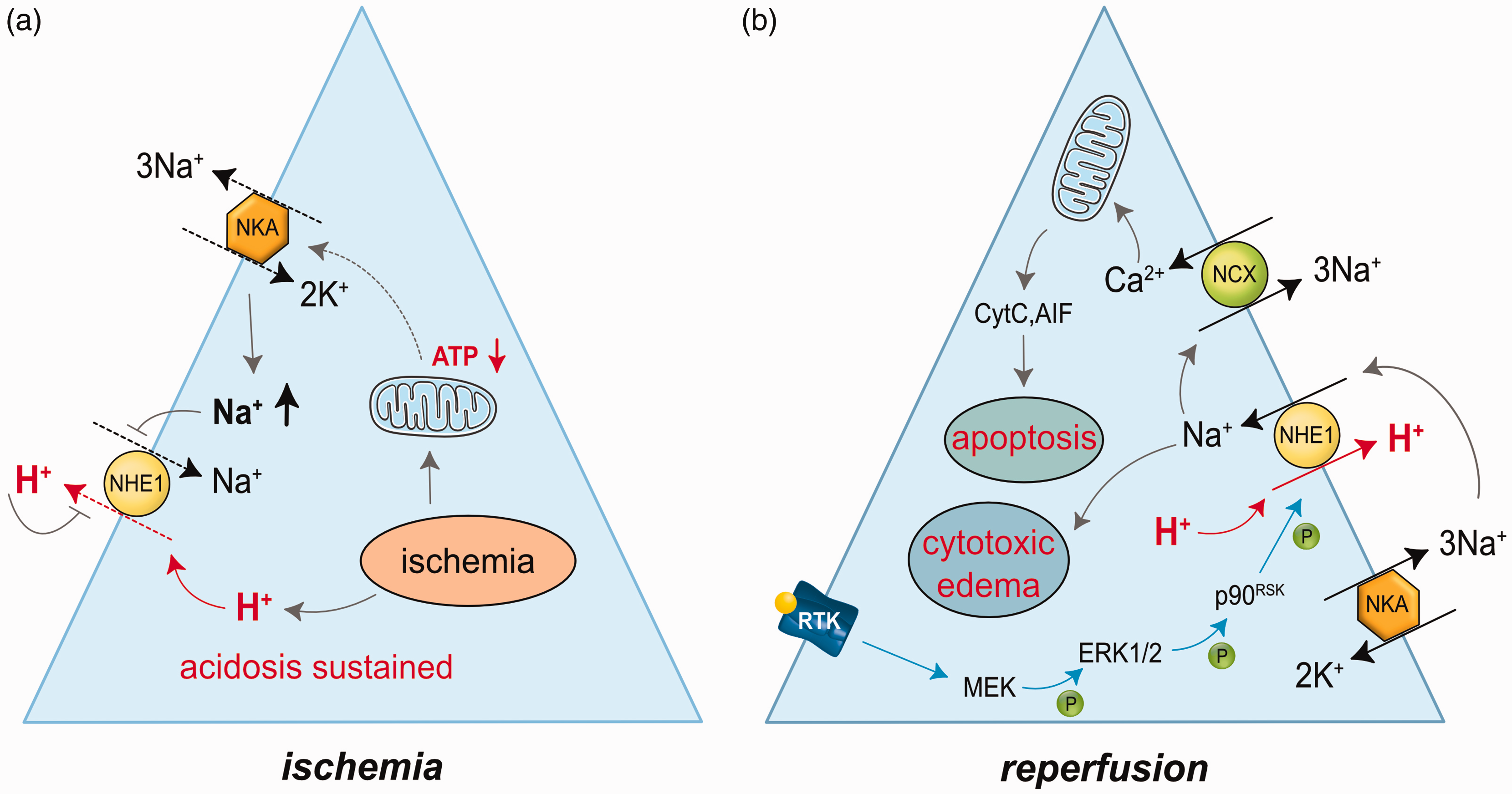
Conceptual neuronal injury cascades linked to impaired Na+/H+ exchanger 1 (NHE1) function under ischemia (a), and enhanced NHE1 activity during reperfusion (b). (a) The transmembrane electrochemical gradients of Na+ and protons gradually decrease and increase, respectively, in ischemic neurons because of the dysfunction of the Na+/K+ ATPase (NKA) and the progressive accumulation of protons in the interstitium. Consequently, NHE1 function becomes impeded, resulting in a sustained intracellular acid load. (b) NHE1 is overactivated in reperfused tissue governed by the re-established Na+ and proton transmembrane gradients. Resumed NKA activity pumps Na+ to the extracellular space, and reperfusion of the tissue removes extracellular protons. Moreover, NHE1 activity is upregulated by a phosphorylation chain leading to the phosphorylation of NHE1 itself. The intracellular signaling cascade is initiated by receptor tyrosine kinase (RTK) activation. Overdriven NHE1 function contributes to intracellular Ca2+ accumulation possibly via reversal of the Na+/Ca2+ exchanger (NCX). The Ca2+ load eventually compromises mitochondrial ion regulation and initiates apoptotic cell death cascades by the release of cytochrome C (CytC) and apoptosis-inducing factor (AIF). Abbreviations: ERK1/2, extracellular signal-regulated kinase 1/2; MEK, mitogen-extracellular kinase; p90RSK, 90-kDa ribosomal S6 kinase.
On the other hand, NHE1 extrudes protons effectively after the resolution of ischemia; 125 in fact, NHE1 function may be overdriven (Figure 5(b)). When tissue perfusion or oxygenation is restored, NHE1 function may overshoot due to renewed NKA activity and restored Na+ gradient, leading to intracellular alkalosis. This, in turn, increases neuronal excitability and is ultimately harmful to nervous tissue. 126 Supporting this concept, pHi of cultured neurons exhibited an alkalotic shift during reoxygenation after oxygen-glucose deprivation (OGD), which was prevented by selective NHE1 inhibition. 125 The sustained operation of NHE1 in reperfused or reoxygenated preparations is widely believed to contribute incessant Na+ uptake because of the exchange of a proton for a Na+. In turn, the elevating [Na+]i will either weaken Ca2+ export by the Na+/Ca2+ exchanger (NCX) or outright reverse NCX, which will then extrude Na+ in exchange for the intake of Ca2+. 127 This can trigger cell death cascades, especially when combined with Ca2+ influx through other routes like ionotropic glutamate receptors and Ca2+ channels. Such Na+-driven reversal of NCX has been recently demonstrated during the passage of an SD in the ischemic penumbra of mice after permanent MCAO, as well as in acutely isolated tissue slices exposed to transient energy deprivation. 124
Supporting this notion, in cortical neuron cultures exposed to OGD, neuronal somata and dendrites showed a progressive increase in [Na+]i from 10–15 mM to 45–55 mM over 60 minutes after reoxygenation.83,125 Pharmacological NHE1 inhibiton or cultivating neurons from NHE1 knock-out mice prevented intracellular [Na+]i accumulation.83,125 These experiments established an NHE1-related Na+ overload of neurons during reoxygenation. Under the same conditions, a marked increase of [Ca2+]i was also seen from about 70 nM to 500–1000 nM in somata and reaching 0.8–1.6 µM in dendrites, which was abolished with NHE1 inhibition.83,125 It was concluded that NCX operating in a reverse mode was concurrent with NHE1 activation in reoxygenated neurons after ischemia. 125 The reverse operation of NCX during reoxygenation remains, however, contentious because NCX may quickly regain its forward mode in neurons with re-established NKA function in reperfused brain tissue. 128
To explore whether the excessive NHE1 activity was implicated in neuronal injury, the degree of cell death was evaluated in neuron cultures. The pharmacologic inhibition of NHE1 or using neurons obtained from NHE1-k.o. mice reduced cell damage induced by OGD-reoxygenation or glutamate.83,129,130 Furthermore, the NHE1 inhibitors, or experimenting with NHE1+/− heterozygous mice reduced infarct size in vivo after experimental focal cerebral ischemia.83,130,131 NHE1 inhibition achieved neuroprotection and relieved striatum-dependent motor and spatial learning impairment after experimental perinatal hypoxia/ischemia. 132 Taken these data together, NHE1 activation after the resolution of ischemia and reperfusion of the tissue appears to be a mechanism responsible for neuronal injury.
Next, intracellular signaling pathways that mediate NHE1-associated neuronal injury were examined. Na+ overload can lead to cytotoxic edema. Indeed, dendritic beading was prominent after OGD and was markedly attenuated by NHE1 inhibition. 125 Another plausible consequence of NHE1 overactivation is mitochondrial Ca2+ dyshomeostasis. Post-ischemic NHE1 activity was found coincident with mitochondrial Ca2+ loading through the mitochondrial Ca2+ uniporter and decreasing mitochondrial membrane potential. 125 Further, transient focal cerebral ischemia in NHE1+/− mice decreased the release of cytochrome C and apoptosis-inducing factor from mitochondria. Active caspase 3 levels and apoptosis were also reduced. 133 These data together suggest that increased [Ca2+]i, generated indirectly by NHE1, contributes to delayed neuronal injury after transient ischemia, due to an alteration of mitochondrial Ca2+ homeostasis and the linked apoptotic cell death cascades (Figure 5(b)).
Finally, based on insights gained from cardiac ischemia models, the overexpression of the NHE1 protein or the upregulation of NHE1 activity emerge as potential mechanisms (Figure 5(b)). Increased NHE1 protein levels were confirmed 1–4 h after reoxygenation of OGD-treated neuron cultures. 133 Yet, the expression of NHE1 did not consistently increase in experimental focal cerebral ischemia. 133 Since NHE1 phosphorylation by the serine/threonine kinase p90RSK (90-kDa ribosomal S6 kinase) enhances NHE1 activity, 134 this pathway was further investigated. The activation of p90RSK by phosphorylation is achieved directly by ERK1/2 (extracellular signal-regulated kinase 1/2) downstream to MEK (the mitogen-extracellular kinase). The transient phosphorylation of ERK1/2, p90RSK, and NHE1 occurs early in cortical and striatal neurons following focal cerebral ischemia, with a peak at 3–10 minutes into reperfusion 135 (Figure 5(b)). These experiments suggest that NHE1 phosphorylation plays a role in cerebral ischemic injury, as demonstrated by the reduction in NHE1 expression and infarct size with the p90RSK inhibitor fluoromethyl ketone. 135
Extracellular acidification and neuronal injury linked to ASICs
Acid sensing ion channels (ASICs) are proton-gated voltage-independent cation channels that allow Na+ influx into cells upon activation. 136 In neurons, the ASIC1a splice variant is the most common of the three known subunit variants, occurring either in homotrimeric form or in heterotrimeric combinations with ASIC2a or ASIC1a/2b. 137 The ASIC1a homotrimeric channel composition is relevant for acidotoxicity in neurons, apparently because ASIC1a conducts the influx of Ca2+ in addition to Na+. 137 – 139 Homotrimeric ASIC1a exhibits the highest sensitivity to protons, becoming activated when the pH drops steeply below ∼6.9. Maximal activation is reached at pHe 6.0, with pH50 at pHe 6.2–6.5.137,139 The pH sensitivity range of ASIC1a is characteristic of tissue acidosis that occurs during cerebral ischemia. Taken together, the Ca2+ permeability and the pH sensitivity of ASIC1a have made it a subject of extensive research into cellular mechanisms and therapeutic targets of acidosis-induced neuronal injury in ischemic stroke.116,139 –141 In contrast to NHE1, ASIC1a activity and related acidotoxic neuronal injury centers on ischemia rather than reperfusion.
The fundamental role of ASIC1a in acidotoxic neuronal injury was demonstrated in neuron cultures exposed to low pH. 139 This work has shown acidosis-induced, ASIC1a-dependent Ca2+ accumulation and related injury in neurons, which was blocked by the non-selective ASIC blocker amiloride and the ASIC1a-selective PcTx1 venom peptide 139 (Figure 6(a)). Pharmacological inhibition of ASIC1a or genetic ablation of ASIC1a has been shown to reduce infarct volume in experimental focal cerebral ischemia.139,142 –144 A compelling aspect of pharmacologic ASIC1a inhibition is that neuroprotection was achieved in experimental stroke when the blockers were administered in clinically realistic time windows, up to 8 hours post-ischemia onset.142 –144 The translational relevance of ASIC1a blockage is increased further by a recent study that evaluated and proved the efficacy of an antibody treatment against ischemic injury in which the treatment successfully kept ASIC1a downregulated in ischemic brain tissue. 145
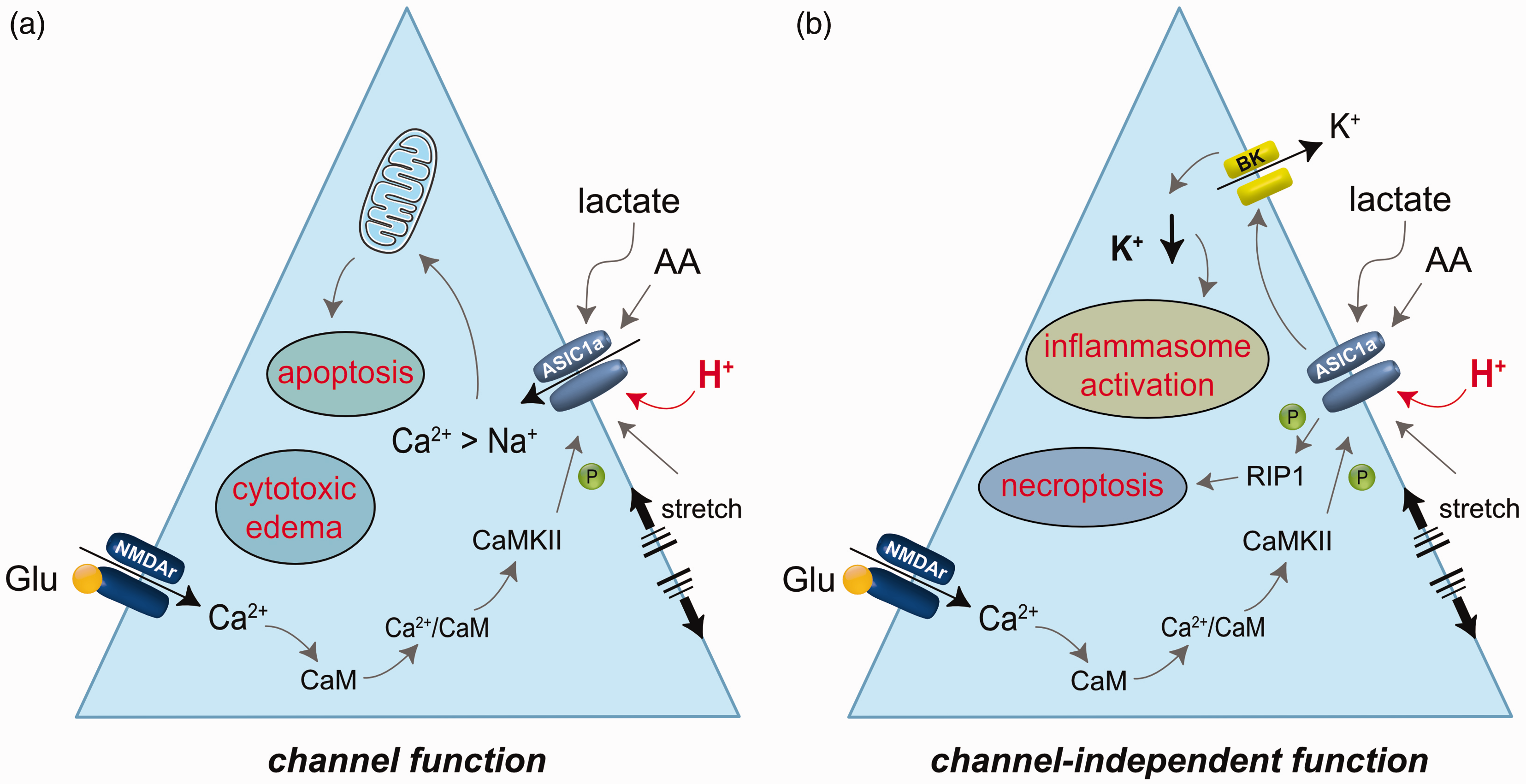
Conceptual neuronal injury cascades linked to pathophysiological activation of acid-sensitive ion channel 1a (ASIC1a). Injury mechanisms can be triggered by inward cation currents (a) or channel-independent function (b). In addition to the activation of ASIC1a by a sharp increase of extracellular proton concentration, ASIC1a currents are potentiated by membrane stretch, lactate and arachidonic acid (AA) accumulation. ASIC1a phosphorylation by Ca2+-calmodulin (CaM)-dependent protein kinase II (CaMKII), which has been linked to the activation of and Ca2+ influx through the NR2B subunit of NMDA receptors (NMDAr) also enhances ASIC1a current. (a) The ASIC1a cation current in response to an abrupt pHe drop is created by the transient influx of Na+, and to a lesser degree, Ca2+ down their concentration gradients. Intracellular Na+ accumulation may attract water and contribute to cellular swelling (cytotoxic edema). A transient intracellular Ca2+ surge may add to intracellular Ca2+ accumulation occurring via ionotropic receptors and contribute to mitochondrium-associated apoptotic cell death. (b) Channel-independent ASIC1a function upon acid exposure may trigger necroptosis by receptor interacting protein 1 (RIP1) recruitment to ASIC1a and RIP1 phosphorylation. Alternatively, ASIC1a may augment K+ efflux via large conductance Ca2+ activated K+ (BK) channels. The resulting decrease of intracellular K+ concentration has been suggested to contribute to NLRP1 inflammasome activation and the linked production of pro-inflammatory cytokines and neuroinflammation.
These studies have suggested the involvement of ASIC1a in ischemic brain injury. However, the original hypothesis that homomeric ASIC1a-mediated calcium influx must be strong enough to trigger acidotoxic cell death cascades 139 (Figure 6(a)) seems to contradict some of the inherent properties of ASICs. First, the Ca2+ permeability of ASIC1a is quite low compared to its Na+ permeability (PNa/PCa >15), or to Ca2+ influx through other ionotropic receptors, such as the NMDA receptor.137,146 Second, ASIC1a activation by a pH drop is followed by rapid desensitization within a few seconds. 147
The observation that ASIC function can be augmented during ischemia may resolve some of the controversy (Figure 6). The amplitude of the ASIC current was found enhanced, and the desensitization of the channel delayed when acidosis concurred with ischemia. 148 For instance, pHe reduction in the presence of high lactate or arachidonic acid concentration typical of ischemia, membrane stretch (neuronal swelling), or OGD itself potentiated the ASIC1a current in cortical neuronal or cerebellar Purkinje cell cultures139,149 (Figure 6). Furthermore, about a third of cultured Purkinje cells showed a sustained, inward current at a level of about 10% of the peak current at reduced pHe. 149 Additionally, during experimental global ischemia, the ASIC current increased due to the phosphorylation of ASIC1a by Ca2+-calmodulin-dependent protein kinase II (CaMKII). 150 ASIC phosphorylation was associated with the activation of the NR2B subunit of NMDA receptors 150 (Figure 6). Collectively, ASIC channels are suggested to conduct more prominent acid-induced cation current under ischemic conditions.
Alternative injury cascades triggered by ASIC1a activation, independent of its channel function, have also been explored (Figure 6(b)). For instance, the serine/threonine kinase RIP1 (receptor interacting protein 1) was shown to be recruited to the intracellular C-terminus of ASIC1a upon acid exposure and became activated by phosphorylation. 131 ,S1 Activated RIP1 plays a crucial role in triggering necroptosis, a form of regulated necrotic cell death. 131 ,S2 In other neuron culture experiments, ASIC1a activation increased large-conductance Ca2+-activated K+ (BK) channel currents. 131 The reduced intracellular K+ concentration was then implicated in NLRP1 inflammasome activation 131 (Figure 6(b)), promoting the release of pro-inflammatory cytokines and contributing to neuroinflammation in ischemic states.S3
Injury induced by astrocytic changes in pH
Compared to neurons, the role of pHi changes in astrocytic injury is less well understood. Despite being generally considered less prone to ischemic injury, astrocytes also undergo massive cell death in the core region of ischemic stroke.S4–S6 Within a few hours astrocytes exhibit hypertrophy and retraction of processes, as well as increased expression of GFAP, forming a persisting glial scar at the border to the infarct. These changes decrease in severity at more distant sites.S 4 ,S7,S8
Astrocytes play a critical role in regulating extracellular neurotransmitters and ion homeostasis, and changes in their function can worsen neuronal damage.S
9
,S10 A decrease in astrocytic ATP, for example, reduces their ability to take up K+, eventually driving
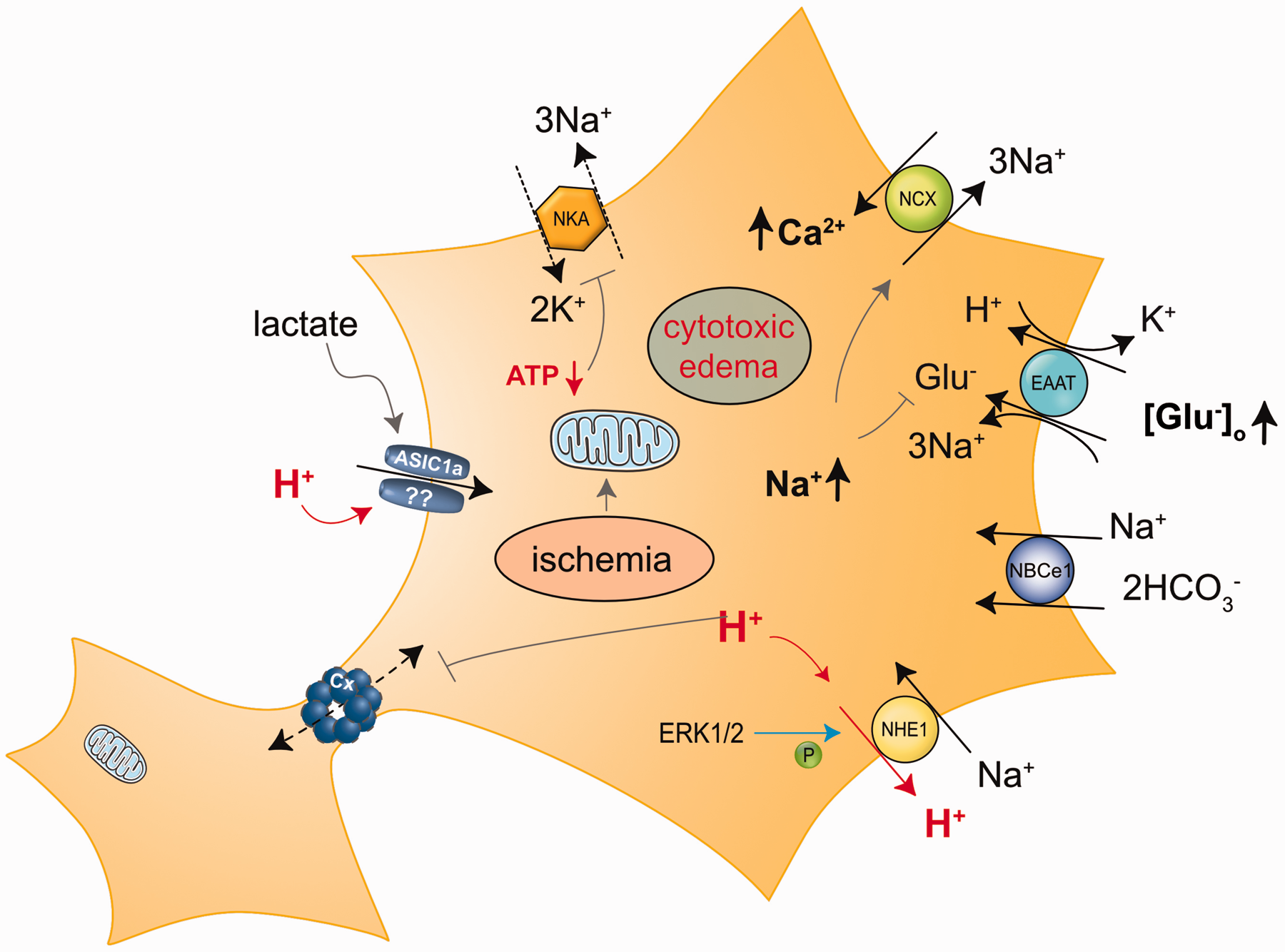
Conceptual injury cascades linked to ischemia-induced acidosis in astrocytes. Reduced cellular ATP attenuates NKA while intracellular acidification activates NHE1 and NBCe1, all leading to an increase in intracellular Na+. Extracellular acidification may putatively also activate ASIC1a, resulting in further Na+ influx and depolarization of astrocytes. Depolarization and Na+ increase reduce the driving force for glutamate uptake through EAAT, resulting in an increase in extracellular glutamate, and cause reversal of NCX, resulting in Ca2+ loading of astrocytes. Cellular acidification and Ca2+ increase decrease the permeability of connexin-based gap junctional coupling between astrocytes. Abbreviations: ERK1/2, extracellular signal-regulated kinase 1/2; p90RSK, 90-kDa ribosomal S6 kinase.
In addition, acidification of astrocytes promotes tissue damage. A decrease in astrocytic pHi reduces their ability to buffer changes in pHe and aggravates the acidification of the ECS.1,2,15,S18 Glial acidification reduces the capacity for glutamate uptake and may induce its release from astrocytes, promoting glutamate-induced excitotoxicity 72 ,S19–S21 (Figure 7). Due to the strong pH-dependence of connexinsS22, the conductance of hemichannels and gap junctions decreases and astrocytes uncouple during prolonged metabolic inhibitionS23–S26 (Figure 7). The consequences of such acidification-induced astrocyte uncoupling for the propagation of SDs in the ischemic brain are still unclear.S27–S29
Notably, astrocyte acidosis also plays an important role in ischemia-induced astrocyte cell death itself. 87 Astrocytes exposed to low pHe are unable to maintain their pHi, leading to swelling, acidification, and eventually to cell death.44,87,97,S30,S31 Cultured astrocytes can survive prolonged periods of severe hypoxia, however, when hypoxia is combined with acidosis, it significantly increases cell death. 96 ,S5,S32,S33 There is also a clear correlation between the degree and duration of changes in pHi and cell damage. While brief and mild acidification may not immediately cause cell damage, astrocytes are highly susceptible to cell death when exposed to short periods of strong acidification, as well as in response to longer periods of less severe acidification. 97 ,S34,S35
The mechanisms that induce acidification-mediated injury to astrocytes include impaired mitochondrial respiration, increased formation of free radicals, oxidative stress, and inhibition of cellular metabolism. 68 In addition, and as established for neurons, both carrier-mediated and channel-mediated injury mechanisms have been reported, with the former clearly taking center stage.90,116,S8
Carrier-mediated injury of astrocytes
As described above, NHE1 is strongly associated with neuronal injury cascades. In addition, NHE1 also plays a pivotal role in acidosis-induced astrocyte cell damage. Pharmacological inhibition of NHE1 prevented cell death in cultured astrocytes exposed to a medium that mimics ischemic conditions in the intact brain. 68 The authors suggested that activation of NHE1 in response to cellular acidification leads to an increase in astrocytic [Na+] and a reversal of the NCX, resulting in Ca2+-loading and Ca2+-induced cell death.68,87,S36 According to this concept, ischemia triggers a series of ion dysregulation events. The initial insult, an increase in astrocytic H+, leads to an increase in [Na+]i, which drives intracellular Na+ accumulation which causes an overload of Ca2+ (Figure 7).
The Sun laboratory's work strongly supports this view. 90 Inhibiting or genetically deleting NHE1 in astrocytes not only prevented recovery from intracellular acidification, but also reduced the increase in astrocytic Na+ and their swelling after 2 hours of oxygen-glucose deprivation and 1 hour of reoxygenation. 82 The group later demonstrated that the NHE1-related Na+ increase caused Ca2+ overload through reverse NCX, resulting in the loss of mitochondrial membrane potential and cytochrome C release.S 19 ,S37 Activation of the ERK1/2 signaling pathway partly contributed to the stimulation of NHE1 in astrocytes exposed to oxygen-glucose deprivationS38 (Figure 7). Global ischemia moreover resulted in a rapid upregulation of astrocytic NHE1 protein levels.S19,S39 Later on it was demonstrated that animals with an astrocyte-specific deletion of NHE1 showed reduced brain edema, reduced astrocyte gliosis, smaller infarct volumes and milder neurological deficits following transient MCAO.S40 Altogether, these results strongly suggest that astrocyte acidification and stimulation of NHE1 in brain ischemia contribute to immediate astrocyte ion dysregulation, reactive astrogliosis, and neurovascular damage 90 ,S40 (Figure 7).
NBCe1 is the second acid/base transporter that is likely to contribute to ischemia-induced ion dysregulation and cellular damage in astrocytes. As previously stated, cellular depolarization, such as that observed with non-ischemic SDs, activates inward NBCe1, resulting in the inward transport of Na+ and
Channel-mediated injury of astrocytes
The involvement of specific ion channels in pH-mediated astrocytic injury is less clear. Mature astrocytes express high levels of inwardly-rectifying Kir4.1 channels. These have a high open probability at rest and are involved in setting the highly negative membrane potential.S43 The conductance of Kir4.1 channels is significantly reduced in acidic conditions, resulting in cellular depolarization.S44 In contrast, TREK-1 channels, which belong to the two-pore domain K+ channels, are activated by a decrease in pH. Based on these opposing effects, astrocytes may maintain overall K+ conductance during brief reduction in pHe.S44,S45
While it is well-established that ASICs play a role in causing damage to neurons during ischemia, the relevance of ASICs in astrocytes is not yet fully understood. Some evidence suggests that ASICs are expressed in various types of glial cells, including astrocytes.S46 In cultured primary rat astrocytes, decreasing pHe to 6.0 induced fast inward ASIC-like currents. In addition, immunolabeling for ASIC1, ASIC2a and ASIC3 was found on astrocytes both in culture and in brain tissue slices, albeit predominately in the nucleus.S47 Additionally, more recent studies have detected ASIC1a expression in a fraction (10–15%) of mouse hippocampal astrocytes.S48 Expression levels of ASIC1a significantly increased in GFAP-positive reactive astrocytes of epileptic mice.S48 The latter study furthermore showed that a decrease in pHe to 6.0 induced an increase in astrocytic Ca2+ levels, which was at least partly mediated by ASCIC1a. Finally, the frequency of spontaneous seizures was reduced by downregulating astrocytic ASIC1a, indicating that its activation plays a role in epilepsy's pathogenesis.S48 These findings suggest that ASICs may contribute to acid-induced damage to astrocytes (Figure 7). Nevertheless, additional studies are necessary to confirm this hypothesis.
Conclusion: the intersection of spreading depolarization and acidotoxicity
A considerable body of knowledge has accumulated on cerebral ischemic tissue acidosis and acidotoxic neuronal injury over the past 50 years. However, providing a concise and accurate mechanistic insight into ischemic tissue acidosis and subsequent toxicity remains challenging due to the wide variation in experimental approaches and results in this area. A greater reduction in pH in ischemic brain tissue has traditionally been associated with a more severe metabolic challenge and higher blood glucose levels.35,69,77,78,108 –110 Here, we propose that the occurrence of SD, especially in a repeated pattern is an additional, strong determinant of acidosis. Importantly, SD is apparently required to lower tissue pH below a threshold necessary to activate acid carriers or proton-activated ion channels implicated in cell damage. This notion is supported by the good correspondence between the low tissue pH levels reached during SD (pH 6.5–6.8) 62 and the activation thresholds of NHE1 (activation below pH 6.8–7.0, maximal activation at pH 6.6) 119 or ASIC1a (activation below pH 6.9, maximal activation at pH 6.0).137,139
The concept that NHE1 becomes activated with SD was supported by the recording of a smaller extracellular acid shift following the pharmacological inhibition of NHE1. 79 Regular NHE1 function would imply Na+ influx in exchange for H+ extrusion. Intracellular Na+ accumulation is a typical feature of SD,40,42,S13 and the contribution of NHE1 is likely, but remains to be determined. Na+ transport in exchange for H+ by NHE1 may osmotically attract water into both neurons and astrocytes or drive reverse NCX and thereby contribute to damaging Ca2+ loading (Figures 4 and 5). Both have been recognized as injury mechanisms. On the other hand, Na+ accumulation within neurons with SD probably occurs via voltage-gated and ligand-gated Na+ channels and is most likely augmented by reduced NKA efficacy under ischemia. 40 ,S49 The increase in [Na+]i may reduce NHE1 activity due to the reduced Na+ gradient that drives NHE1.S13 This may cause suboptimal NHE1 function and acid retention in cells with potentially damaging consequences linked to SD (Figure 4). Future work on this topic could reveal whether the stimulation or the inhibition of NHE1 activation is dominant with SD under ischemia, and how NHE1 manipulation may modulate subsequent injury.
Regarding ASIC1a, no direct experimental evidence has been reported yet for its function in the context of SD. Intraneuronal Ca2+ concentration increases with SD in brain slices exposed to OGD and in experimental focal cerebral ischemia,S13,S50,S51 and is thought to surge in neurons via ionotropic glutamate receptors,S 50 ,S51 L-type voltage-gated Ca2+ channels,S52 reverse NCXS13 and conceivably also ASIC1a (Figure 5). This latter possibility is intriguing, unexplored and deserves further investigation.
It has been suggested that recurrent SDs have significant potential to exacerbate ischemic injury. 53 Proposed mechanisms of injury include ATP depletion,S53 high concentration glutamate release and/or failure of glutamate uptake,S14 intraneuronal Ca2+ accumulation,S50 cytotoxic oedema formationS14,S54 and neuroinflammation.S55 In conclusion, we argue that SD emerges as a key condition for igniting acidotoxicity. In turn, acidotoxicity is proposed as an additional and potent contributor to SD-related injury in cerebral ischemia.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X241289756 - Supplemental material for A dangerous liaison: Spreading depolarization and tissue acidification in cerebral ischemia
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X241289756 for A dangerous liaison: Spreading depolarization and tissue acidification in cerebral ischemia by Eszter Farkas and Christine R Rose in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the German Research Foundation (DFG), Research Unit RU2795: “Synapses under stress” (CRR: Ro2327/13-2, EF: Mercator Fellowship); the EU’s Horizon 2020 research and innovation program No. 739593; the National Research, Development and Innovation Office of Hungary (No. K134377); and the University of Szeged Open Access Fund (Nr. 6939).
Acknowledgements
We thank Claudia Roderigo and Dr. W. Karl Kafitz (HHU) for help with the figures.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
EF and CRR wrote the manuscript and provided funding.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.