
Abstract
Cerebral ischemia resulting from a disruption of blood flow to the brain initiates a cascade of events that causes neuron death and leads to neurologic dysfunction. Reactive oxygen species are thought, at least in part, to mediate this disease process. The authors recently cloned and characterized an antioxidant protein, SAG (sensitive to apoptosis gene), that is redox inducible and protects cells from apoptosis induced by redox agents in a number of in vitro cell model systems. This study reports a neuroprotective role of SAG in ischemia/reperfusion-induced brain injury in an in vivo mouse model. SAG was expressed at a low level in brain tissue and was inducible after middle cerebral artery occlusion with peak expression at 6 to 12 hours. At the cellular level, SAG was mainly expressed in the cytoplasm of neurons and astrocytes, revealed by double immunofluorescence. An injection of recombinant adenoviral vector carrying human SAG into mouse brain produced an overexpression of SAG protein in the injected areas. Transduction of AdCMVSAG (wild-type), but not AdCMVmSAG (mutant), nor the AdCMVlacZ control, protected brain cells from ischemic brain injury, as evidenced by significant reduction of the infarct areas where SAG was highly expressed. The result suggests a rather specific protective role of SAG in the current in vivo model. Mechanistically, SAG overexpression decreased reactive oxygen species production and reduced the number of apoptotic cells in the ischemic areas. Thus, antioxidant SAG appears to protect against reactive oxygen species–induced brain damage in mice. Identification of SAG as a neuroprotective molecule could lead to potential stroke therapies.
Stroke resulting from the disruption of blood flow to the brain is the third most common cause of mortality and the major cause of disability in developed countries. Blood flow disruption induces focal cerebral ischemia that initiates a cascade of molecular events leading to neuronal death followed by neurologic dysfunction. Among several mediators of ischemic cell death, reactive oxygen species (ROS) play an important role (Chan, 1994, 1996; Facchinetti et al., 1998).
Reactive oxygen species are a group of very reactive and short-lived chemicals, consisting of superoxide anion, hydrogen peroxide, hydroxyl radicals, organic peroxides, and others. They are produced in the reduction of oxygen to water during normal respiration and after oxidative insults, such as exposure to irradiation, UV, and chemicals (Sun, 1990). During cerebral ischemia/reperfusion, oxygen supply is first significantly limited, followed by an oxygen overflow. Reoxygenation during reperfusion provides an excess of oxygen that not only sustains neuronal viability but also catalyzes numerous enzymatic oxidation reactions to produce ROS. In addition, the antioxidant defense system is perturbed during ischemia/reperfusion (Chan, 1994, 1996). Overproduction of ROS with failure to adequately replenish antioxidants in the ischemic brain tissue induces DNA damage, lipid peroxidation, protein degradation, and contributes to cellular dysfunction and apoptotic neuronal death (Knight, 1997; Love, 1999; Simonian and Coyle, 1996; Sun, 1990).
Apoptosis is a genetically programed active process for maintaining homeostasis under physiologic conditions and for responding to various stimuli (Thompson, 1995; Vaux, 1993). This form of cell death is characterized by cell membrane blebbing, cytoplasmic shrinkage, nuclear chromatin condensation, and DNA fragmentation (Wyllie et al., 1980). Apoptosis can be initiated in various cells by a wide variety of physical, chemical, and biologic stimuli including diverse anticancer drugs, reagents that cause oxidative DNA damage, cytokines, and ROS (Kroemer, 1997; Thompson, 1995; Vaux and Strasser, 1996; White, 1996). Reactive oxygen species cause cellular death by randomly damaging macromolecules (Fiers et al., 1999; Sun, 1990). At the molecular level, it is thought that ROS induce apoptosis in an indirect manner by modifying cellular redox-sensitive molecules (Mignotte and Vayssiere, 1998). Examples include apoptosis inducer p53 and apoptosis inhibitor nuclear factor kappa B (NF-κB) (Beg and Baltimore, 1996; Ko and Prives, 1996; Mayo et al., 1997; Sun and Oberley, 1996; Verhaegh et al., 1997). In addition, ROS also induce necrosis, another type of cell death (Fiers et al., 1999).
Recently, the authors found that 1,10-phenanthroline (OP), a metal chelator and redox-sensitive agent, is a potent apoptosis inducer (Sun, 1997). Through differential display, the authors cloned SAG (sensitive to apoptosis gene), an OP-inducible gene (Duan et al., 1999; Sun, 1997). Further characterization revealed that SAG is a redox-sensitive protein, subject to redox regulation both in vitro and in intact cells (Duan et al., 1999; Swaroop et al., 1999). When overexpressed, SAG protects cells from apoptosis induced by redox compounds (Duan et al., 1999; Sun, 1999). The authors now have extended this observation from tissue culture cells to intact animals. The current article reports that SAG is an inducible protein that appears in neuronal cells and astrocytes upon ischemic insult. SAG expression through recombinant adenovirus injection attenuates ischemia-induced brain injury by reducing ROS production and inhibiting apoptotic cell death.
MATERIALS AND METHODS
Animal model
The institutional animal care and use committee approved the procedures for the use of laboratory animals. Male CD-1 mice (Charles River, Wilmington, MA, U.S.A.), weighing 30 to 35 g were anesthetized with 1.5% isoflurane in 70% N2O/30% O2. The animals were ventilated to maintain Pao2 at ≥90 mm Hg. Body temperature was monitored and maintained at 37.0°C ± 0.5°C using a heating pad during the operation. The middle cerebral artery occlusion (MCAO) procedure was performed as described previously (Yang, 1994). Briefly, the left common carotid artery was exposed through a midline incision. The internal carotid artery then was isolated and its branch, the pterygopalatine artery, was ligated close to its origin. A 2-cm length of 5–0 rounded tip dermalon suture then was advanced from the external carotid artery through the common carotid artery and then up to the internal carotid artery for a distance of 11.0 ± 0.5 mm. Body temperature was maintained at 37°C ± 0.5°C until the operated mice recovered from the surgery.
Surface cerebral blood flow (sCBF) was measured using a laser–Doppler flowmetry monitor (LDF; Model BPM2 System, Vasamedics, St. Paul, MN, U.S.A.) equipped with a small caliber probe of 0.7 mm diameter (P-433; Vasamedics). The laser–Doppler probe was advanced steadily and perpendicularly to rest on the skull surface 3.5 mm lateral to the sagittal suture and 1 mm posterior to the coronal suture, the ischemic core. Baseline LDF recordings were made 10 minutes before MCAO. Laser–Doppler flowmetry value was recorded 5 minutes after MCAO to verify success of the occlusion. Cerebral blood flow values were calculated and expressed as a percentage of baseline values.
Immunoprecipitation and Western blot analysis
Immunoprecipitation and Western blot analysis were performed to measure SAG expression in mouse brain tissues after ischemic injury. Seven groups of mice (n = 4 to 6 in each group) were killed after 0, 2, 6, 12, 24, 48, and 72 hours of permanent MCAO. The ischemic hemisphere (≈50 to 80 mg) was dissected, homogenized, and lysed in a buffer containing 150 mmol NaCl, 50 mmol Tris-Cl, NaCl (pH 8.0), 1% NP-40, 0.5% deoxycholic acid, (Sigma) 0.1% sodium dodecyl sulfate (SDS), and 100 μg/mL polymethylsulfonylfluoride, as well as the proteinase inhibitors pepstatin, leupstatin, and chymostain (2 μg/mL each). Tissue lysates were spun at 14,000 g for 10 minutes at 4°C, and the supernatants were measured for protein concentration with a Bio-Rad assay (Bio-Rad Laboratories, Hercules, CA, U.S.A.). An equal amount of protein from each sample was immunoprecipitated as follows. Normal rabbit IgG (5 μg/μL) and protein A/G-sephorase-4B beads were applied to the supernatant and mixed for 2 hours at 4°C. Nonspecific binding proteins were discarded by centrifugation at 8000 g for 20 minutes at 4°C. The rabbit anti-SAG polyclonal antibody and proteinA/G-sephorase-4B beads then were added into the supernatant and rotated overnight at 4°C. The pellet was spun-down and washed with PBST (100 mmol/L phosphate-buffered saline (PBS) solution with 1% Tween 20, pH 7.4) three times. Proteins were dissociated from the complex by treatment with Glycin-HCI (100 mmol. pH 3.0). The samples then were boiled at 100°C in SDS sample buffer (100 mmol/L Tris-Cl, 4% SDS, 0.2% bromophenol blue and 20% glycerol) for 10 minutes and loaded onto a 15% SDS-polyacrylamide gel. Western blots were performed as described previously using an enhanced ECL kit (Amersham, Piscataway, NJ, U.S.A.) (Duan et al., 1999).
Immunohistochemistry
Tissue slides were first fixed with Carnoy fixation (60% methanol, 30% chloroform, and 10% acetate) for 20 minutes then rinsed 3 × 5 minutes using 0.1 mol/L PBS (pH 7.4, Sigma). Nonspecific binding was blocked by incubating tissue slides with 15% normal goat serum for 30 minutes at room temperature. Slides were incubated in a 1:200 dilution of rabbit polyclonal anti-SAG antibody or preimmune serum (for negative control) overnight at 4°C. After treatment with 1% H2O2 in 30%/70% methanol/PBS solution, the slides were incubated with biotinylated goat anti-rabbit secondary antibody for 90 minutes at room temperature followed by an ABC process (ABC-Elite Kit, Vector, Burlingame, CA, U.S.A.). Finally, the slides were treated with stable 3,3′-diaminobenzidine tetrahychloride (DAB; Research Genetics, Huntsville, AL, U.S.A.) as a peroxidase substrate.
Double immunofluorescent staining was used to identify cells that express SAG and to reveal subcellular localization of SAG. Sections first were incubated with 5% normal donkey serum containing 2% bovine serum albumin (BSA) and 0.1% Saponin in PBS for 30 minutes at room temperature. Sections then were incubated with 1:100 dilution of rabbit anti-SAG antibody and sheep antineuron specific enolase (NSE 1:100; Capricorn Products, ME, U.S.A.) or goat anti-glial fibrillary acidic protein (GFAP 1:100, for astrocyte; Santa Cruz, Santa Cruz, CA, U.S.A.) overnight at 4°C. After washing, sections were incubated with both secondary antibodies: donkey anti-rabbit IgG-fluorescein and donkey anti-sheep IgG-rhodamine (Cortex Biochem, CA, U.S.A.) or donkey anti-goat IgG-rhodamine (Santa Cruz) for 1 hour at room temperature. Double-labeled immunostaining was evaluated with a fluorescence microscope (Nikon Microphoto-SA, Melville, NY, U.S.A.) with a filter cube (excitation filter = 450 to 490 nm; suppression filter = 515 to 560 nm) for fluorescein isothiocyanate labeling and another filter cube (excitation filter = 515 to 560 nm; suppression filter = 590 nm) for rhodamine. Photomicrographs were obtained by changing the filter cube without altering the position of the section or focus.
Construction of recombinant adenovirus expressing human SAG and intracerebral injection
Adenovirus encoding either AdCMVSAG or AdCMVmSAG (with no antioxidant activity) (Swaroop et al., 1999) were constructed by homologous recombination in 293 cells. The hSAG cDNA (Duan et al., 1999; Swaroop et al., 1999) was placed in an expressing vector pCA13 (Microbix, Toronto, Canada) under the control of a CMV promoter. Positive plaques were identified by polymerase chain reaction, and SAG expression was confirmed by Western blot analysis. Positive plaques were subjected to two rounds of plaque purification. AdCMVSAG was expanded in 293 cells and purified by CsCl gradients. The adenovirus titer, plaque-forming units (pfu), was determined in 293 cells by plaque assay. Recombinant adenoviral vector AdCMVlacZ was obtained from the Institute for Human Gene Therapy at the University of Pennsylvania.
Mature male CD-1 mice (Charles River) weighing 30 to 35 g were anesthetized with 4% chloral hydrate (400 mg/kg, intraperitoneal). Mice were placed in a stereotactic frame with a mouse holder (Kopf Model 921; David Kopf Instruments, Tujunga, CA, U.S.A.), and a burr hole was drilled to the pericranium 1.0 mm lateral to the sagittal suture and 1.0 mm posterior to the coronal suture. A 10-μL Hamilton syringe (Hamilton, Reno, NV, U.S.A.) was slowly inserted into the left caudate putamen (3.0 mm deep from the dura). A volume of 0.5 μL of adenoviral suspension containing 1 × 1012 particles/mL of either AdCMVSAG or AdCMVmSAG was injected stereotactically into the lateral caudate putamen at a rate of approximately 0.1 μL/min. The needle then was pulled up 2 mm and another dose of the same volume was injected at the same rate. Control animals were injected with the same amount of AdCMVlacZ or saline at the same injection rate. The needle then was withdrawn over a course of 10 minutes, the hole was sealed with bone wax, and the wound was closed with suture. After 24 hours of adenoviral transducton, animals underwent 30 minutes of temporary MCAO.
Infarct volume measurement
Four groups of mice injected with AdCMVSAG, AdCMVmSAG, AdCMVlacZ, or the saline (n = 6 to 8 in each group) were killed at 24 hours of reperfusion after 30 minutes of MCAO. The brains were removed and immediately frozen in 2-methylbutane at −42°C. Twenty-micrometer-thick cryostat sections distal from the frontal pole were cut and mounted on slides. The sections were dried and then stained with Cresyl violet. Using NIH Image 1.62 software, the ischemic lesion area was calculated as the difference between the area of the nonischemic hemisphere and the normal area in the ischemic hemisphere. Infarct volume was calculated by multiplying the infarct area by the thickness of the section. The measurement of infarct volume was performed in a blinded fashion.
In situ detection of superoxide anion production
The production of O2 was determined at 2 hours of reperfusion after 30 minutes of MCAO using hydroethidine (HEt; Molecular Probe, Eugene, OR, U.S.A.), a fluorescent dye taken up by living cells and oxidized to ethidium selectively by O2 (Bindokas et al., 1996; Fujimura et al., 1999). Mice were anesthetized with 2% isoflurane in 30% O2/70% N2O mixed gases. HEt (200 l) was administered 15 minutes before MCAO through the jugular vein in each mouse. Animals were killed after 2-hour reperfusion and the brains were removed and cut into sections (50-μm-thick) at the level of the anterior commissure and the hippocampus using a cryostat (CM1800; Leica, Nussloch, Germany). Ethidium was detected through a fluorescent microscope with excitation 510 to 550 nm and emission >580nm (red fluorescent dye). Microphotographs of the expression pattern of oxidized HEt in the ischemic core area and contralateral hemisphere were compared with control nonischemic brain. To analyze the fluorescence signal of HEt, photomicrographs were scanned with NIH image 1.62 software and then the signal intensity was measured in 16 individual cells per section.
TUNEL staining for apoptotic cells
Four groups of mice injected with AdCMVSAG, AdCMVmSAG, AdCMVlacZ, or the saline (n = 6 to 8 in each group) were killed at 24 hours of reperfusion after 30 minutes of MCAO. TUNEL staining was performed with an ApopTag Plus in situ Apoptosis Detection Kit (Intergen, Purchase, NY, U.S.A.; see ApopTag manual for details). Briefly, frozen brain sections were fixed with 2% paraformadehyde and washed three times in PBS solution. Tissue sections were postfixed with ethanol: acetic acid (2:1) for 5 minutes at −20°C, washed, and immediately incubated in equilibration buffer for 10 seconds at room temperature. The sections then were incubated with terminal deoxynucleotidyl transferase (TdT) enzyme for 1 hour at 37°C. A stop/wash buffer then was applied, and the sections were visualized with a DAB substrate solution. Sections then were cleared and mounted. Sections treated with the DNAse I enzyme (Amersham, Cleveland, OH, U.S.A.) were used as positive controls; sections not treated with TdT were used as negative controls. Total TUNEL-positive staining cells were counted semiquantitatively. Three coronal sections measured at 0.56, 0.36, 0.16 mm related to Bregma in each mouse were selected to calculate the TUNEL staining area. The following four scores were used in the current study: (1) indicates few positive staining cells within the caudate putamen; (2) indicates positive staining cells are less than 50% of the caudate putamen; (3) indicates positive staining cells are more than 50% but not beyond caudate putamen; and (4) indicates positive staining cells are beyond caudate putamen and extend to the ischemic cortex.
Statistical analysis
All data are expressed as mean and standard deviation. Parametric data among the AdCMVSAG, AdCMVmSAG, AdCMVlacZ, and saline control groups were evaluated using analysis of variance followed by Scheffe's between group comparison (Statview; Abacus Concepts, Berkeley, CA, U.S.A.). P < 5% was considered statistically significant.
RESULTS
Induction of SAG expression in mouse brain upon ischemia insult
The authors have shown previously that SAG is inducible by redox compounds in cultured mouse cells (Duan et al., 1999). To determine whether SAG can be induced by ischemic injury in vivo, immunoprecipitation-coupled Western blot analysis was performed with mouse brain samples collected from mice that underwent permanent MCAO for time periods of 0, 2, 4, 6, 12, 24, 48, and 72 hours. SAG was expressed at a very low level in the sham and the contralateral hemisphere after MCAO (data not shown). However, in the ischemic hemisphere, expression of SAG, shown as a major band at approximately 13 kDa, gradually increased at 2 hours of MCAO, peaked at 6 and 12 hours, and then decreased gradually to the basal level (Fig. 1, top panel, as a representative experiment). Densitometric quantitative data (n = 5 at each time point) is shown in Fig. 1 (bottom panel). Statistically significant induction of SAG occurred at 6 to 12 hours after ischemia (P < 0.05), demonstrating that SAG is an intermediate responsive gene, induced by ischemic insult in the mouse brain.
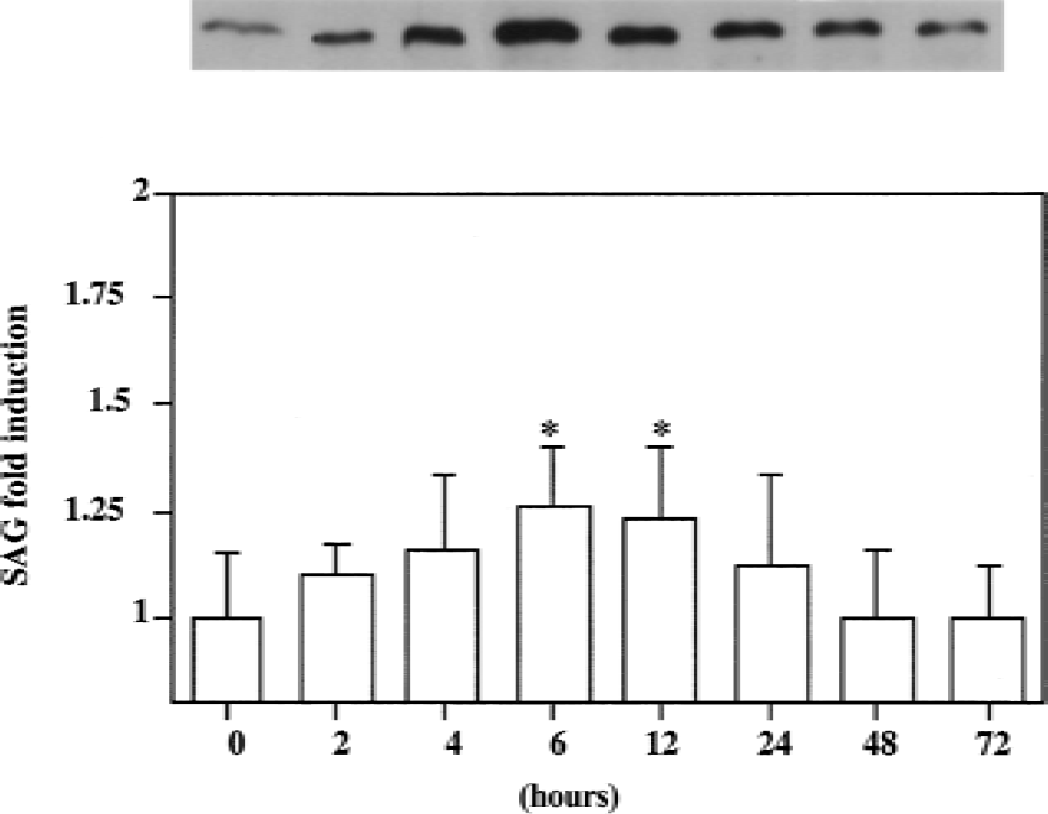
Time course of SAG expression during ischemia in the mouse brain. Total soluble protein extracts were prepared from the ischemic hemisphere in mouse brain after 0, 2, 4, 6, 12, 24, 48, and 72 hours of permanent middle cerebral artery occlusion.
Cellular localization of SAG to neuronal cells and astrocytes
To localize SAG expression, immunostaining was performed with brain sections prepared from mice that underwent MCAO for different lengths of time. The current results demonstrated that few SAG-positive staining cells can be detected in brain sections from normal and sham-operated mice or in the contralateral hemisphere (as a control). However, SAG staining developed in the ischemic cortex and basal ganglia area from 2 hours of MCAO, radiated to the perifocal region after 12 to 24 hours of MCAO, and then gradually reduced. This result parallels the Western blot analysis. As shown in Fig. 2, most SAG-positive cells were distributed in the ipsilateral cortex (Fig. 2B), striatum (Fig. 2D), hippocampus (Fig. 2F), and corpus callosum (Fig. 2H) after 24 hours of MCAO, but not in the contralateral hemisphere (Fig. 2A, 2C, 2E, and 2G). No immunoreactivity was detected when a preimmune serum from the same rabbit was used (data not shown).
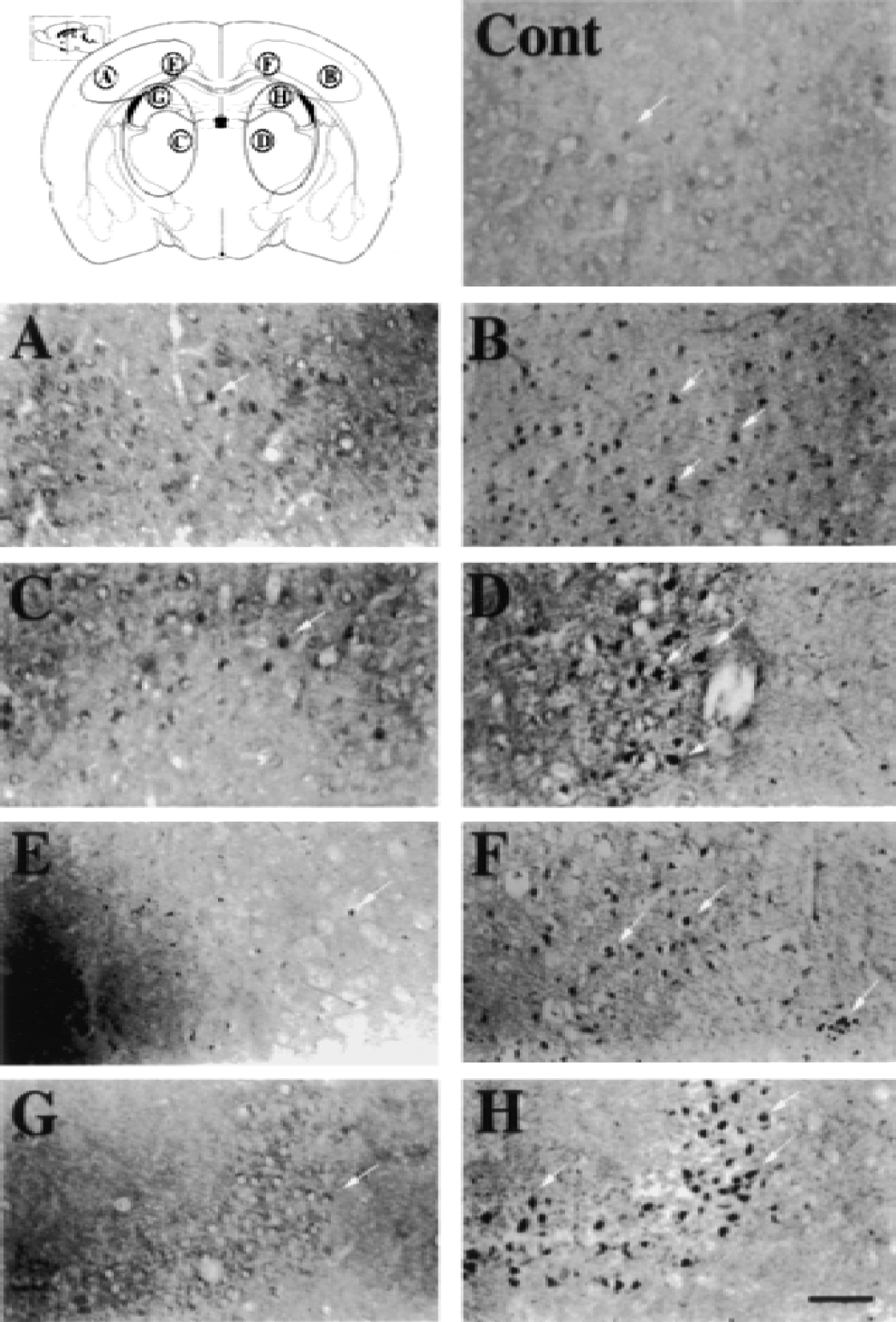
Photomicrographs show SAG expression in the mouse brain after 24 hours of permanent middle cerebral artery occlusion (MCAO). Illustration (top left) shows the sampling of brain tissue from contralateral
Double-labeled immunohistochemical studies revealed that SAG was colocalized with NSE, a neuron-specific marker (Fig. 3A: SAG; 3B: NSE; and 3C: SAG and NSE). Immunopositive staining appeared mainly in the cytoplasm of the SAG-positive cells. Double-labeled fluorescence for SAG and GFAP, an astrocyte specific marker in nerve fibers, revealed that SAG immunoreactivity also was colocalized with GFAP (Fig. 3D: SAG; 3E: GFAP; and 3F: both SAG and GFAP). These results demonstrate that SAG is expressed mainly by neuronal cells and astrocytes and is located in the cytoplasm.
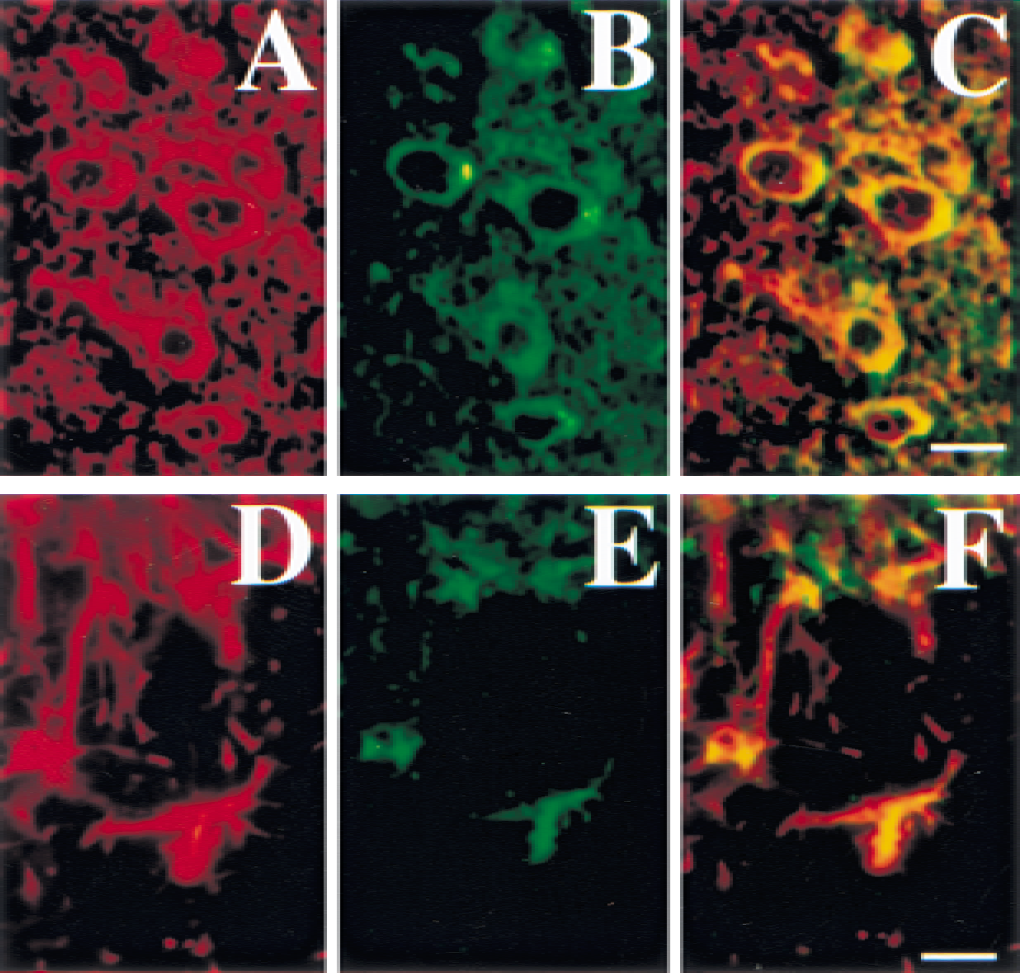
Photomicrographs showing double-labeled immunofluorescence staining in the mouse brain after 12 hours of middle cerebral artery occlusion (MCAO). SAG-stained cells
Protection of ischemia-induced brain injury by AdCMVSAG transduction
The authors found that in response to redox-induced apoptosis, SAG was induced to protect rather than promote apoptosis (Duan et al., 1999; Sun, 1997). The authors next examined whether in vivo SAG overexpression through AdCMVSAG injection would attenuate ischemia-induced brain damage in mice. It was observed that the mice were overtly normal and no difference in behavior or body weights appeared after injection of AdCMVSAG, AdCMVmSAG, AdCMVlacZ, and saline, indicating no visible toxicity associated with the adenovirus administration. SAG expression 24 hours after recombinant adenoviral SAG injection with immunofluorescence was examined next. Figure 4 shows strong SAG immunopositive staining in the adenoviral transduced hemisphere, whereas the contralateral hemisphere exhibited no positive staining (Fig. 4A). In the areas adjacent to the needle track, many cells displayed SAG immunoreactivity (Fig. 4A and 4C). SAG immunopositive staining was observed in the AdCMVmSAG transduced mouse brain (Fig. 4D), but not in the sections from the AdCMVlacZ transduced mice (Fig. 4B). These results indicate that transduction of AdCMVSAG induces an overexpression of SAG protein in mouse brain cells.
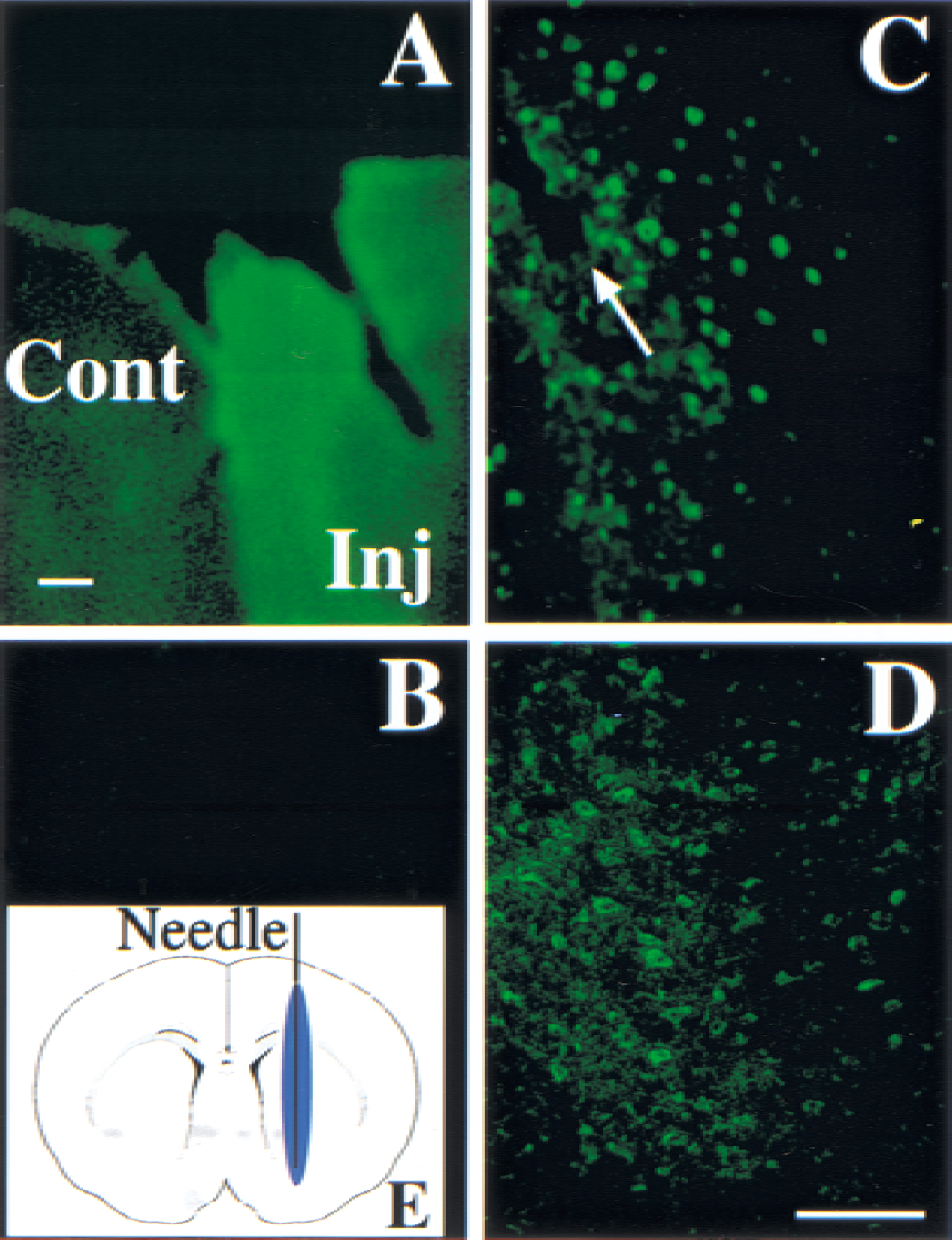
One microliter of adenoviral suspension containing 1 × 1012 particles/mL was injected stereotactically into the left caudate putamen region in the mouse brain followed by fluorescence positive staining for SAG expression.
The authors further confirmed ischemia and reperfusion in all four groups of animals by sCBF measurement. After MCAO, sCBF level in the contralateral hemisphere during occlusion was ≈90% of baseline CBF in all 4 groups of mice receiving AdCMVSAG, AdCMVmSAG, AdCMVlacZ, and saline injection; sCBF was reduced both in the core area (≈8% to 10% of baseline CBF) and the perifocal area (≈25% of baseline CBF) in all 4 groups of animals at 5 minutes of occlusion. sCBF recovered to more than 80% of baseline flow after 5 to 15 minutes of reperfusion in the ischemic core and perifocal area in all mice. There was no difference between 30 minutes after reperfusion and 24 hours after reperfusion (P > 0.05;Fig. 5).
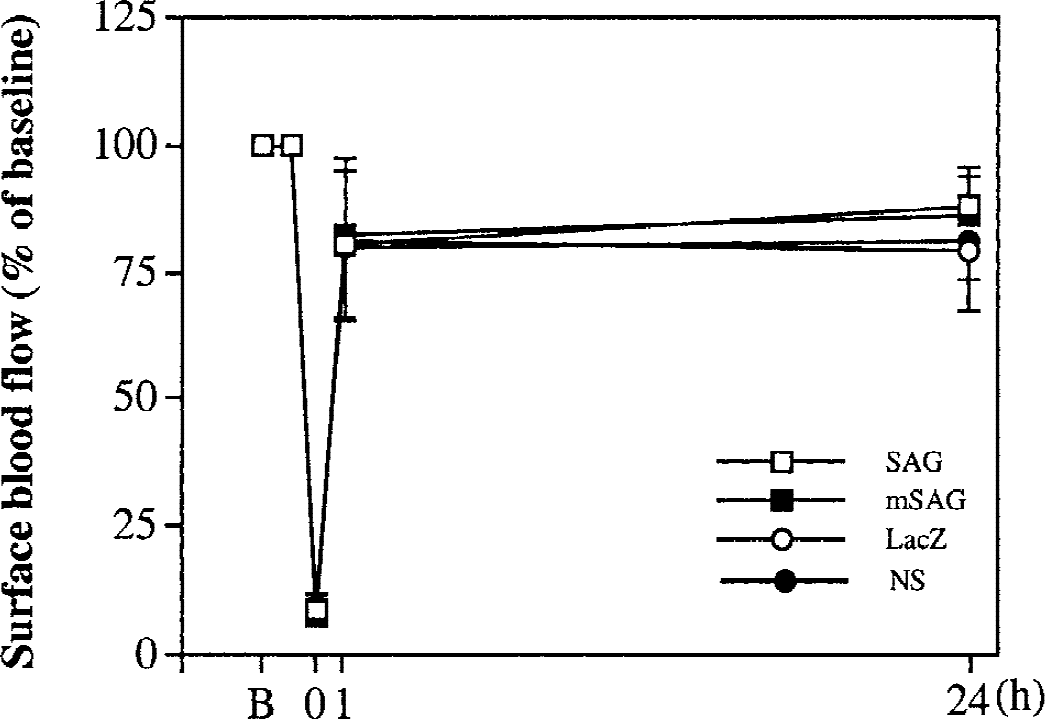
Plots showing the percentage changes of surface CBF in the ischemic hemisphere during 30 minutes of middle cerebral artery occlusion (MCAO) and 24 hours of reperfusion, measured by laser–Doppler flowmeter. Values are expressed as a percentage of baseline blood flow (100%) measured before MCAO. SAG, AdCMVSAG transduced; mSAG, AdCMVmSAG transduced; lacZ, AdCMVlacZ transduced; NS, saline-injected mice. Values are mean ± SD. n = 6 to 12 in each group.
To examine the potential protective activity of SAG against ischemia-induced brain damage, ischemia/reperfusion-induced infarct lesions were measured in 4 groups of mice (n = 6 in each group) receiving injections of AdCMVSAG, AdCMVmSAG, AdCMVlacZ, or saline. After 24 hours of recombinant adenovirus transduction, the mice underwent 30 minutes of MCAO followed by 24 hours of reperfusion. Figure 6 (top panel) showed one representative coronal section of mouse brain with either an injection of saline (Fig. 6A), AdCMVlacZ (Fig. 6B), AdCMVmSAG (Fig. 6C), or AdCMVSAG (Fig. 6D). The infarct area in the AdCMVSAG transduced mice on section numbers 9, 10, and 11 (0.16 mm, 0.36 mm, and 0.56 mm posterior to the bregma) were significantly smaller than that in the AdCMVmSAG, AdCMVlacZ, or saline-treated mice (Fig. 6, middle panel;P < 0.05). The overall infarct volume in the AdCMVSAG, AdCMVmSAG, AdCMVlacZ transduced, and the saline control group was 16 ± 1 mm3, 22 ± 3 mm3, 25 ± 3 mm3, and 25 ± 2 mm3, respectively (Fig. 6, bottom panel). Although the infarct volume in the AdCMVSAG transduced mice appeared smaller, no statistical difference was found among these 4 groups (P > 0.05), reflecting limited SAG expression around the injected areas in large brain tissues. These results suggest that wild-type SAG, not its mutant or the lacZ control, protects against ischemia-induced brain injury in areas where SAG is expressed.
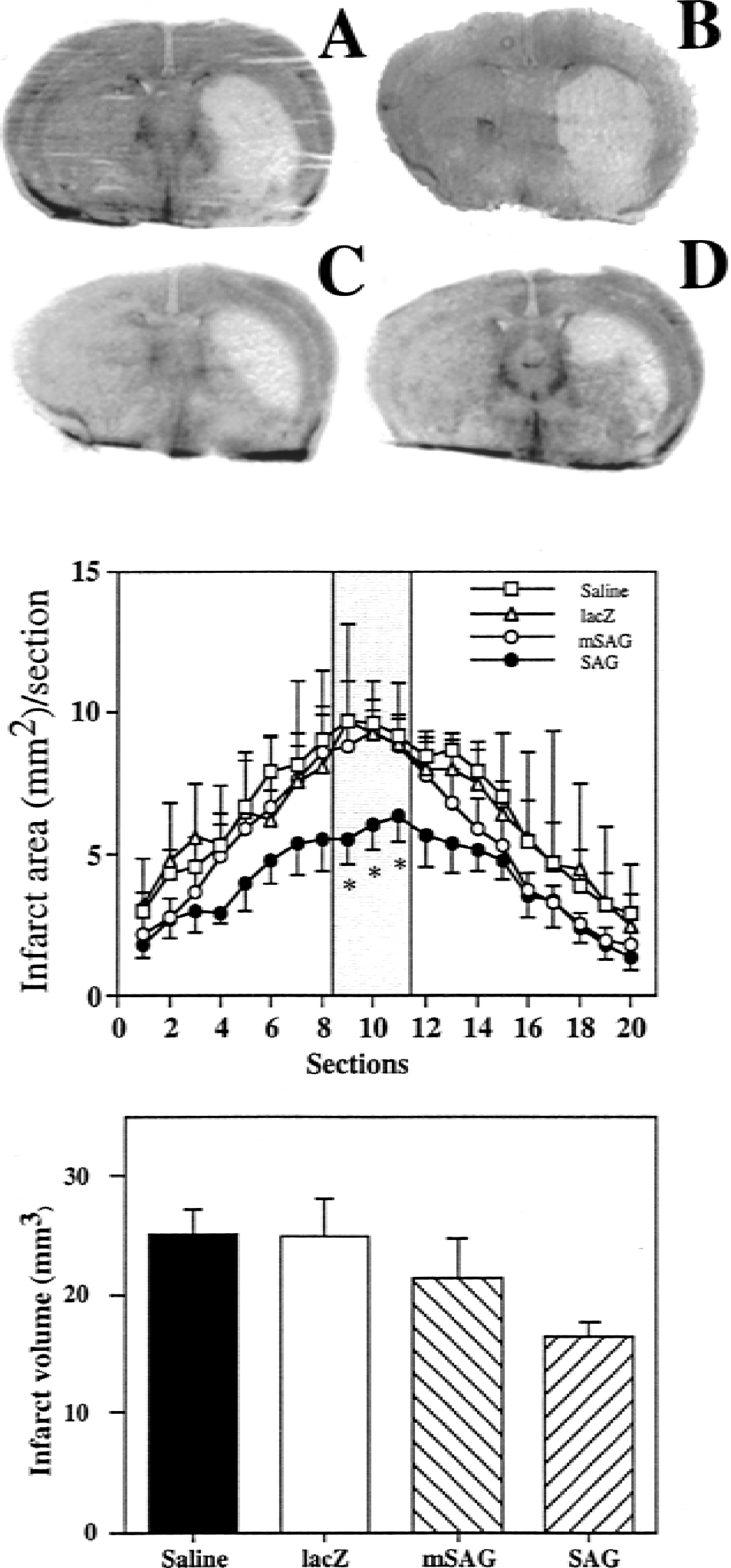
Mice were injected with 1 μL (1 × 1012 particles/mL) of AdCMVSAG, AdCMVmSAG, AdCMVlacZ, and saline. Twenty-four hours thereafter, mice underwent 30 minutes of MCAO followed by 24 hours of reperfusion. The mice were killed, brain sections were prepared, and infarct area was measured as detailed in Materials and Methods.
SAG expression reduces local superoxide anion production
The authors have shown previously that SAG can bind metal ions, such as zinc and copper, and prevent copper or ROS-induced lipid peroxidation in vitro (Duan et al., 1999; Swaroop et al., 1999). To determine the mechanism by which SAG acts as a protector against ischemia-induced damage, in situ production of superoxide anion was measured in the current MCAO model after recombinant adenovirus transduction. HEt, a fluorescent dye that can be taken by living cells, was used as an indicator and oxidized to red fluorescent dye, ethidium, specifically by superoxide anion, but not by other ROS (Bindokas et al., 1996; Fujimura et al., 1999). Increased production of superoxide anion recently was observed in the vascular lumen after transient focal cerebral ischemia in rats (Mori et al., 1999). As shown in Fig. 7, the oxidized HEt signal was weak in the contralateral hemisphere after 30 minutes of transient MCAO (tMCAO) and 2 hours of reperfusion in the adenoviral transduced group and saline-treated group (Fig. 7A, 7C, 7E, and 7G). The red ethidium signals were markedly increased in the ipsilateral hemisphere, especially in the ischemic core (Fig. 7B, 7D, 7F, and 7H). They appeared as small particles in the cytoplasm, suggesting mitochondrial production of O2.−. Some microvessels displayed strong ethidium signals, suggesting that endothelial cells may produce O2.−. As shown in the bar graph with semiquantitative data (Fig. 7, bottom panel), the mean intensity of the HEt signal was significantly less in AdCMVSAG transduced mice (Fig. 7H, 8.9 ± 0.7; mean ± SD, P < 0.05) than in AdCMVmSAG (Fig. 7F, 16.2 ± 2.2) or AdCMVlacZ (Fig. 7D, 16.5 ± 3.2) or saline- (Fig. 7B, 20.9 ± 2.2) treated mice. Thus the neuronal protective role of SAG appears to be mediated by scavenging superoxide ion. However, the possibility that SAG works at many other more proximal mechanisms cannot be excluded. For example, transduction of Bcl-2 has been shown to decrease the production of ROS (Hockenbery et al., 1993). However, it is now known that Bcl-2 does not work as a free radical scavenger, rather it works at regulating mitochondrial permeability (Kroemer and Reed, 2000).
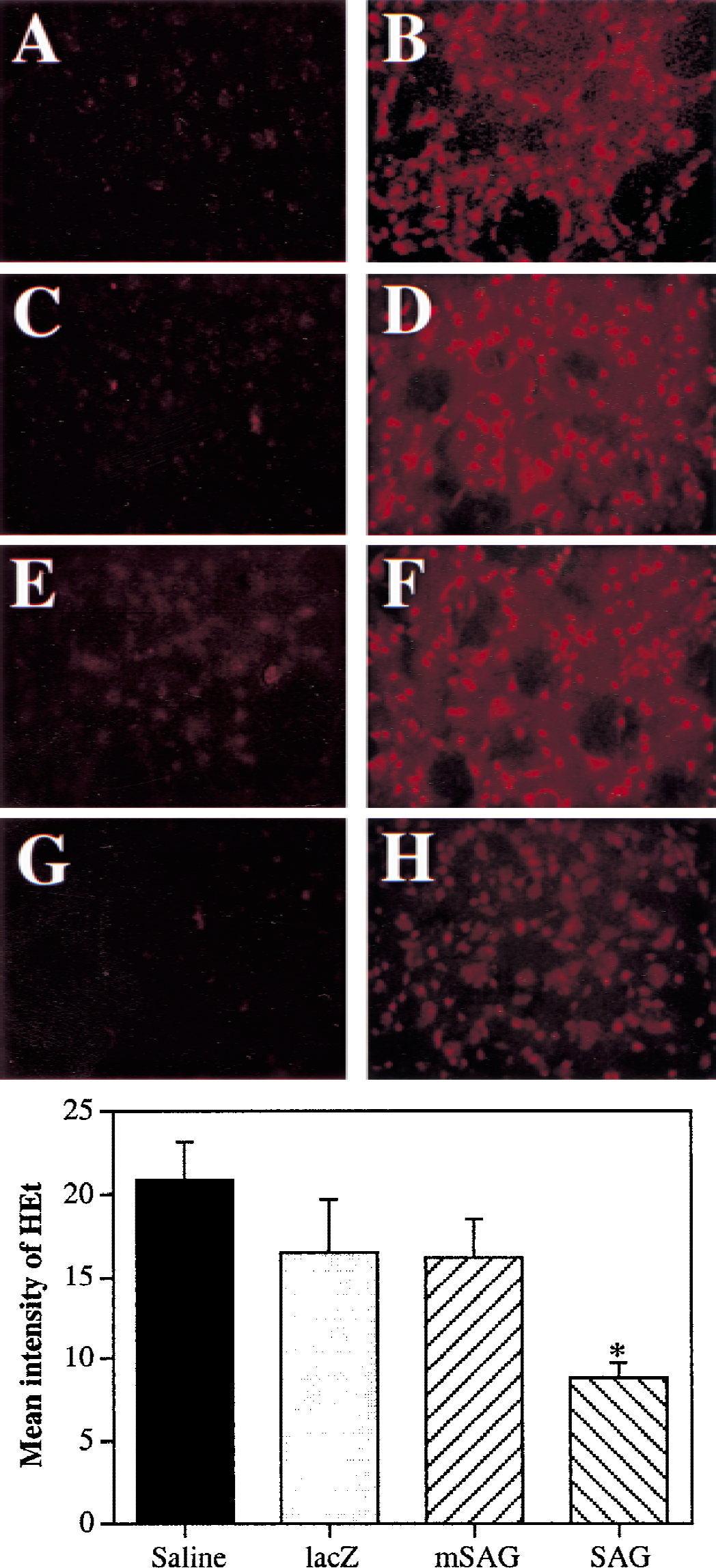
Mice were injected with recombinant adenovirus followed by 30 minutes of tMCAO and 2 hours of reperfusion. Hydroethidine (HEt) was administered 10 minutes before middle cerebral artery occlusion (MCAO). Brain sections were prepared and O2.− production was measured as detailed in Materials & Methods. Representative photomicrographs show expression of oxidized HEt in the ischemic hemisphere in saline-injected
SAG expression decreases apoptotic cell death
The authors have shown previously that overexpression of SAG can protect SY5Y neuroblastoma cells from apoptosis induced by metal ions, zinc and copper (Duan et al., 1999). To determine whether reduced infarct size seen in the AdCMVSAG transduced mouse brain is attributable to decreased apoptotic cell death, a TUNEL assay to stain the apoptotic cells was performed. As shown in Fig. 8, TUNEL-labeled cells were distributed mainly in the ischemic core areas at the inner boundary zone after 24 hours of temporary ischemia in all 4 groups of mice (Fig. 8A to 8D). These cells were densely labeled in the nuclei and showed morphologic signs of apoptosis (Fig. 8E and 8F, arrows), an observation consistent with previous studies (Li et al., 1995; Murakami et al., 1997a). Compared with the saline-treated (Fig. 8A), AdCMVlacZ transduced (Fig. 8B), or AdCMVmSAG transduced (Fig. 8C) groups of mice, TUNEL-positive staining cells were greatly reduced in the AdCMVSAG transduced (Fig. 8D) mice. No TUNEL-positive–stained cells were detected in the contralateral hemisphere (data not shown). The semiquantitative data by scoring TUNEL-positive staining cells was shown on the bar graph (Fig. 8, bottom panel). The AdCMVSAG transduced mice (1.3 ± 0.2, mean ± SD) had much less TUNEL-positive staining cells, as compared with the AdCMVmSAG (2.3 ± 0.3), or AdCMVlacZ (2.1 ± 0.2), or saline- (2.1 ± 0.2) treated mice. The current results demonstrate that SAG can inhibit ischemia/reperfusion-induced DNA fragmentation.
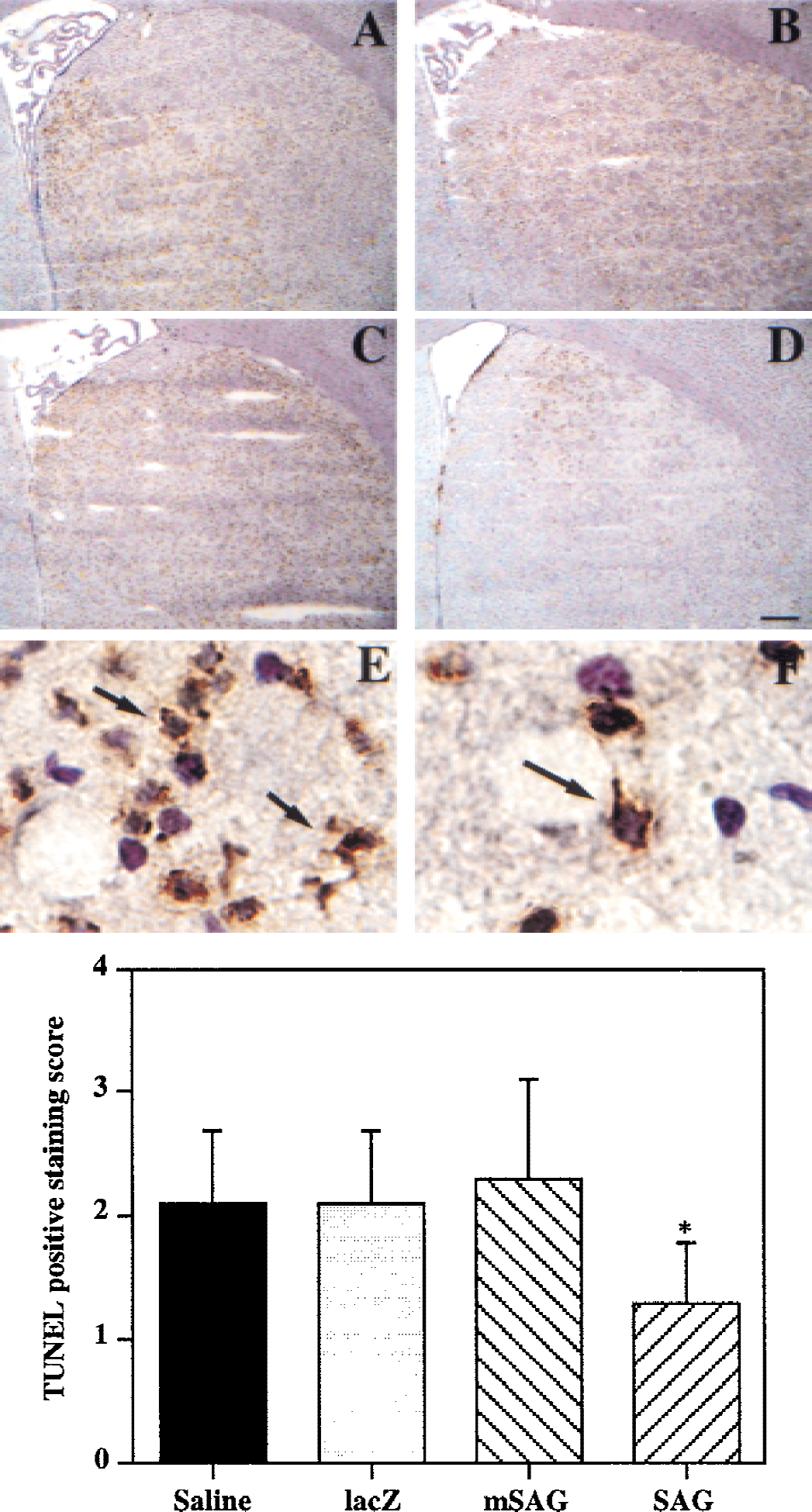
The mouse in vivo MCAO model was established 24 hours after recombinant virus injection. Brain sections were prepared and stained with a TUNEL assay. Photomicrographs show the TUNEL-positive staining cells in the ischemic core after 30 minutes of tMCAO followed by 24 hours of reperfusion.
DISCUSSION
The authors previously have shown in vitro that SAG, a redox-inducible antioxidant protein, can chelate metal ions and scavenge ROS. Therefore, overexpression of SAG protects cells from apoptosis induced by redox compounds (Duan et al., 1999; Swaroop et al., 1999). Here, using a mouse in vivo MCAO model, the authors showed that (1) SAG is up-regulated after cerebral ischemia; (2) SAG can be locally overexpressed through caudate putamen injection of AdCMVSAG; (3) infarct areas are reduced in AdCMVSAG transduced mice; and (4) ROS production and apoptotic cell death are reduced in AdCMVSAG transduced mice. The protective effect of SAG appears to be rather specific because a SAG mutant, SAG-MM14, that failed to prevent copper-induced lipid peroxidation (Swaroop et al., 1999) showed no protective activity. Furthermore, the protection was only seen in the injection sites where SAG was expressed. Because SAG expression is limited to the injection sites that account for a small fraction of the ischemic zone, no significant protection could be seen when the entire infarct volume was measured.
The current study demonstrates that recombinant adenovirus administration has no measurable toxic effect. The animal behaviors and body weights observed after adenoviral injection in all four groups were identical. Physiologic parameters after tMCAO also showed no differences among the four groups (data not shown). In this animal model, the degree of which blood flow is occluded influences the degree of severity of the stroke insult. Therefore, sCBF measurement is an important assay for validating the extent of occlusion (Yang and Betz, 1994). Surface CBF in the ischemic hemisphere of the AdCMVSAG, AdCMVmSAG, AdCMVlacZ transduced, and saline control mice all decreased to ≈10% of baseline CBF after tMCAO, and no significant differences were detected between these groups. This result suggests that adenovirus transfection does not affect CBF in the mouse brain and that the protective effect of SAG during ischemia is not because of higher CBF from inadequate occlusion of the MCAO.
Antioxidant defense systems that scavenge ROS consist mainly of antioxidant enzymes (for example, superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase), antioxidant proteins (for example, thioredoxin, metallothionein), and small molecular antioxidants (for example, GSH, N-acetyl-L-cysteine, vitamin C, and vitamin E) (Sun, 1990). Almost all antioxidant molecules have been shown to protect cells against apoptosis induced by redox-sensitive reagents (Baker et al., 1997; Kayanoki et al., 1996; Kondo et al., 1997c; Manna and Aggarwal, 1998). Numerous studies also have shown that antioxidant proteins can protect against neuronal death induced by ischemia/reperfusion. Copper zinc superoxide dismutase (Cu,ZnSOD), when overexpressed or postischemically infused, protects neurons against ischemic injury and reduces cerebral infarct size after global cerebral ischemia/reperfusion (Chan, 1996; Chan et al., 1998; Francis et al., 1998; Murakami et al., 1997b). When the Cu,ZnSOD gene was knocked out, mice exhibited exacerbated neuronal cell injury and edema formation after cerebral ischemia (Kawase et al., 1999; Kondo et al., 1997a, b ). Likewise, manganese superoxide dismutase (MnSOD) was found to prevent neural apoptosis and reduce ischemic brain injury by scavenging ROS, suppressing lipid peroxidation, and inhibiting cytochrome c release and DNA fragmentation (Fujimura et al., 1999; Keller et al., 1998; Murakami et al., 1998). Extracellular SOD, when overexpressed, increased the animals' resistance to focal cerebral ischemia; when deficient, the outcome worsened (Sheng et al., 1999a, b ). Other antioxidant enzymes such as catalase and glutathione peroxidase also showed protection against ischemia-induced injury (Truelove et al., 1994; Weisbrot-Lefkowitz et al., 1998). Metallothionein is a gene family that binds to metal ions and protects against zinc toxicity and ROS-induced damage (Nordberg, 1986). Some family members were found to be induced by ischemia (Neal et al., 1996; Yanagitani et al., 1999). Metallothionein I was recently found to protect against focal cerebral ischemia and reperfusion in a transgenic mouse model (van Lookeren Campagne et al., 1999). Other antioxidant proteins such as thioredoxin, and small antioxidant molecules such as vitamin E, also provided protection (Tagami et al., 1998; Takagi et al., 1999). Finally, Bcl-2, a well-known protein that inhibits apoptosis in an antioxidant pathway in certain type of cells (Hockenbery et al., 1993), also reduces neuronal cell death in focal cerebral ischemia when injected (Linnik et al., 1995). In the current article, the authors provide evidence that SAG, a zinc RING finger protein, also protects against ischemia-induced brain damage in vivo. Like those antioxidant proteins, the neuroprotective role of SAG appears to be achieved mainly through reduction of superoxide anion production and apoptotic cell death. In addition, it has been shown that zinc released after transient global cerebral ischemia selectively induced neuronal cell death, which could be inhibited by the intraventricular injection of zinc chelating agent (Koh et al., 1996). Zinc also was associated with the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat (Tonder et al., 1990). Thus, as a zinc-binding protein (Duan et al., 1999; Swaroop et al., 1999), SAG may also function as a protector by chelating excess zinc released during ischemia/reperfusion.
The authors have shown previously that SAG was localized in both the cytoplasm and nucleus in mammalian culture cells (Duan et al., 1999). Here, the authors show that in mouse neurons and astrocytes, SAG is mainly localized in cytoplasm. This is probably because of SAG's small size (13 kDa) that allows it to shuttle between cytoplasm and nucleus. Subcellular localization of a protein determines its biologic function. MnSOD, a mitochondrial protein, and Bcl-2, mainly localized in mitochondria and the endoplasmic reticulum, are most likely responsible for scavenging ROS produced in these organelles. SAG, along with other cytoplasmic antioxidant proteins, may therefore scavenge the ROS produced in cytoplasm and nucleus.
Recently, a zinc RING finger protein named ROC1 (Regulator of Cullins), Rbx1 (RING box), or Hrt1 has been cloned and characterized (Kamura et al., 1999; Ohta et al., 1999; Seol et al., 1999; Skowyra et al., 1999; Tan et al., 1999; Tyers and Willems, 1999). Roc1/Rbx1/Hrt1 is a component of SCF (Skp1-Cullin-F box protein) E3 ubiquitin ligase complex that degrades cyclins/cyclin-dependent kinase inhibitors and signal molecules to regulate cell cycle progression and signal transduction (Carrano et al., 1999; Krek, 1998; Maniatis, 1999; Sutterluty et al., 1999; Tsvetkov et al., 1999; Tyers and Willems, 1999). SAG turns out to be the second member of this ROC/Rbx/Hrt family (ROC2/Rbx2/Hrt2) based on direct sequencing comparison. Like Roc1/Rbx1/Hrt1, SAG has E3 ubiquitin ligase activity when complexed with Cullin-1 (Swaroop et al., 2000). It has been shown in vitro that ROC1/Rbx1/Hrt1 promotes IκBα ubiquitination and degradation (Ohta et al., 1999; Tan et al., 1999). Upon degradation of IκBα, an inhibitor of NFκB, NFκB was released and translocated into the nucleus, where it transactivates a list of target genes (Beg and Baldwin, 1993; Maniatis, 1999; Thanos and Maniatis, 1995). It has been shown that transient global ischemia induced a marked activation of NFκB (Stephenson et al., 2000), and activation of NFκB promotes cell death (Clemens et al., 1997a, b ; Schneider et al., 1999). However, inhibition of NFκB activity in the rat CNS by injection of proteasome inhibitor induced apoptosis (Taglialatela et al., 1998). Whether activation of NFκB leads to neuroprotection or neurodegeneration remains an important question that should be address (O'Neill and Kaltschmidt, 1997). Nevertheless, given that ROC/SAG family members may regulate NFκB activity through IκBα ubiquitination/degradation at least in vitro, the authors determined whether SAG overexpression would promote IκBα degradation and NFκB activation in both the MCAO model described in the current article and cultured HeLa cells upon tumor necrosis factor-α exposure. No significant difference was observed in SAG-infected cells compared with the lacZ controls in Hela cells (Sun et al., 2001), or in MCAO model (unpublished observations) indicating that SAG is not a rate-limiting factor for IκBα degradation in these models. Thus, the protective role of SAG in ischemic-induced injury appears to be mediated mainly through its metal ion binding and ROS scavenging activity. However, the possibility cannot be excluded that SAG associated E3 ubiquitin ligase activity targeting other apoptosis related proteins, which could also contribute to its protection.
Footnotes
Acknowledgments:
The authors thank Dr. Richard F. Keep, Director of the Crosby Neurosurgical Laboratory, for valuable discussion, and Ms. Kathleen Donahoe for editorial assistance.