
Abstract
We investigated the expression of cyclin D1 and its kinase, cdk4, after induction of focal cerebral ischemia in the rat. Brain from rats (n = 6) subjected to 2 hours of transient middle cerebral artery occlusion and 46 hours of reperfusion, and control sham-operated (n = 3) and normal (n = 2) rats were processed for dual label immunohistochemical study for cellular identification of the expression of these cell cycle proteins. Antibodies raised against microtubule-associated protein 2 and neuronal specific enolase for neurons, glial fibrillary acidic protein for astrocytes, myelin basic protein for oligodendrocytes and lectin histochemical study with the B4-isolectin for microglia were used for cell type identification. Double staining for DNA fragmentation detection (TUNEL) and expression of cyclin D1 and cdk4 also was performed. Cyclin D1 and cdk4 were selectively expressed in morphologically intact or altered neurons and oligodendrocytes localized to the ischemic tissue. Apoptotic cells were not immunoreactive to cyclin D1 and cdk4 at 46 hours after 2 hours of middle cerebral artery occlusion. The selective expression of cell cycle proteins observed in nonapoptotic ischemic postmitotic neurons and oligodendrocytes suggests a role for these proteins in cell survival after transient focal cerebral ischemia.
In normal cells, cellular proliferation follows an orderly progression controlled by protein complexes that are composed of cyclins and cyclin-dependent kinases (cdk). Cyclins are a family of proteins that are the regulatory proteins for the cdk family members and are differentially synthesized and degraded at specific points during the cell cycle (Murray et al., 1989; Hunter and Pines, 1992; Pagano et al., 1992; Baldin et al., 1993; Ohtsubo and Roberts, 1993; Quelle et al., 1993). Cyclin D1 is upregulated early in G1 and subsequently peaks by mid-G1, interacts with its kinase cdk4, and usually decreases as cells approach S phase (Baldin et al., 1993). Recent evidence indicates that downregulation of cyclin D1 is necessary for nuclear relocation of the proliferating cell nuclear antigen protein and for DNA repair, as well as for initiation of DNA replication (Pagano et al., 1994).
Cyclin D1 has been associated with apoptosis. Cyclin D1 is selectively induced in vitro in postmitotic sympathetic neurons undergoing programmed cell death (Freeman et al., 1994). Also, cdk4 promotes neuronal apoptosis (Kranenburg et al., 1996). Thus, cyclin D1 and cdk4 have multiple functions, controlling cell proliferation, death, or survival in various cell types.
Cell proliferation, death, or survival may be highly regulated and interrelated processes. In the current study, we describe cyclin D1 and cdk4 expression in ischemic brain. Cyclin D1 and cdk4 are expressed in postmitotic neurons and oligodendrocytes, but not in astrocytes and microglia within the ischemic areas of rats subjected to 2 hours of middle cerebral artery occlusion (MCAO) and 46 hours of reperfusion. Apoptotic cells were not immunoreactive to cyclin D1 and cdk4 in these rats. These data indicate cell type-specific expression of cyclin D1 and cdk4 after experimental stroke, and suggest that these proteins may have functions in postmitotic neurons unrelated to cell division and may be associated with cell survival.
MATERIALS AND METHODS
Animal model
Experiments were performed on 11 male Wistar rats weighing 260 to 300 g. Transient MCAO was induced using a method of intraluminal vascular occlusion (Zea Longa et al., 1989). Briefly, rats (n = 6) were anesthetized with 3.5% halothane and maintained with 1.0% halothane in 70% N2O and 30% oxygen using a face mask. The rectal temperature was controlled at 37°C with a feedback-regulated water heating system. The right femoral artery and vein were cannulated for measuring pH and blood gases (Po2, Pco2) before ischemia and for monitoring blood pressure during the surgery. A length of 18.5 to 19.0 mm 4-0 surgical nylon suture, with its tip rounded by heating near a flame, was advanced from the external carotid artery into the lumen of the internal carotid artery until it blocked the origin of the middle cerebral artery. Two hours after MCAO, rats were reanesthetized with halothane, and reperfusion was performed by withdrawal of the suture until the tip cleared the internal carotid artery. Experimental rats were given an overdose of ketamine and xylazine and killed at 46 hours after withdrawing the intraluminal suture. Three rats served as a sham-operated control population in which a 15-mm nylon monofilament was inserted into the internal carotid artery for 2 hours, and the rats were sacrificed at 46 hours. This length of nylon monofilament was too short to occlude the middle cerebral artery.
Rat brains (n = 11) were fixed by transcardial perfusion with heparinized saline, followed by perfusion and immersion in 10% buffered formalin phosphate. A standard section, corresponding to coronal coordinates interaural 7.6 ∼ 9.6 mm, bregma −1.4 ∼ −0.6 mm (Paxinos and Watson, 1986), was obtained using a rodent brain matrix. Blocks were embedded in paraffin. Separate adjacent 6-µm thick sections were obtained from paraffin tissues for neuropathologic evaluation. Two normal rats served as a control for detection of immunoreactive proteins and for in situ detection of DNA fragmentation.
Analysis of hematoxylin and eosin staining
Histologic features used to identify the ischemic lesion included vacuolation (sponginess) of the neuropil, diffuse pallor of the eosinophilic background, and alterations in the shape and stainability of cellular perikarya. By light microscopic study, distinct histologic abnormalities defined reversible and irreversible cellular injury. Morphologic features defining the acute reversible ischemic cellular injury included scalloped or shrunken dark neurons and swollen neurons (Garcia et al., 1994). Morphologic abnormalities defining irreversible cellular injury were necrosis and apoptosis. We applied criteria developed by Farber and associates (1981) and Trump and coworkers (1984), who outlined morphologic features (light and electron microscopic study) of necrotic cells (Farber et al., 1981). Necrotic injury included pyknotic nuclei exhibiting an intense eosinophilic cytoplasm (red neurons) and nuclei lacking cellular structures (ghost neurons). Apoptotic cells are characterized by the protuberances on the cell surface separated with plasmalemma sealing, which produce membrane-bounded apoptotic bodies of roughly spherical or ovoid shape (Wyllie et al., 1980). We therefore identified apoptotic cells by the presence of at least two membrane associated apoptotic bodies.
Detection of cell type proteins and cell cycle proteins by immunohistochemical and histochemical study
After deparaffinizing, brain sections (n = 55 sections from 11 animals using 5 specific antibodies) were put in boiled citrate buffer (pH 6) within a microwave oven (650 to 720 W) for cyclin D1, cdk4, microtubule-associated protein 2 (MAP2), neuronal specific enolase (NSE), and myelin basic protein (MBP) antibody identification. Sections (n = 11) were treated with pepsin at 37°C for 15 minutes for glial fibrillary acidic protein (GFAP) antibody identification. After blocking in normal serum, sections were treated successively with primary antibodies against specific proteins (Table 1). Second antibodies conjugated with fluorescein isothiocyanate (FITC, Calbiochem, LaJolla, CA, U.S.A.) were bound to the primary antibodies against cyclin D1 and cdk4. Fluorescent intensity was detected by immunofluorescent microscopic study. Peroxidase conjugated secondary antibodies were bound to the primary antibodies against MAP2, NSE, MBP, and GFAP. Diaminobenzidine then was used as a sensitive chromogen for light microscopic study. Microglial cells were identified by means of histochemical study with the B4-isolectin (IB4) as described by Streit (1990). deparaffinized sections (n = 11) were incubated with peroxidase labeled IB4 in phosphate-buffered saline containing divalent cations at room temperature for 3 hours and overnight at 4°C. Then the sections were reacted with diaminobenzidine and hydrogen peroxide for color development. Negative control sections from each animal received identical staining preparation, except that the primary antibodies or the secondary antibody were omitted. Finally, the immunostaining pattern between the lesioned versus the nonlesioned hemisphere was used as an internal control within the same animal.
Antibodies for detection of cell type proteins and cell cycle proteins
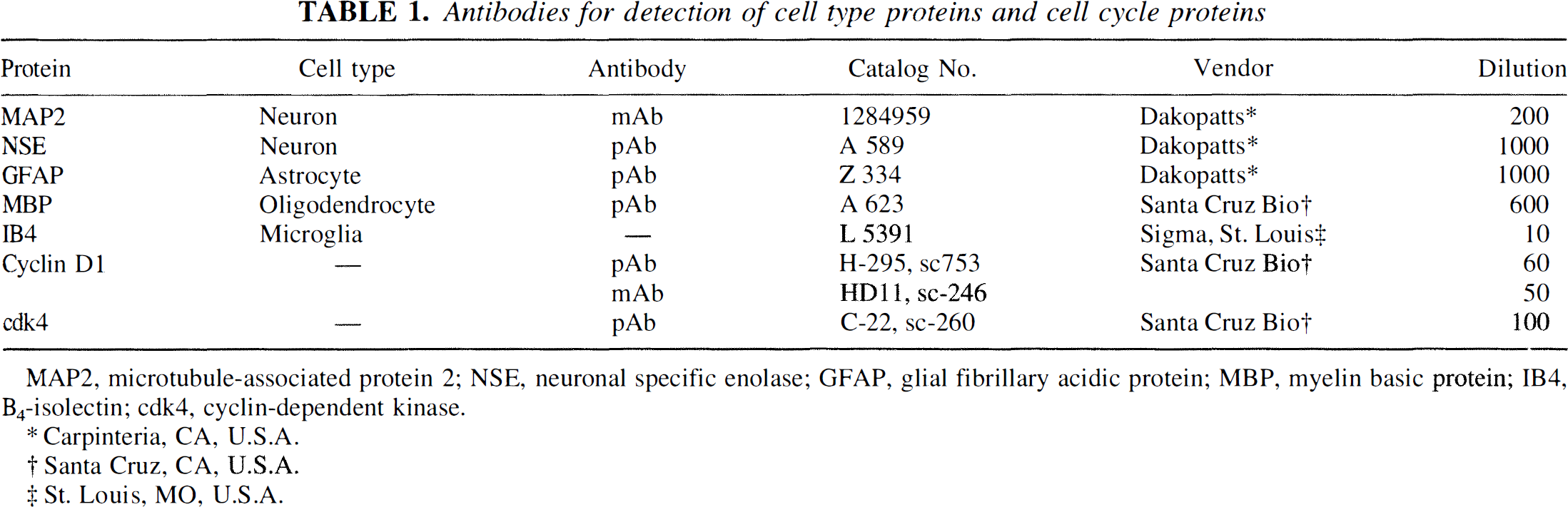
MAP2, microtubule-associated protein 2; NSE, neuronal specific enolase; GFAP, glial fibrillary acidic protein; MBP, myelin basic protein; IB4, B4,-isolectin; cdk4, cyclin-dependent kinase.
Carpinteria, CA, U.S.A.
Santa Cruz, CA, U.S.A.
St. Louis, Mo, U.S.A.
Detection of apoptosis by analysis of TUNEL staining
The TUNEL method (ApopTag kit, Oncor, Gaithersburg, MD, U.S.A.) was used for the detection of DNA fragmentation and apoptotic bodies in cells, as described by Gavrieli et al (1992). After deparaffinizing proteins within brain sections (n = 11) were digested using proteinase K, endogenous peroxidase activity was quenched with 2% hydrogen peroxide in phosphate-buffered saline. Then sections were incubated in working terminal deoxynucleotidyl transferase (TdT) enzyme. After two drops of Anti-digoxigenin-peroxidase were applied to the slides, peroxidase was detected with diaminobenzidine. Negative controls were performed using distilled water for TdT enzyme in the preparation of working TdT. The labeling target of the TUNEL method was the new 3'-OH DNA ends generated by DNA fragmentation, which typically were localized to morphologically identifiable nuclei and apoptotic bodies. Only cells containing more than two apoptotic bodies were referred to as apoptotic cells (Li et al., 1995a,1995b,1995d). This criteria is essential for identification of apoptotic cells because necrotic cells (some dark-shrunken cells, but not red or ghost cells) also may be TUNEL positive, albeit with diffuse staining. Coronal sections stained with the TUNEL method also were counter-stained with hematoxylin.
Detection of cyclin D1 and cdk4 in different cell types or apoptotic cells by dual immunoreactivity or histochemical methods and TUNEL/immunofluorescence
Dual immunohistochemical and histochemical staining were used to visualize the expression of cyclin D1 and cdk4 in different cell types. Each coronal section (n = 121 sections, from 11 animals using specific double stainings: MAP2/cyclin D1, NSE/cyclin D1, GFAP/cyclin D1, MBP/cyclin D1, IB4/cyclin D1, MAP2/cdk4, NSE/cdk4, GFAP/cdk4, MBP/cdk4, IB4/cdk4, cyclin D1/cdk4) was first stained by markers of cell type-specific proteins and then stained with diaminobenzidine for brown color development. Subsequently, immunofluorescein staining for cyclin D1 and cdk4 antibody identification was performed on the same sections. Negative control sections from each animal received identical preparations for double staining, except that both primary antibodies were omitted.
Double staining was performed on coronal sections for analysis of cyclin D1, cdk4, and apoptosis. Each coronal section (n = 22 sections, n = 11 animals × 2 double staining) was first stained by the TUNEL method for apoptotic cell identification. Immunohistochemical staining (FITC) for cell cycle protein identification was performed on the same sections. Double-stained sections were counterstained with hematoxylin.
A semiquantitative scale for immunohistochemical study and histochemical cellular reactivity was employed. Grading of immunohistochemical and histochemical staining intensity consisted of three levels: –, no staining of cells; +, weak staining; ++, moderate staining.
RESULTS
The blood gases were within normal ranges for all animals and did not differ between experimental rats (pH, 7.41 ± 0.02; Po2, 123.9 ± 14.2 mm Hg; Pco2, 39.9 ± 3.0 mm Hg) and sham-control rats (pH, 7.42 ± 0.03; Po2, 122.9 ± 13.0 mm Hg; Pco2, 37.0 ± 6.0 mm Hg). Within 6-µm thick coronal sections stained with hematoxylin and eosin in each hemisphere of all normal and sham-operated rats, as well as the contralateral hemisphere of ischemic rats, no cells were classified as necrotic; however, a few scattered apoptotic cells were found in each hemisphere. After MCAO, grossly visible swelling, as suggested by compression of the ventricles and marked pallor of the middle cerebral artery territory in coronal sections of the ipsilateral hemispheres, were clearly detected. Most necrotic cells and scattered apoptotic cells were seen in the ischemic core. Scattered necrotic and apoptotic cells were seen in the outer boundary zone to the ischemic core. Clustered necrotic and apoptotic cells were observed in the inner boundary zone of the ischemic lesion. These data are consistent with previous observations (Li et al., 1995d).
In normal brain and nonischemic brain, cyclin D1 (Figs. 1A, 1C, 1M, 1Q, 1U, and 3B) was localized to the cellular cytoplasm, whereas cdk4 (Figs. 2A, 2C, 2I, 2N, 2R, and 3A) was primarily localized to the cytoplasm of some cells and present in the nuclei of others. At 46 hours after 2 hours of MCAO, expression of cyclin D1 (Figs. 1E, 1G, and 1S) and cdk4 (2E, 2G, and 2P) was detected primarily in nuclei of scattered cells within the ischemic core of both the striatum and the cortex. In the boundary zone to the ischemic core, cyclin D1 and cdk4 was overexpressed in nuclei, cytoplasm, or both (Figs. 1I, 1K, 1O, 1W, and 2L) in morphologically damaged cells.
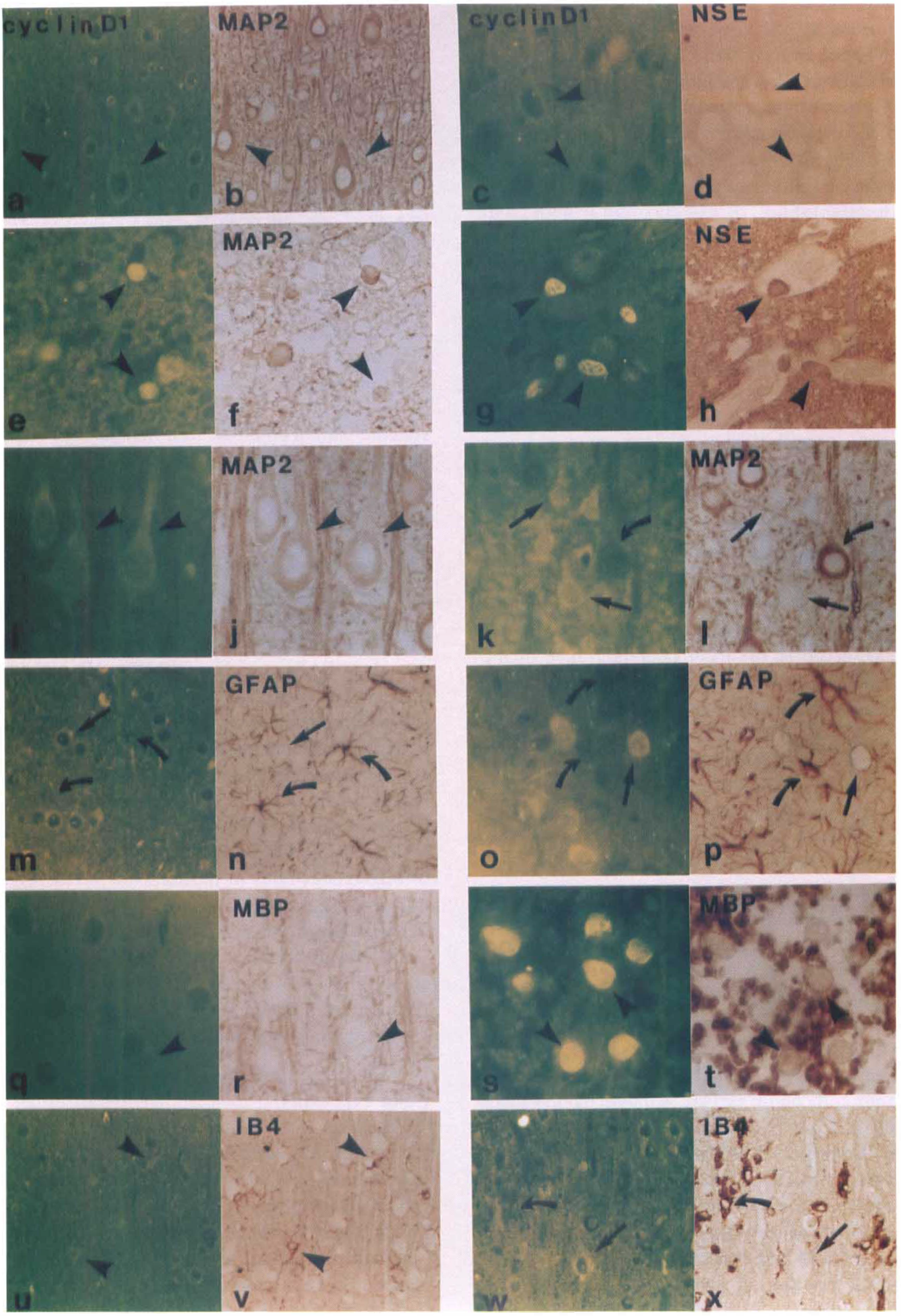
A series of adjacent sections from a representative rat subjected to 2 hours of middle cerebral artery occlusion (MCAO) and killed at 46 hours are dual immunoreactively stained for cyclin D1 (FITC) along with anti-microtubule-associated protein 2 (MAP2) and anti-neuronal specific enolase (NSE) for neurons, anti-glial fibrillary acidic protein (GFAP) for astrocytes, anti-MBP for oligodendrocytes, and IB4 for microglia, respectively (diaminobenzidine).
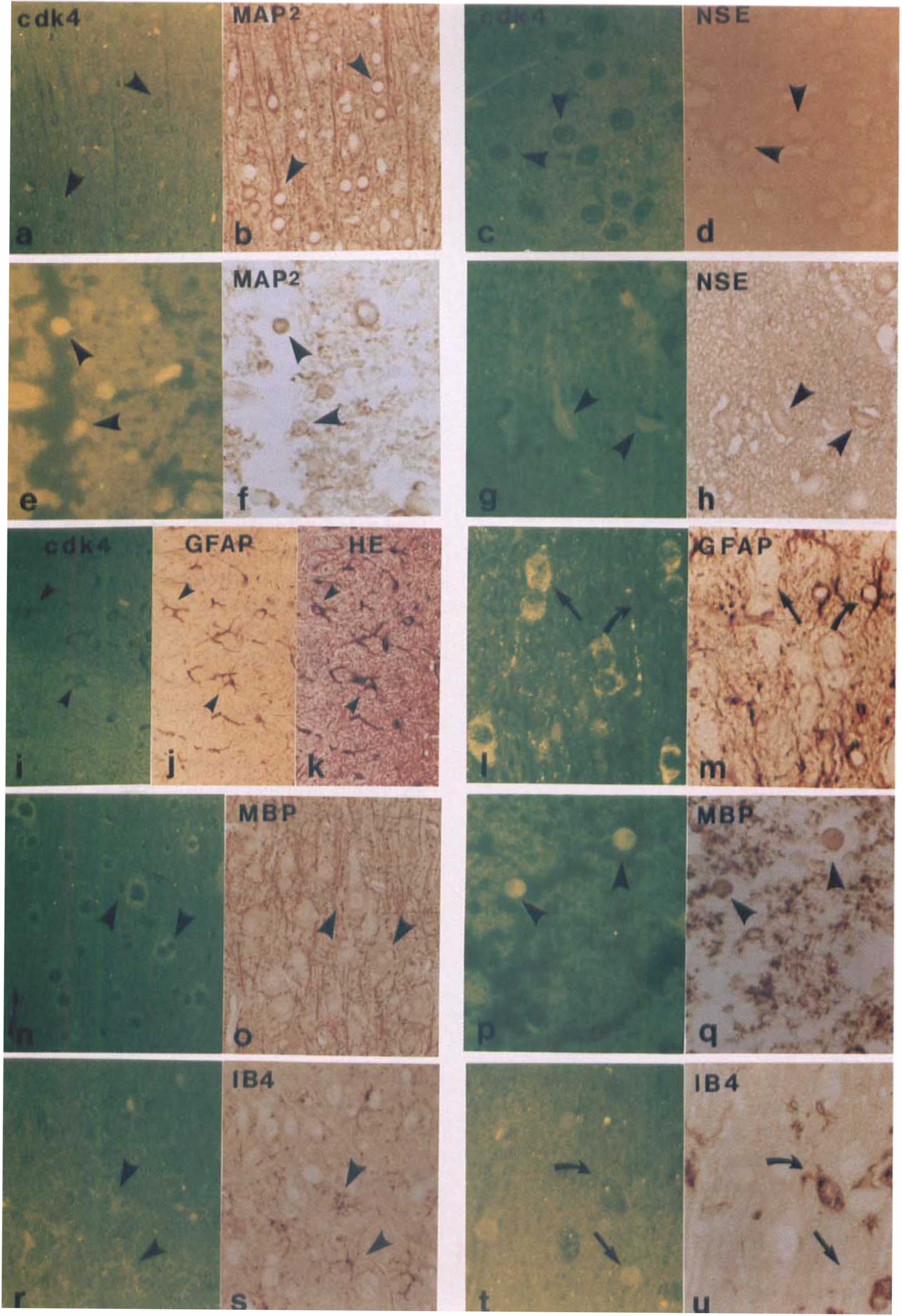
A series of adjacent sections from the same rat as in Fig. 1 are dual immunoreactivity stained by cdk4 (FITC) along with anti-MAP2 and anti-NSE for neurons, anti-GFAP for astrocytes, anti-MBP for oligodendrocytes, and IB4 for microglia, respectively (diaminobenzidine).
Immunoreactivity was used to identify different cell types. In normal brain tissue, immunoreactivities of MAP2 (Figs. 1B and 2B) and NSE (Figs. 1D and 2D) in the neurons were characterized by uniform labeling of both soma and dendrites. Glial cells in nonischemic tissue exhibited positive staining of processes. Clear processes of GFAP (astrocytes) (Figs. 1N and 2J), MBP (oligodendrocytes) (Figs. 1R and 2O), and IB4 (microglia) (Figs. 1V and 2) labeling were observed in the normal glial cells. In the ischemic core, decreased immunoreactivity of MAP2, NSE, and MBP, as well as an almost complete loss of GFAP, were observed in both the striatum and cortex at 46 hours after 2 hours of MCAO (data not shown). However, immunoreactivity of MAP2 (Figs. 1F and 2F), NSE (Figs. 1H and 2H), and MBP (Figs. 1T and 2Q) was expressed in nuclei of scattered cells. The MBP staining of processes of oligodendrocytes was discontinuous and spotty (Figs. 1T and 2Q). In the boundary zone to the ischemic core, immunoreactivity of NSE was not obviously increased. Interestingly, selective accumulation of a beaded MAP2 immunostaining pattern in cytoplasm and dendrites was observed in both outer and inner lesion boundary zones (Figs. 1J and 1L). Selective loss of MAP2 immunoreactivity was observed in the inner boundary zone of the lesion (Fig. 1L). These neuronal changes were accompanied by overexpression of GFAP (Figs. 1P and 2M) and IB4 (Figs. 1X and 2U). In contrast to sham-operated controls, rats with MCAO showed striking GFAP reactivity in astrocytic processes in perifocal regions (Fig. 1P) and white matter tracts (Fig. 2M), concomitant with transformation of GFAP negative into GFAP positive astrocytes. These data are consistent with our previous observation (Li et al., 1995c). Most microglia transformed into lipid-laden macrophages (Figs. 1X and 2U), and microglia obviously increased in number at 46 hours after 2 hours of MCAO.
Cyclin D1 expression
Double immunoreactivity measurements of cyclin D1 expression and cell type in a series of adjacent sections from ischemic and control rats are summarized in Fig. 1 and Table 2.
The expression of cyclin D1 and cdk4 proteins in different cell types after MCAO
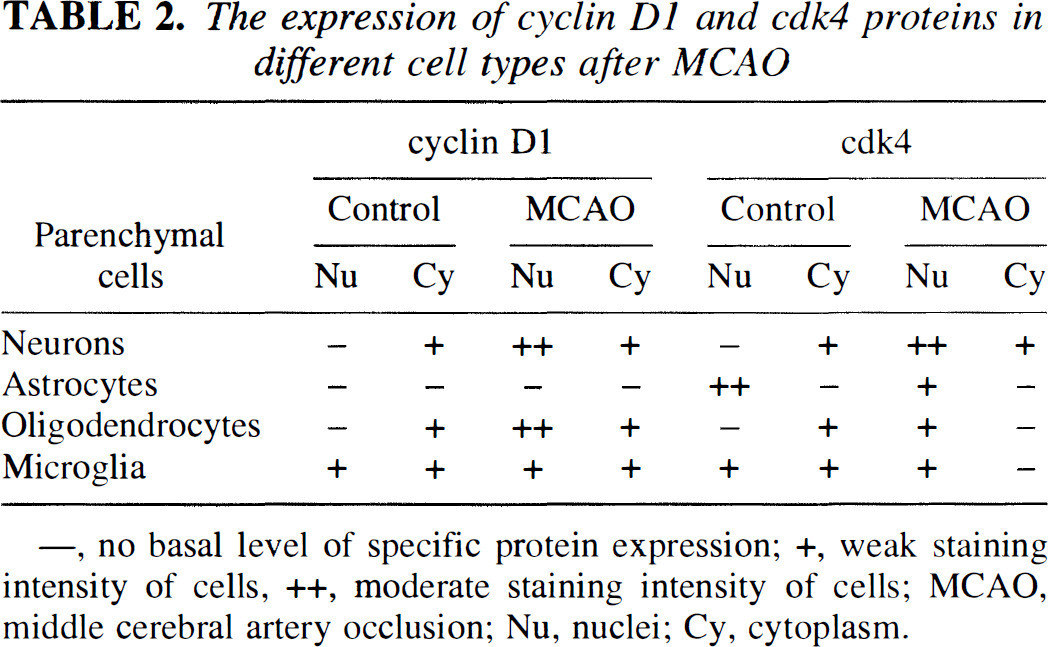
—, no basal level of specific protein expression; +, weak staining intensity of cells, ++, moderate staining intensity of cells; MCAO, middle cerebral artery occlusion; Nu nuclei; Cy, cytoplasm.
Neurons.
Cyclin D1 was detected in neuronal cytoplasm and dendrites (Figs. 1A and 1C) in nonischemic brain. Morphologically intact neurons were indicated by immunoreactivity for the neuronal specific markers of MAP2 (Fig. 1B) and NSE (Fig. 1D). At 46 hours after 2 hours of MCAO, cyclin D1 was present in the nuclei of neurons in the ischemic core (Figs. 1E and 1G), as indicated by MAP2 (Fig. 1F) and NSE (Fig. 1H) immunohistochemical study. In the boundary zone of the lesion, both cyclin D1 and NSE were slightly increased in soma of morphologically intact neurons. The immunoreactivities of both cyclin D1 and MAP2 were increased in soma of morphologically relatively intact neurons (Figs. 1I and 1J) in the outer boundary zone, but a reciprocal staining pattern could be seen in adjacent neurons in the inner boundary zone (i.e., loss of MAP2 and accumulation of cyclin D1 in some neurons, and loss of cyclin D1 in other neurons, which were overexpressed MAP2) (Figs. 1K and 1L).
Astrocytes.
Cyclin D1 was not present in astrocytes (Fig. 1M), as indicated by GFAP staining (Fig. 1N) in nonischemic tissue. After MCAO, cyclin D1 (Fig. 1O) was not detected within the reactive astrocytes, as indicated by GFAP overexpression (Fig. 1P) in the boundary zone of the lesion.
Oligodendrocytes.
Cyclin D1 was present in the cytoplasm of morphologically intact oligodendrocytes (Fig. 1Q), as indicated by light brown stained MBP immunoreactivity (Fig. 1R) and by morphologic features, abutting or indenting the soma of larger neurons. After MCAO, cyclin D1 was intensely expressed in the nuclei of oligodendrocytes in the ischemic areas (Fig. 1S), as indicated by the MBP staining (Fig. 1T).
Microglia.
Cyclin D1 was weakly expressed in the nuclei and the cytoplasm of morphologically intact microglia (Fig. 1U), as indicated by IB4 staining (Fig. 1V). After MCAO, cyclin D1 was detected in microglia but did not obviously increase from preischemic levels (Figs. 1W and 1X).
Cdk4 expression
Comparison of cdk4 expression with different cell types using double immunoreactivity in a series of adjacent sections from rats subjected to 2 hours of MCAO and killed at 46 hours and control rats are summarized in Fig. 2 and Table 2.
Neurons.
In nonischemic tissue, cdk4 was detected in neuronal cytoplasm (Figs. 2A and 2C), as indicated by immunoreactivity for the neuronal specific markers of MAP2 (Fig. 2B) and NSE (Fig. 2D). Cdk4 was present in the nuclei and the cytoplasm of some neurons in the ischemic core (Figs. 2E and 2G), as indicated by MAP2 (Fig. 2F) and NSE (Fig. 2H) staining.
Astrocytes.
Cdk4 was present in nuclei of astrocytes (Fig. 2I), as indicated by well-defined star-shaped processes, rich in GFAP staining (Fig. 2J) as well as by hematoxylin counterstaining (Fig. 2K). However, cdk4 immunoreactivity did not increase in astrocytes after MCAO ischemic areas (Figs. 2L and 2M).
Oligodendrocytes.
Cdk4 was present in the cytoplasm of morphologically intact oligodendrocytes (Fig. 2N), as indicated by MBP immunoreactivity (Fig. 2O) and by observations with hematoxylin and eosin staining of oligodendrocyte abutment of neuronal soma. After MCAO, cdk4 was expressed in the nuclei of the oligodendrocyte soma (Fig. 2P), as indicated by the MBP staining (Fig. 2Q).
Microglia.
Cdk4 was weakly expressed in the nuclei and the cytoplasm of morphologically intact microglia (Fig. 2R), as indicated by IB4 staining (Fig. 2S). After MCAO, cdk4 was weakly detected in microglia and did not obviously increase (Figs. 2T and 2U).
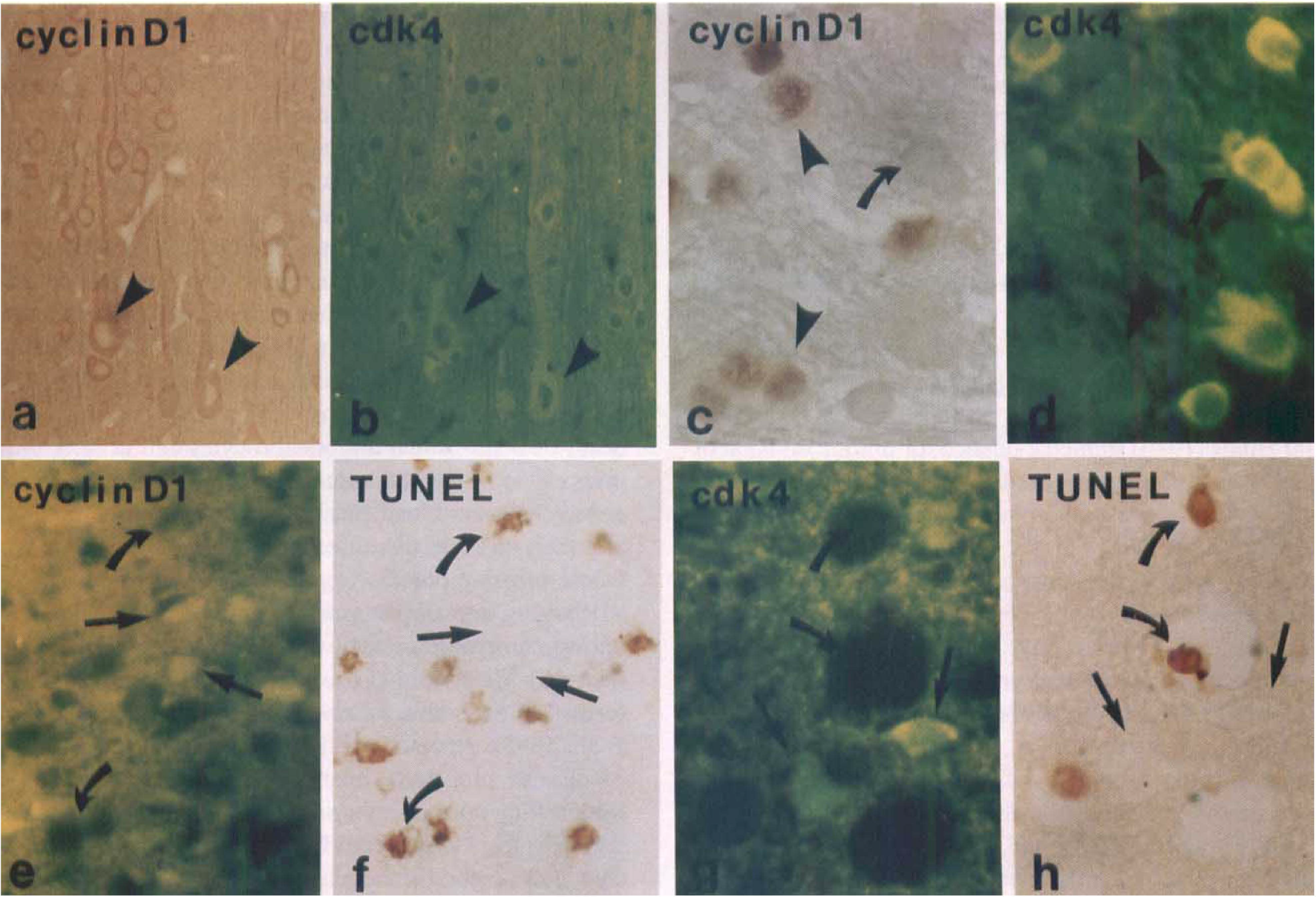
Dual immunohistochemical staining of cyclin D1 and cdk4
Dual immunohistochemical staining of cyclin D1 (Fig. 3A) and cdk4 (Fig. 3B) showed the expression of the two proteins colocalized to the cytoplasm and dendrites of nonischemic cells. After MCAO, co-localization of cyclin D1 (Fig. 3C) and cdk4 (Fig. 3D) was present in some cells, but not in all cells displaying increased levels of the cdk4 protein (Fig. 3D). Comparison of cyclin D1 and cdk4 expression with hematoxylin counterstaining indicated that the expression cyclin D1 and cdk4 was mostly localized to the morphologically relatively intact cells or dark-shrunken cells, but not in red or ghost cells. To determine the expression of cyclin D1 and cdk4 in apoptotic cells induced by MCAO, we employed double staining (TUNEL/FITC immunohistochemical study). No cyclin D1 (Fig. 3E) and cdk4 (Fig. 3G) were found in apoptotic cells (Figs. 3F and 3H). Cyclin D1- and cdk4-positive cells were not apoptotic.
DISCUSSION
The current results obtained from both immunohistochemical and histochemical methods provide direct evidence that cyclin D1 and cdk4 are present in normal and ischemic rat brain. Cellular distribution of these cell cycle proteins is cell type specific. Immunoreactivity to cyclin D1 and cdk4 is expressed in neurons and oligodendrocytes, as well as microglia, in normal tissue. At 46 hours after 2 hours of MCAO, there is an increased expression or intracellular redistribution of cell cycle proteins in morphologically intact and injured neurons and oligodendrocytes within ischemic tissue. Immunoreactive cyclin D1 (Figs. 1E, 1G, and 1S) and cdk4 (2E, 2G, and 2P) proteins are preferentially localized to nuclei after MCAO. In contrast, they are present in cytoplasm of neurons and oligodendrocytes in control tissue (Figs. 1A, 1C, 1Q, 2A, 2C, and 2N). These two proteins are highly expressed in the boundary zone to the ischemic core. However, ischemia does not affect the expression of cyclin D1 and cdk4 in astrocytes and microglia compared with normal tissue. Cyclin D1 and cdk4 may play distinct roles in different cell populations in the CNS.
We identified the cellular expression of cyclin D1 and cdk4 proteins by morphologic features. However, technical problems associated with immunohistochemical study prevent accurate quantitation of the numbers of different cells within the ischemic areas. Korr and others (1994) reported that immunohistochemical identification of oligodendroglia (anti-carbonic anhydrase II, anti-galactocerebraside, anti-MBP) and astrocytes (anti-GFAP, anti-S-100 protein) in mouse brain is not quantitative. Anti-galactocerebraside and anti-MBP stained almost only the oligodendrocytic processes and, thus, cannot be used in measuring somas in brain. Anti-carbonic anhydrase II stained only a portion of the differentiated oligodendrocytes but not proliferating cells. Anti-S-100 protein recognized all of the astrocytes but also many (interfascicular) oligodendrocytes. Anti-GFAP stained only a few astrocytes in the unlesioned mouse. All astrocytes may become GFAP-immunopositive only after brain damage. Thus, immunohistochemical studies with these antibodies on sections of adult animals cannot provide firm quantitative data of cell numbers.
Cytoskeletal proteins play important roles in maintaining normal neuronal function. Among these cytoskeletal proteins, MAP2 is located mainly in dendrites (Caceres et al., 1984; Morales and Fifkova, 1989). MAP2 is a calpain substrate, and degradation of this protein precedes neuronal degeneration. MAP2 is an early indicator of ischemia-induced neurodegeneration in the gerbil forebrain (Matesic and Lin, 1994; Blomgren et al., 1995). The loss of MAP2 may participate in the initial phase of neuronal dysfunction and dendritic breakdown and may be a first sign of neurodegeneration. Consistent with the gerbil study of forebrain ischemia, our study indicates that the expression of MAP2 decreases in the ischemic core after MCAO in the rats. Moreover, we find selectively overexpressed MAP2 in the morphologically relatively intact and reversibly injured swollen neurons in the ischemic areas. After MCAO, cyclin D1 is primarily overexpressed in morphologically intact and dark-shrunken and angular cells, but not in obviously swollen cells, suggesting the existence of different cell death pathways of damaged shrunken and swollen cells after focal cerebral ischemia.
Generally, among parenchymal cell populations, neurons are the most vulnerable, whereas the glial cells are relatively insensitive to ischemic injury (Duchen, 1992). Our observations support the hypothesis that ischemic death-survival mechanism of brain parenchymal cell types may be distinct and may correlate with cellular specific transcriptional activity and protein expression. After MCAO, changes in the expression of cyclin D1 and cdk4 are present in neurons and oligodendrocytes but not in astrocytes and microglia. Overexpression of cyclin D1 in PC12 cells is sufficient to arrest these cells in G1 phase (Yan and Ziff, 1995). Nonrenewable postmitotic neurons may express their cyclin D1 and cdk4 in an attempt to repair DNA, rather than to reenter cell cycle or apoptosis. Oligodendrocytes, the myelin-forming cells of the CNS, proliferate slowly in the normal adult rat brain (Kaplan and Hinds, 1980). Oligodendrocyte proliferation, and repair and remyelination after injury of myelin sheaths in CNS were reviewed by Blakemore (1982). Oligodendrocytes increase at the periphery of lesions in multiple sclerosis. Cell cycle and proliferation dynamics of adult rat oligodendrocytes stimulated with pituitary extract suggest that 53% of the oligodendrocyte population enters into the proliferative phase (Vick et al., 1992). After transient cerebral ischemia, oligodendrocyte up-regulation and nuclear translocation of their cyclin D1 and cdk4 may suggest an effort to reenter the cell cycle in adult rat brain.
In mammalian brain, initiation of astrocyte proliferation is important during development and may play a role after injury (Kimelberg and Norenberg, 1989). Kaplan and Hinds (1980) studied evidence for mitotic division of glial cells in the normal adult rat brain using [3H]thymidine. Astrocytes respond to nearly every type of injury or disease of the CNS. Gliosis is an astrocyte proliferative response and possibly a repair process (Kimelberg and Norenberg, 1989). Astrocyte reactions taking the form of degeneration, hypertrophy, or hyperplasia have been well documented after cerebral ischemia (Piteto and Halaby, 1993). Microglia also proliferate and contribute to gliosis (Matsumoto et al., 1992). At 46 hours after 2 hours of MCAO, changes of cyclin D1 and cdk4 immunoreactivity in both astrocytes and microglia are not detected. However, intense GFAP or IB4 reactivity are observed in response to ischemic injury. Taken together, these findings suggest that cyclin D1 and cdk4 induction is not involved in the onset of astrocyte and microglial cell proliferation after transient focal cerebral ischemia. We found that cyclin D1 protein was primarily localized to neurons and oligodendrocytes. However, Wiessner and coworkers (1996) report that cyclin D1 messenger RNA is induced in microglia rather than in neurons after transient cerebral ischemia in Wistar rats. In addition to differences between measurements of protein and mRNA, this apparent inconsistency may be attributed to the different models of ischemia employed. They employed a model of global cerebral model produced by four-vessel occlusion (30 minutes); however, we employed a model of transient (2-hour) focal cerebral ischemia.
Using the in situ nick translation procedure, Tobita and colleagues (1995) examined whether ischemia causes DNA damage. Single-strand breaks were detected in the CA1 subfield of the hippocampus 1 hour after 15 minutes of forebrain ischemia in gerbils. These strand breaks were three to five times more frequent than in control brain, indicating that DNA single-strand breaks were accelerated by ischemia. Four hours after recirculation, DNA stand breaks returned to control levels, suggesting that structural damage of DNA had been repaired. We find that at 46 hours after 2 hours of transient focal cerebral ischemia, cell cycle proteins are expressed in “morphologically intact” neurons and oligodendrocytes in ischemic tissue. The expression or relocalization of cellular nuclear immunofluorescence signals of cyclin D1 and cdk4 proteins suggests that cell cycle proteins may be expressed and localized to the nucleus in response to DNA damage and repair. Our observations must be interpreted with caution, since cyclin D1 can have a dual role, that is, in cell proliferation (Baldin et al., 1993) and in programmed cell death (Freeman et al., 1994). In addition, data obtained from a single time point after ischemia do not provide information on the expression of cell cycle proteins in different cell types at earlier or later stages of stroke. We also cannot exclude the possibility of cell cycle protein expression promoting death of neurons and oligodendrocytes cell at later time points. It is of interest to measure the temporal profile of cell cycle proteins and elucidate the role of these proteins and their cascade of expression in parallel with the dynamic ongoing process of ischemic cell damage.
Roughly half of postmitotic neurons die by apoptosis during embryonic or fetal development of the mammalian nervous system (Lo et al., 1995). Many of newly formed oligodendroglia also are destined to die (Yasuda et al., 1995). Apoptotic cell death is relevant to a variety of disease processes, apart from its more established roles during normal embryogenesis and tissue homeostasis. Apoptosis is involved in the neuronal death that occurs after cerebral ischemia (Heron et al., 1993; MacManus et al., 1994, 1995; Sei et al., 1994; Hill et al., 1995; Iwai et al., 1995; Tobita et al., 1995; Volpe et al., 1995; Du et al., 1996). Apoptosis also has been hypothesized to be the result of aberrant cell cycle control (Colombel et al., 1992; Chiarugi et al., 1994; Dou et al., 1995). In vitro data suggest that cyclin D1 and cdk4 regulate a physiologic apoptotic process (Kranenburg et al., 1996). Thus, activation of the cell cycle proteins in terminally differentiated neurons may be important for the induction of apoptosis. Nerve cell death has been reported in several experimental situations where neurons have been forced to reenter the cell cycle after entering the G0, nonmitotic stage. Cyclin D1 is an essential mediator of apoptotic neuronal cell death (Kranenburg et al., 1996). To determine whether an association between cell death and unscheduled cell cycling might be found in conjunction with ischemic events, we examined cyclin D1 and cdk4 in rat brain. In situ terminal transferase end-labeling of fragmented DNA (TUNEL) failed to label cyclin D1- and cdk4-positive cells and suggests that cyclin D1 and cdk4 have different apoptotic functions in vitro and in vivo. Our data suggest that in brain tissue in vivo the expression of cyclin D1 and cdk4 proteins is associated with morphologically intact or reversibly damaged cells in the ischemic areas at 2 days after ischemia and may indicate DNA repair after transient focal cerebral ischemia. However, we cannot exclude the possibility that apoptotic cells without cell cycle protein expression at 2 days may have expressed these proteins at earlier time points.
In summary, our data indicate that cell cycle (cyclin D1 and cdk4) proteins are expressed in postmitotic neurons and oligodendrocytes of nonischemic normal brain. Cyclin D and cdk4 proteins also are expressed in ischemic damaged tissue at 46 hours after 2 hours of transient focal cerebral ischemia. These proteins are not present within morphologically identified apoptotic and necrotic cells in the adult rat. Cyclin D1 and cdk4 immunoreactive cells are primarily localized to the boundary zone of ischemic damaged areas. The expression of cyclin D1 and cdk4 in cellular nuclei of morphologically intact and reversible cells suggests that these cells may be associated with DNA repair. Alternatively, the effect of cyclin D1 and cdk4 on mature neurons could function during cell damage in a way unrelated to cell cycle progression. An intriguing possibility suggested by the expression of cyclin D1 and cdk4 is that after an ischemic insult, the “machinery” for cell cycle is present within parenchymal cells, including postmitotic neurons. Capitalizing on the expression of these proteins may be an avenue to promote brain plasticity.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Ms. Xiuli Zhang and Ms. Qing Wang for technical assistance and Ms. Denice Janus for secretarial support.