
Abstract
Amyloid β peptide (Aβ), a 39 to 43 amino acid fragment of the β-amyloid precursor protein (βAPP), forms insoluble fibrillar accumulation in neurofibrillary tangles and vascular plaques. Aβ has been implicated in neuronal and vascular degeneration in brain regions susceptible to plaque formation because of its cytotoxic effect on neurons and endothelial cells (ECs). The authors used a murine cerebral endothelial cell (CEC) line and primary cultures of bovine CECs to explore the cytotoxic mechanism of Aβ. Aβ 1–40 and Aβ 25–35 peptides caused cell death in a dose-dependent and time-dependent manner. Exposure to either Aβ 25–35 or Aβ 1–40 at 10 μmol/L for 48 hours caused at least 40% cell death. Cerebral endothelial cell death was characterized by nuclear condensation, mitochondrial dysfunction, and nuclear and mitochondrial DNA damage. Aβ 25–35 activated both caspase-8 and caspase-3 in murine CECs. zVAD-fmk, a broad-spectrum caspase inhibitor, prevented Aβ 25–35-induced increase in caspase-3 activity and CEC death. N-acetyl-cysteine, an antioxidant, also prevented Aβ-induced cell death. Together, these findings indicate that Aβ-mediated CEC death is an apoptotic process that is characterized by increased oxidative stress, caspase activation, mitochondrial dysfunction, and nuclear and mitochondrial DNA damage.
Alzheimer's disease (AD) has a characteristic neuropathology consisting of senile plaques, neurofibrillary tangles, cerebrovascular β-amyloidosis, and selective neuronal loss (Wisniewski and Wegiel, 1995; Yankner, 1996; Selkoe, 1999). One of the main constituents of the senile plaques and cerebrovascular amyloid deposits is amyloid β peptide (Aβ), a hydrophobic peptide fragment 39 to 43 amino acids in length derived from an ubiquitously expressed transmembrane glycoprotein β-amyloid precursor protein (βAPP) (Glenner and Wong, 1984; Masters et al., 1985). Differential accumulation of fibrillar Aβ aggregates have been linked to neuronal degeneration in AD brains (Yankner, 1996; Selkoe, 1999). In addition to depositions in the brain parenchyma, Aβ accumulates in cerebral blood vessels in AD (Perlmutter, 1994; Wisniewski et al., 2000). Amyloid β peptide deposition in the cerebral vasculature is a major cause of hemorrhagic and ischemic strokes in the elderly with or without AD (Walker, 1997). Amyloid β peptide has been shown to induce significant damage to endothelial cells (ECs) (Thomas et al., 1996). Collectively, Aβ deposition in cerebrovascular walls and Aβ cytotoxicity to ECs may contribute to the age-dependent degeneration of cerebral vasculature and development of cerebral amyloid angiopathy (CAA).
In vitro studies have shown that Aβ is neurotoxic, inducing metabolic and oxidative stress in cultured neurons (Yankner et al., 1989; Behl et al., 1994). A synthetic truncated Aβ corresponding to amino acids 25–35 of βAPP also forms fibrils and is cytotoxic to neurons by a mechanism similar to that of Aβ 1-40/42 (Yankner et al., 1990; Pike et al., 1993). Although it has been shown that Aβ is cytotoxic to cerebral endothelial cells (CECs) in culture (Price et al., 1997; Preston et al., 1998) and contributes to the alterations in cerebral blood flow and neuronal dysfunction in Alzheimer's dementia (Iadecola et al., 1999), the molecular mechanism of Aβ-induced toxicity to CECs has not been fully explored but may involve formation of oxygen radicals (Harris et al., 1995; Thomas et al., 1996), inhibition of nitric oxide (NO) production (Sutton et al., 1997), and/or disruption of calcium homeostasis (Suo et al., 1997; Crawford et al., 1998). In the current study, the authors characterized the mechanism by which Aβ 25–35 and Aβ 1–40 induced cell death in a murine CEC line and in bovine CECs grown as primary cultures. Results show that Aβ disrupted the integrity of nuclear and mitochondrial DNA, activated caspase-8 and caspase-3, and caused apoptosis in CECs. Aβ-induced cell death could be prevented by zVAD-fmk, a broad-spectrum caspase inhibitor, or N-acetyl-cysteine (NAC), an antioxidant.
MATERIALS AND METHODS
All chemicals and reagents were purchased from Sigma (St. Louis, MO, U.S.A.), and all cell culture supplies were from GIBCO/BRL (Gaithersburg, MD, U.S.A.) unless otherwise specified.
Murine CEC line and bovine CEC culture
The murine CECs (a generous gift from Dr. William Frasier, Department of Pathology, Washington University School of Medicine) were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum at 37°C under 5% CO2.
Bovine CECs were prepared and characterized as previously described (Xu et al., 1998). Briefly, meninges and superficial blood vessels were removed from fresh bovine brains in ice-cold Hank's balanced salt solution with antibiotics. Brain gray matter was homogenized, filtered, and the resulting microvessel fraction then was sequentially digested with 4 mg/mL collagenase B for 2 hours and 1 mg/mL collagenase/dispase (Boehringer Mannheim, Indianapolis, IN, U.S.A.) for 8 hours. After centrifugation over a 40% Percoll solution, the second band containing microvessels was collected and washed before plating onto collagen-coated dishes. Bovine CECs migrating from the microvessels were pooled to form a culture of proliferating ECs, which were propagated in DMEM supplemented with 10% fetal bovine serum, 0.5 mg/mL heparin, and 75 μg/mL EC growth factor (complete culture medium). Bovine CECs of passages 4–15, uniformly positive for factor VIII and vimentin (>95% ECs) and exhibited characteristic bradykinin receptors (Xu et al., 1992), were grown to 80% to 90% confluence before use.
CEC death assessment
Cerebral endothelial cell viability was quantitated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and trypan blue exclusion method as well as measuring the lactate dehydrogenase (LDH) released into the culture medium after Aβ treatment (Koh and Choi, 1987; Shaikh et al., 1997; Xu et al., 1998). The amount of LDH released by cells killed with Triton X-100 was considered maximal cell death or “full kill” (Xu et al., 1998).
Quantification of cytoplasmic histone-associated DNA fragments by ELISA
A Cell Death Detection enzyme-linked immunosorbent assay (ELISA) kit (Boehringer Mannheim) was used to quantitate DNA damage after the induction of cell death. This assay determines the levels of histone-associated DNA fragments including mono-and oligonucleosomes in cell lysates (Duke and Cohen, 1986; Leist et al., 1994), based on a sandwich enzyme-immunoassay protocol using a monoclonal mouse-anti-DNA and histone antibody. The increase in DNA fragmentation compared with the control was determined by calculating the enrichment factor, based on the following mathematical formula:

Assessment of DNA fragmentation by agarose gel electrophoresis
A DNA isolation kit (Promega, Madison, WI, U.S.A.) was used for DNA extraction after the induction of cell death. Cerebral endothelial cells in 100-mm culture dishes in DMEM were treated with 20 μmol/L Aβ 25–35 for 48 hours. After washing with phosphate-buffered saline (PBS, pH 7.4), the cells were harvested and lysed by adding 1.5 mL of cell lysis solution containing RNase A. The high molecular weight chromosomes and proteins were removed by the precipitation solution. DNA in the supernatant was isolated by ethanol precipitation and resuspended in buffer (10 mmol/L Tris-HCl, 1 mmol/L ethylenediamine tetraacetic acid (EDTA), pH 8.0). Ten micrograms per lane of DNA was electrophoresed at 75 V for 2 hours in a 1.5% agarose gel with 0.4 μg/mL ethidium bromide in a Tris-acetate buffer (0.4 mol/L Tris, 0.25 mol/L sodium acetate, 0.22 mmol/L EDTA, pH 7.8), and the separated DNA bands were visualized by ultraviolet transillumination and photographed (Xu et al., 1998).
TUNEL staining
Confluent CECs on coverslip were treated with 20 μmol/L Aβ 25–35 for 24 hours followed by fixation with 4% paraformaldehyde. TDT-mediated dUTP-biotin nick end-labeling (TUNEL) staining was performed according to the protocol provided by ONCOR (Catalog # S7100-kit; Gaithersburg, MD, U.S.A.) (Xu et al., 1998).
Immunocytochemical staining for cytochrome c
Cerebral endothelial cells were treated with 20 μmol/L Aβ 25–35 for 24 hours and then fixed with 4% paraformaldehyde. After 3 washes with PBS, the fixed cells were stained with a primary monoclonal mouse anti-cytochrome c antibody (1:100; PharMingen, San Diego, CA, U.S.A.) followed by a secondary tetra-rhodamine-conjugated goat anti-mouse IgG antibody (1:100, Promega). Optical sections through cultured cells were imaged in z-series with a Noran-Odyssey laser confocal (Noran Instrument, Middleton, WI, U.S.A.) with 568 nm excitation and >590 emission using a 60X oil objective with a N.A. of 1.4 (Nikon, Melville, NY, U.S.A.). Images were collected with a confocal slit aperture of 50 μm and analyzed with an image analysis system (Metamorph; Universal Imaging, West Chester, PA, U.S.A.) (Xu et al., 2000). Nuclei were visualized with 1 μg/mL DAPI (4′, 6-diamidino-2-phenylindole dihydrochloride; Molecular Probes, Eugene, OR, U.S.A.).
Western blotting
Cerebral endothelial cells were treated with 20 μmol/L Aβ 25–35 for 24 hours. The cytosolic protein fraction was isolated and immunoblotted as previously described (Xu et al., 2000). For cytochrome c blotting, cells were homogenized in cytosol extraction buffer (20 mmol/L HEPES-KOH, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L ethylene glycol-bis(beta-aminoethyl ether)-N,N,N′,N′-tetraacidic acid (EGTA), 1 mmol/L dithiothreitol (DTT), 250 mmol/L sucrose, 10 mmol/L succinate, 5 μg/mL leupeptin and 5 μg/mL aprotinin, pH 7.4) and centrifuged at 20,000 g for 15 minutes. For caspase-3 and caspase-8 Western blots, cells were homogenized in buffer containing 10 mmol/L HEPES, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.1% NP40, 0.5 mmol/L DTT, 5 μg/mL leupeptin, and 5 μg/mL aprotinin, pH 7.4. Twenty micrograms of protein from each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred onto polyvinylidene difluoride membranes. Cytochrome c blots were detected using a primary monoclonal mouse anti-cytochrome c antibody (1:1000, PharMingen) followed by a secondary alkaline phosphatase-conjugated goat anti-mouse IgG (1:5,000, Promega). Caspase-3 and caspase-8 were detected with a primary polyclonal rabbit anti-mouse caspase-3 or caspase-8 antibody (both used at 1:200; Santa Cruz Biotechnology, Santa Cruz, CA) followed by a secondary alkaline phosphatase-conjugated goat anti-rabbit IgG (1:5000, Promega) antibody.
Caspase-3 and caspase-8 activity
Cerebral endothelial cells grown to confluence in 100-mm were pretreated for 2 hours with 0, 2, 10, and 50 μmol/L zVAD-fmk or 0, 0.1, 1 and 10 mmol/L NAC, followed by 20 μmol/L Aβ 25–35 for up to 48 hours. The CECs were lysed in lysis buffer (10 mmol/L Tris-HCl, 1% Triton X-100, 0.32 mol/L sucrose, 5 mmol/L EDTA, 1 mmol/L phenylmethyl sulfonyl difluoride (PMSF), 1 μg/mL aprotinin, 1 μg/mL leupeptin, 2 mmol/L DTT; pH 8), sonicated, and centrifuged at 20,000 g for 20 minutes at 4°C. Enzyme activity was detected in supernatants by measuring the proteolytic cleavage of fluorogenic substrates of caspase-3 (Ac-DEVD-7-amido-4 trifluoromethylcoumarin) or caspase-8 (Ac-IETD-7-amido-4 trifluoromethylcoumarin) in assay buffer (100 mmol/L HEPES, 10% sucrose, 0.1% CHAPS, 1 mmol/L PMSF, 1 μg/mL aprotonin, 1 μg/mL leupeptin, 2 mmol/L DTT, pH 7.5) using an excitation wavelength of 400 nm and an emission wavelength of 505 nm. One unit is defined as the amount of enzyme required to produce 1 pmol of amido-4-trifluoromethylcoumarin/min at 25°C at saturating substrate concentration.
Long polymerase chain reaction analysis of mtDNA
Total DNA from murine CECs was isolated using a DNA isolation kit (Qiagen, Chatsworth, CA, U.S.A.). The extent of mtDNA damage was assessed by a modified long polymerase chain reaction (PCR) method (Barnes, 1994; Xu et al., 2001). The 10-μl PCR reaction mixture contained 0.4 ng total DNA, 4 pmol of the oligonucleotide primer pair, 400 μmol/L dNTP, and 0.5 unit of TaKaRa LA Taq (Fisher Scientific, Pittsburgh, PA). An equal amount of total rat brain DNA was added to each reaction as an internal standard. The primers used for amplification of the 14.3-kb rat and mouse mtDNA were as follows: 5-ATATTTATCACTGCTGAGTCCCGTGG-3′ and 5-AATTTCGGTTGGGGTGACCTCGGAG-3′. Samples were first denatured for 1 minute at 94°C and then amplified for 26 cycles, with each cycle consisting of denaturation at 94°C for 15 seconds and primer annealing/extension at 68°C for 10 minutes. The final extension was at 72°C for 10 minutes. The PCR conditions described above were within the linear portion of the curves for both the number of cycles and the amount of total input DNA. The PCR products were treated with NcoI (Promega) at 37°C for 2 hours to specifically digest only the long PCR product derived from the mouse mtDNA template into 7.0-kb and 7.3-kb fragments. The 14.3-kb rat long PCR product was separated from the smaller 7.0/7.3-kb mouse entities by 1% agarose gel electrophoresis. Ethidium bromide–stained DNA bands were quantitated by a phosphoimager (Molecular Dynamics, Sunnyvale, CA, U.S.A.). Reduction in quantity of the mouse long PCR product relative to that of rat was used to assess the extent of mtDNA damage.
Statistical analysis
Quantitative data are expressed as mean ± SD based on at least three separate experiments of triplicate samples. Difference among groups was statistically analyzed by one-way analysis of variance followed by Bonferroni's post hoc test. Comparison between two experimental groups was based on two-tailed t-test. P < 0.05 was considered significant.
RESULTS
Aβ 25–35-induced cell death
Treatment of murine CECs with 0.01 to 50 μmol/L Aβ 25–35 for 48 hours caused cell death in a dose-dependent manner as determined by LDH (Fig. 1A), MTT (Fig. 1B), and trypan blue exclusion assays (Fig. 1C). Ten μmol/L Aβ 25–35 caused more than 40% cell death. The LDH and MTT assays showed that Aβ induced murine CEC death in a time-dependant manner with substantial cell death noted as early as 24 hours after Aβ exposure (Fig. 1D). Aβ 25–35 also induced death of bovine CECs in a dose-dependent manner (Fig. 1E). In most experimental systems the biologic effect of Aβ 25–35 and Aβ 1–40 were comparable. Aβ 1–40 and to a lesser extent Aβ 1–42 are the major components of Aβ deposits (composed of peptides varying from 39 to 43 amino acids in length) in the brain. Aβ 1–42, Aβ1–40, and active fragments of Aβ, such as Aβ 25–35, have been shown to be directly toxic to neurons in culture at high micromolar concentrations (Loo et al., 1993; Behl et al., 1994). Aβ 1–40 has approximately the same potency as Aβ 25–35 in inducing cell death in murine CECs (Fig. 1F).
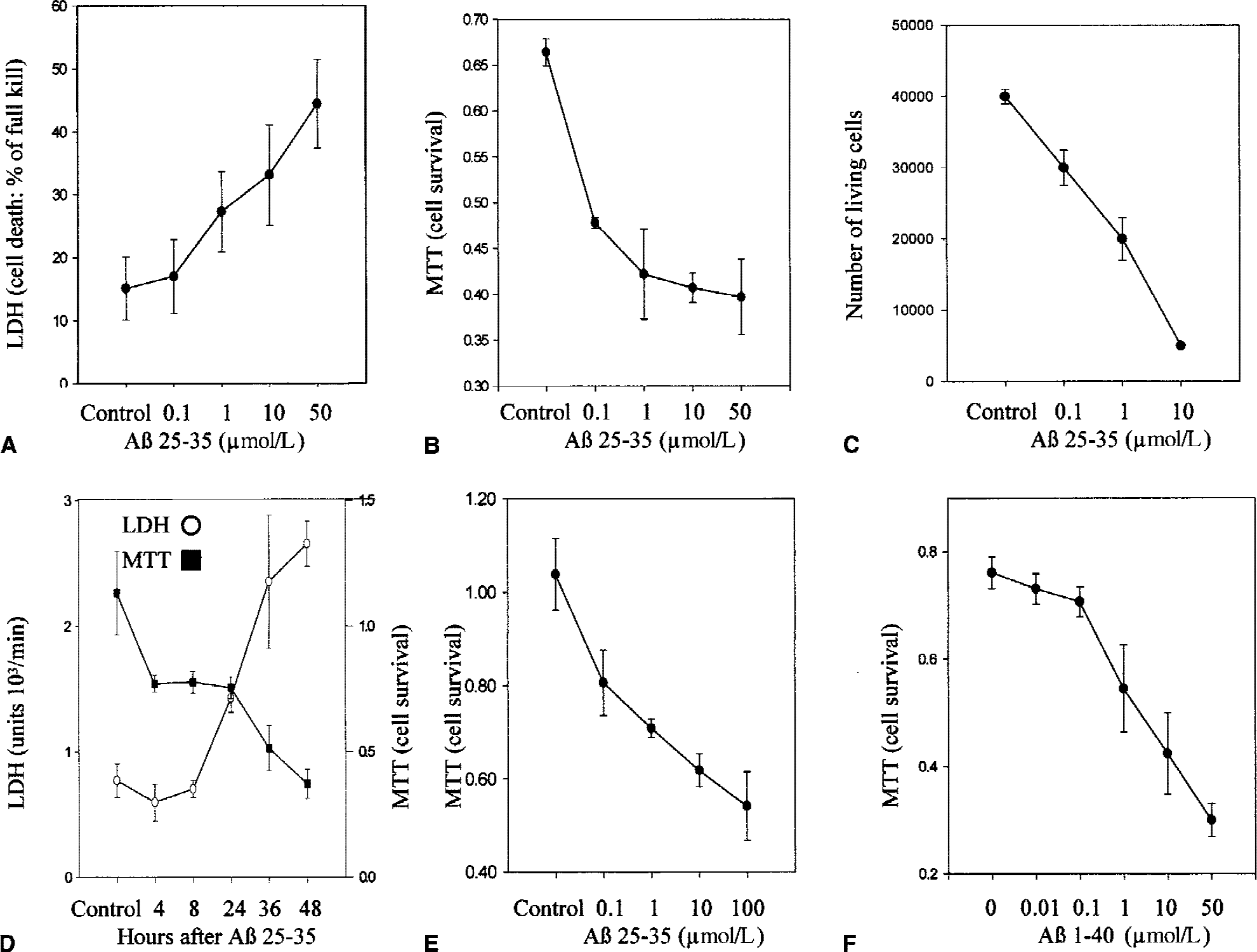
Aβ-induced CEC death. Murine CECs in DMEM without FBS were incubated without (control) or with Aβ 25–35 (0.1 to 50 μmol/L) for 48 hours. The extent of cell death was determined by LDH
Aβ 25–35-induced cell death determined by ELISA, TUNEL, and DNA laddering
Treatment with either 1 or 10 μmol/L Aβ 25–35 for 24 hours caused a significant increase in the cytosolic content of DNA strand breaks as measured by ELISA in murine CECs (Fig. 2A) and in primary bovine CECs culture (data not shown). Gel electrophoretic analysis of DNA extracted from murine CECs treated with 20 μmol/L Aβ 25–35 for 48 hours showed the characteristic DNA laddering (Fig. 2B). Moreover, increased numbers of TUNEL-positive cells were detected in murine CECs treated with 20 μmol/L Aβ 25–35 for 24 hours when compared with the control cultures (Fig. 2C). Together these findings indicate that Aβ induced DNA damage in CECs.
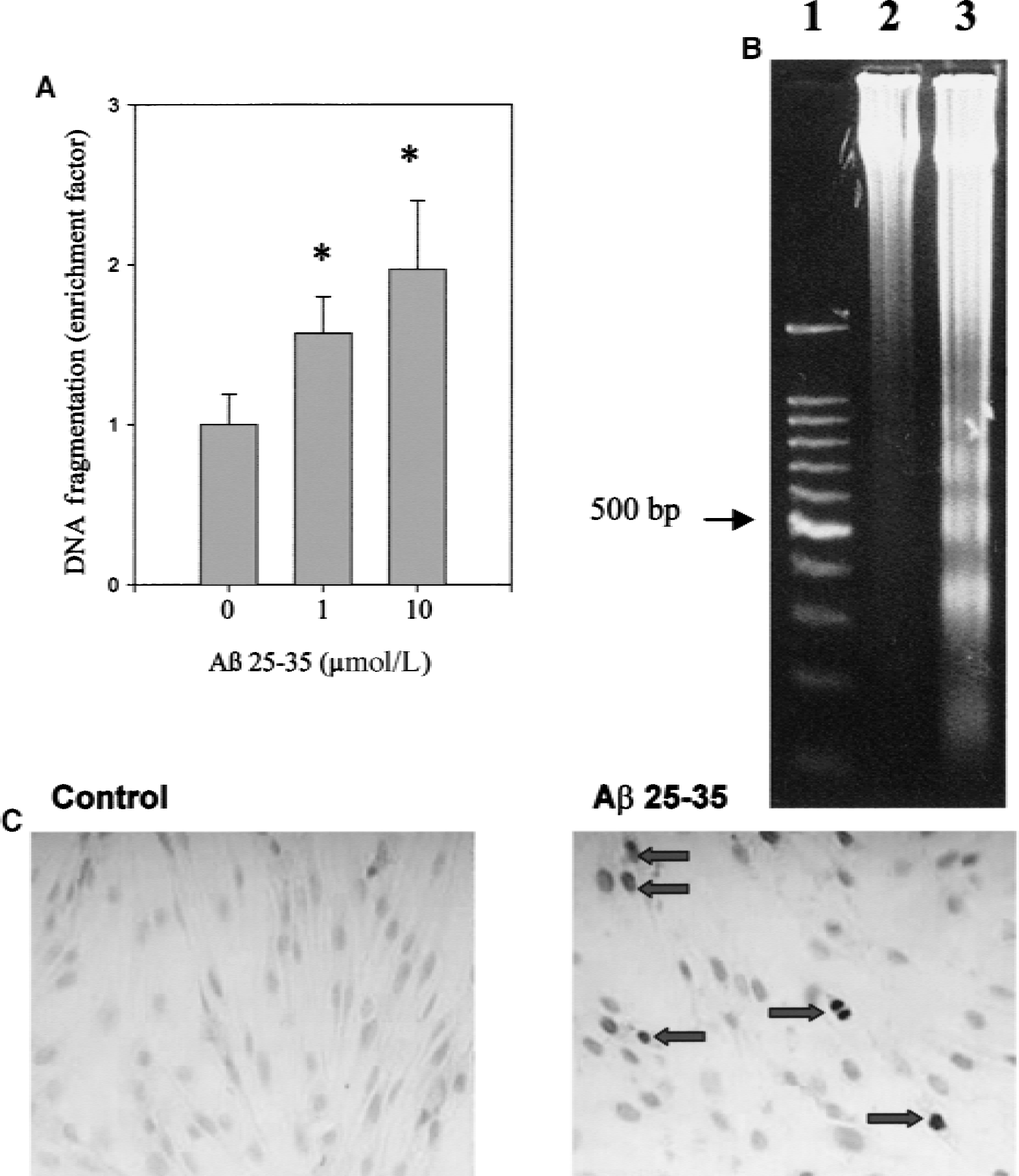
Aβ-induced DNA damage in murine CECs.
Aβ 25–35-induced mitochondrial dysfunction in CECs
The findings that Aβ-induced CEC death was associated with extensive DNA damage suggests an apoptotic cell death mechanism. Mitochondrion is an organelle implicated as the execution center in apoptotic cells (Green and Reed, 1998). Aβ also caused substantial mtDNA damage in murine CECs as demonstrated by a reduction in the quantity of the long PCR products (Fig. 3A and 3B). Another key feature of apoptosis is the release of cytochrome c from mitochondria into the cytosol. Early after the induction of apoptosis, a loss of mitochondrial transmembrane potential can be measured in conjunction with the formation of permeability transition pores (Zamzami et al., 1996). At the same time, cytochrome c is inactivated and then released into the cytoplasm (Krippner et al., 1996; Adachi et al., 1998). Exposure of murine CECs to 20 μmol/L Aβ 25–35 for 24 hours resulted in cytochrome c release as demonstrated by Western blotting (Fig. 3C) and immunohistochemistry (Fig. 3D and 3E). A significant increase in the amount of the 15 kD band corresponding to cytochrome c was detected in the Aβ-treated murine CECs (Fig. 3C). Immunohistochemistry showed in control CECs a punctate pattern of cytochrome c staining and normal appearance of nuclei detected by DAPI counterstain (Fig. 3D), which was easily distinguishable from dying cells with diffuse redistribution of cytochrome c in the cytosol and condensed nuclei (Fig. 3E).
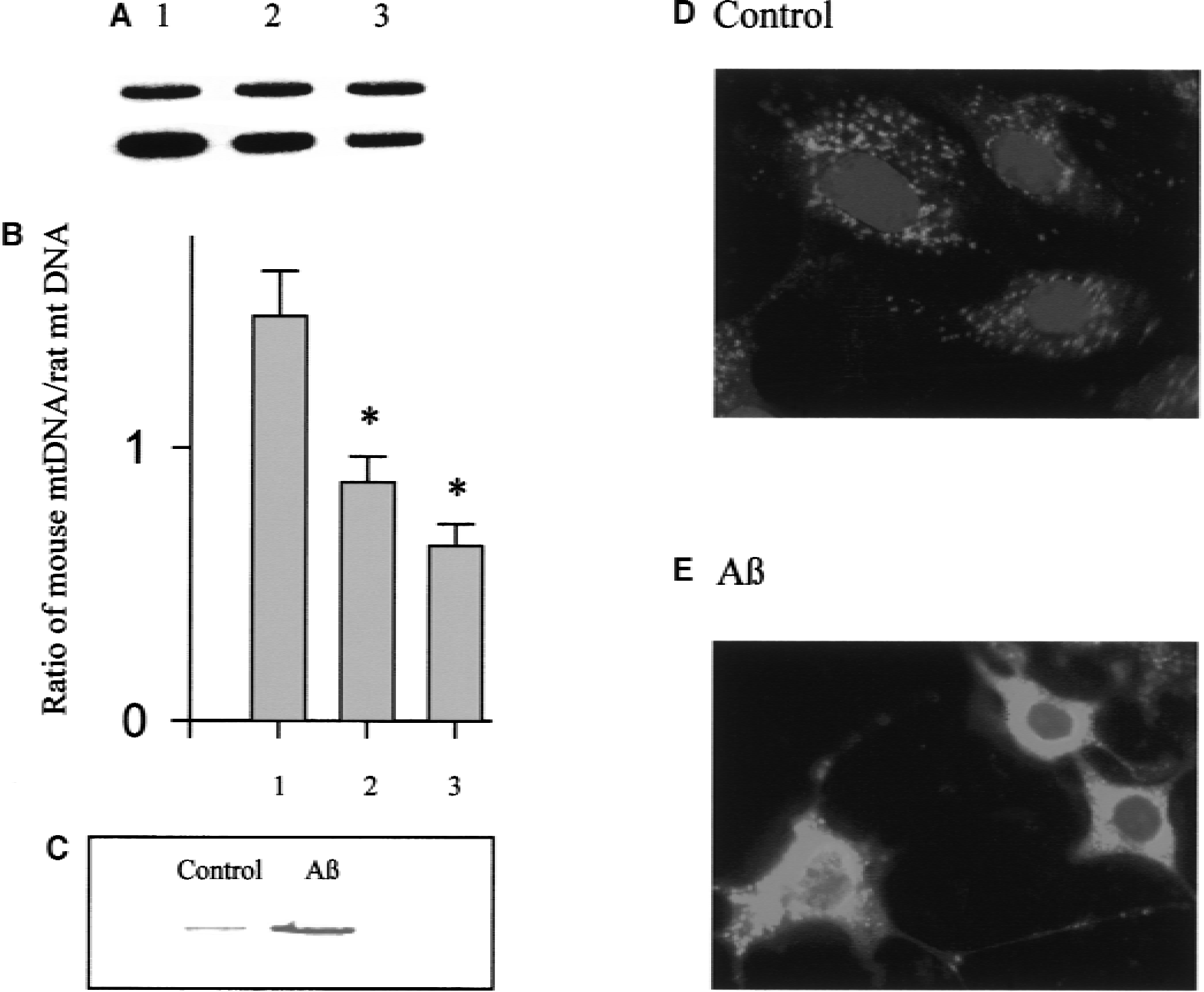
Aβ-induced mitochondrial dysfunction in murine CECs.
Aβ 25–35-induced activation of caspases and zVAD-fmk inhibition of caspase-3 activation
Apoptosis involving mitochondrial dysfunction is frequently accompanied by activation of caspases. CD95-mediated apoptosis in certain cell types is initiated by large amounts of active caspase-8 formed at the death-inducing signaling complex that disrupts mitochondria function and cytochrome c release and subsequent cleavage of caspase-3 (Scaffidi et al., 1999). Thus, Western blot analysis and enzymatic activity assays for caspase-8 and caspase-3 were used to determine if the caspase-8 → mitochondrial dysfunction → caspase-3 pathway was operative in Aβ-induced CEC death. Western blotting for caspase-8 and caspase-3 showed that both the active or 18 kD forms of caspase-8 and caspase-3 protease were expressed at 24 hours in murine CECs treated with 20 μmol/L Aβ 25–35 (Fig. 4). In addition, an increase in caspase-8 and caspase-3 enzymatic activity was detected after Aβ exposure (Fig. 5A). Pretreatment with zVAD-fmk (50 μmol/L) inhibited Aβ-induced increase in caspase-3 activity in murine CECs (Fig. 5B). These results suggest that Aβ exposure resulted in caspase-8 activation and subsequent mitochondrial dysfunction that led to cytochrome c release and the activation of caspase-3.
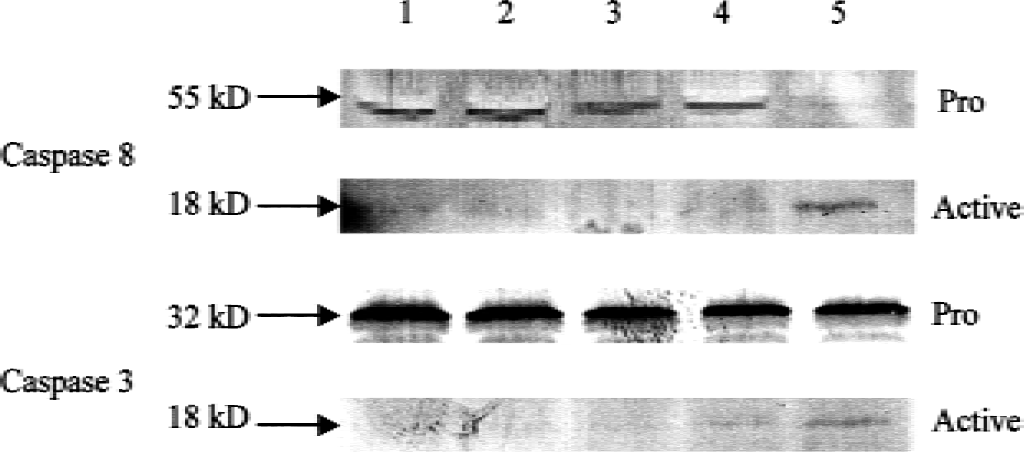
Western blot analysis shows expression of the active form of caspase-3 and caspase-8. The upper band in each panel represents the 55 and 32 kD pro-caspase-8 and −3, respectively, and the lower band in each panel the 18 kD active form. Lane 1: Murine CECs in DMEM plus FBS (normal); lane 2: CECs in DMEM without FBS (control); lane 3: CECs incubated in DMEM without FBS and 20 μmol/L Aβ 25–35 for 4 hours; lane 4: Aβ for 8 hours; lane 5: Aβ for 24 hours. Note the reduction in signal intensity in the pro-caspase bands was accompanied by the appearance of the active forms.
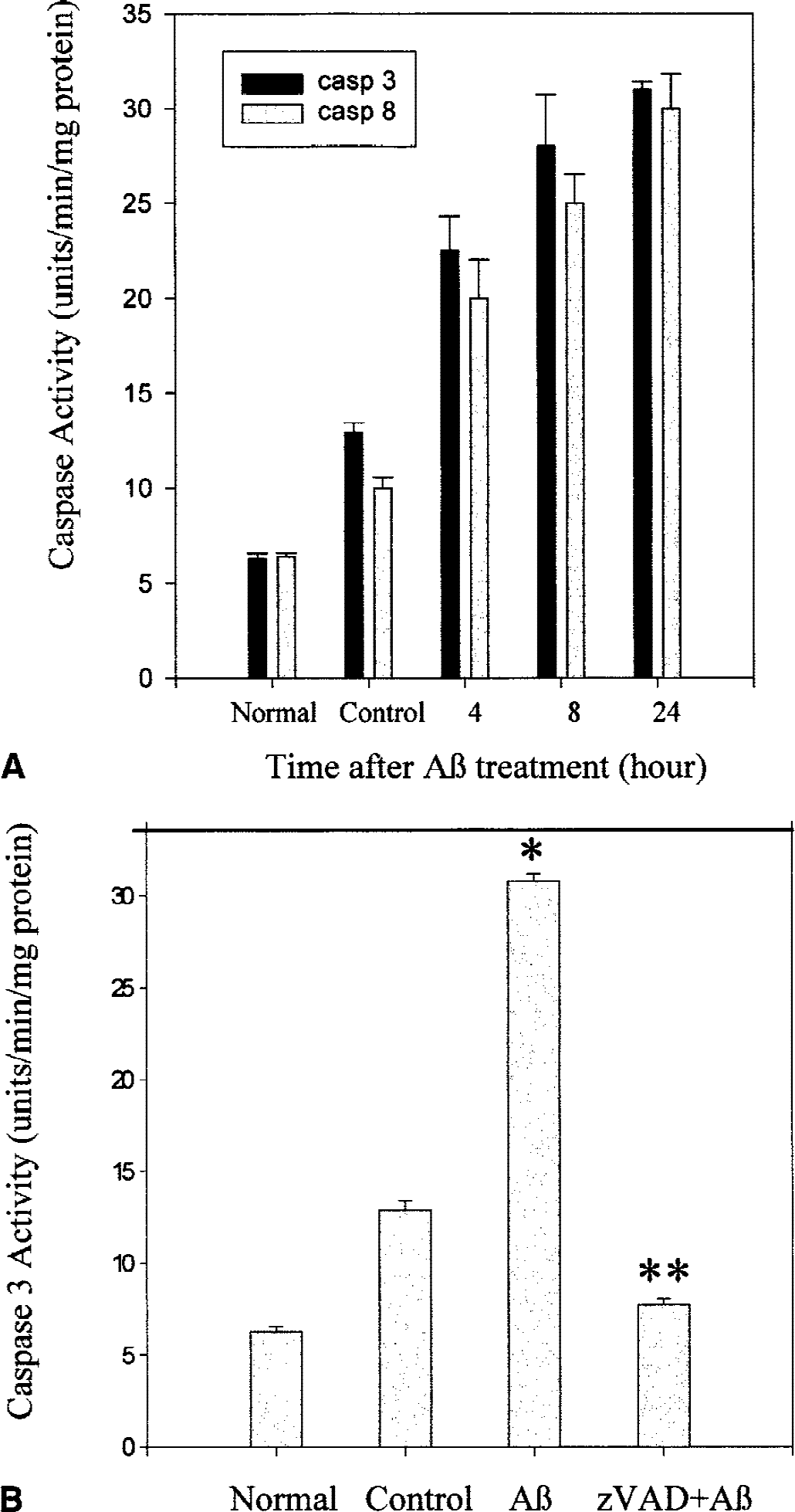
Aβ-induced increase in caspase activity.
Aβ 25–35-induced cell death protected by zVAD-fmk and NAC
To determine whether the activation of caspase-3, a common final pathway of apoptosis, contributes to Aβ-induced CEC death, the authors tested the effect of zVAD-fmk, a broad-spectrum caspase inhibitor. zVAD-fmk, in a dose-dependent manner, prevented Aβ 25–35-induced cell death in murine CECs (Fig. 6). NAC, an antioxidant, also showed a similar protective effect (Fig. 6).
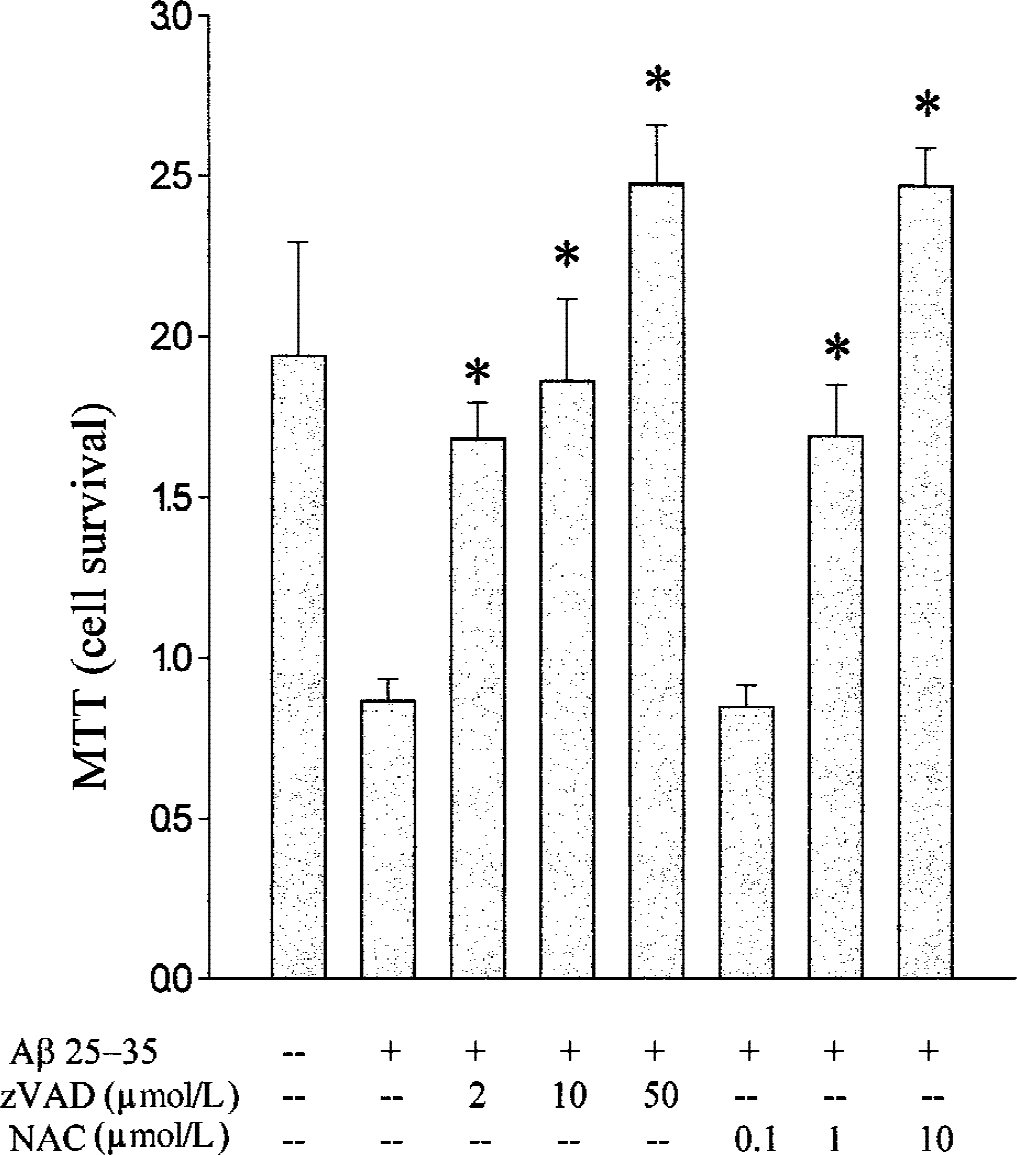
Inhibition of Aβ-induced CEC death by zVAD-fmk and NAC. Murine CECs in DMEM without FBS were pretreated with zVAD-fmk (2, 10, and 50 μmol/L) or NAC (0.1, 1, and 10 mmol/L) for 2 hours before exposure to 20 μmol/L Aβ for 24 hours. Data are expressed as mean ± SD from three separate experiments in triplicate. *Denotes difference from Aβ treatment alone is significant.
DISCUSSION
Endothelial cells lining the cerebral capillaries have tight junctions forming the blood–brain barrier (BBB) when intact protects the brain by rendering it impermeable to cells and many biologically active macromolecules. In the AD brain, the BBB may be breached because of cerebrovascular EC damage (Mattila et al., 1994; Perlmutter, 1994; Mecocci et al., 1995), which could lead to leakage of blood-borne Aβ (Maness et al., 1994), other plasma constituents, and/or neurotoxic substances (Choi, 1995). Aβ-induced endothelial dysfunction may also contribute to the pathology of vascular disorders associated with aging, hypertension, coronary artery disease, cerebral ischemia, vasospasm, and stroke (Sutton et al., 1997; Thomas et al., 1997b).
Aβ 25–35 has been shown to be cytotoxic to ECs (Thomas et al., 1996, 1997a; Blanc et al., 1997; Suo et al., 1997; Sutton et al., 1997). Specifically, Aβ inhibits EC replication (Grammas et al., 1995) and is directly toxic to peripheral and cerebral vascular endothelium (Thomas et al., 1996, 1997a; Price et al., 1997). The exact mechanism(s) that brings about the growth inhibition and cellular toxicity is being studied extensively. Oxidative injury may be one mechanism by which Aβ exerts its cytotoxic effects. Studies have shown that Aβ induces reactive oxygen species (ROS) production, which reacts with the lipids of the cell membrane causing dysfunction of ion-motive ATPases. This ATPase dysfunction leads to membrane depolarization and excessive calcium influx causing cell death (Mark et al., 1995, 1996). An imbalance between NO and ROS may also be responsible for Aβ 25–35-induced EC death (Sutton et al., 1997). In addition, free radical scavengers such as superoxide dismutase and carnosine have been shown to be protective against Aβ 25–35 cytotoxicity (Thomas et al., 1996; Preston et al., 1998).
Both bovine and murine CECs were used to demonstrate the cytotoxic effect of Aβ to CECs manifests across different species. In most experimental systems, the biologic effect of Aβ 25–35 and Aβ 1–40 were comparable. Aβ 1–40 and to a lesser extent Aβ 1–42 are the major components of Aβ deposits (composed of peptides varying from 39 to 43 amino acids in length) in the brain. Aβ 1–42, Aβ1–40, and active fragments of Aβ, such as Aβ 25–35, have been shown to be directly toxic to neurons in culture at high micromolar concentrations (Loo et al., 1993; Behl et al., 1994). The authors have shown previously that Aβ 25–35 and Aβ 1–40 had similar dose effects inducing death in oligodendrocytes (Xu et al., 2001). Using a murine CEC line and primary cultures of bovine CECs as model systems, the authors characterized Aβ-induced CEC death. Both Aβ 1–40 and its truncated fragment, Aβ 25–35, induced CEC death in vitro in a dose-dependent manner. Aβ at a concentration of 10 μmol/L caused more than 40% cell death according to the LDH, MTT, and trypan blue assays. Aβ-induced CEC death was associated with nuclear chromatin condensation, DNA fragmentation, and DNA laddering. These results suggest that CEC exposed to Aβ die by an apoptotic mechanism. Mitochondrial dysfunction has been implicated as a key mechanism in apoptosis in a number of cell death paradigms (Green and Reed, 1998). Mitochondrial dysfunction leading to apoptosis may be initiated by the activation of a caspase upstream of the death cascade, caspase-8 (Gross et al., 1999). In the current study, the authors demonstrated Aβ-induced activation of caspase-8 by Western blotting and an enzymatic assay.
At least two major events have been noted in apoptosis involving mitochondrial dysfunction. One event is the change in the membrane permeability transition and the subsequent release of a 50 kD protein apoptosis-inducing factor from the intermembrane space of mitochondria (Susin et al., 1997). The other is the release of cytochrome c, an extrinsic protein found on the outer surface of the inner mitochondrial membrane, into the cytosol (Liu et al., 1996; Kluck et al., 1997; Yang et al., 1997). Cytochrome c is released into the cytosol of murine CECs after Aβ 25–35 treatment and thus is a reliable indicator of mitochondrial dysfunction in the Aβ-induced CEC death paradigm. The Aβ-induced mtDNA damage noted in the current study provides further support for mitochondrial dysfunction in CEC death.
Downstream events after cytochrome c release involve a cytosolic protein fraction known as apoptosis activating factor 1 (Apaf1) found in apoptotic cells (Liu et al., 1996). Apaf1 in the presence of cytochrome c and dATP binds to the N-terminus of pro-caspase-9 and formation of this complex in vitro result in activation of caspase-9, which in turn cleaves and activates caspase-3 (Li et al., 1996; Skulachev, 1998; Susin et al., 1999). Caspase-3 is the common final event in numerous apoptotic pathways, being responsible wholly or partially for the proteolytic cleavage of many key proteins involved in apoptosis (Cohen, 1997). The authors also noted Aβ activation of caspase-3 by Western blotting and an enzymatic assay.
Together, these findings are in agreement with the contention that Aβ-induced CEC death entails an apoptosis paradigm involving a number of sequential events including caspase-8 activation, mitochondrial dysfunction, and caspase-3 activation. The fact that caspase-3 activation is consequent to Aβ-induced CEC death is supported by the observation that zVAD-fmk reduced caspase-3 activity as well as cell death.
Another possible role for mitochondria in the apoptotic pathway is the alteration of the redox state. An increase in intracellular concentration of oxidized glutathione (GSSG), an event that precedes DNA damage, is caused partially by excessive peroxide production by mitochondria in apoptotic cells. Within the mitochondrial matrix, an essential cellular enzyme manganese-containing superoxide dismutase converts superoxide radical to hydrogen peroxide and molecular oxygen (Li et al., 2000). A direct relation between GSSG formation and mtDNA damage has been described previously (Esteve et al., 1999), thus supporting the role of mitochondria-related oxidative stress in apoptosis induction. In Aβ-induced CEC death, mtDNA damage was detected and may reflect mtDNA oxidation occurring during the CEC apoptotic process. Oxidative stress can lead to the release of cytochrome c from the mitochondria into the cytosol in cells induced to die with NO (Ushmorov et al., 1999). Moreover, activation of caspase-3 by oxidative stress is differentially regulated, with involvement of p38, a mitogen-activated protein kinase family member, and caspase-8 acting upstream of Bid in the singlet oxygen-mediated apoptotic pathway (Zhuang et al., 2000). In line with the possible involvement of oxidative stress in mitochondrial dysfunction and apoptosis (Fujimura et al., 2000; Hurn and Macrae, 2000), the authors noted that NAC, a potent antioxidant that replenishes glutathione stores in cells, protected CECs against Aβ 25–35-induced cell death. It remains to be determined whether the two mechanisms—caspase activation and oxidative stress—that mediate Aβ-induced EC death act sequentially or independently of each other.
In summary, Aβ 25–35-induced cell death in CEC is characterized by a number of biochemical and morphologic features indicative of apoptosis. Cerebral amyloid angiopathy is characterized by amyloid deposition in the wall of small cerebral and meningeal blood vessels. The notion that Aβ is cytotoxic to CECs suggests Aβ may play a causal role contributing at least partly to the pathogenesis of CAA. Understanding the molecular mechanism of this death paradigm may aid in future development of therapeutic strategies to counteract age-dependent degeneration of the cerebrovascular system and CAA caused by Aβ deposition.