
Abstract
Patterns of hypoxic-ischemic brain injury in infants and children suggest vulnerability in regions of white matter development, and injured patients develop defects in myelination resulting in cerebral palsy and motor deficits. Reperfusion exacerbates the oxidative stress that occurs after such injuries and may impair recovery. Resuscitation after hypoxic-ischemic injury is routinely performed using 100% oxygen, and this practice may increase the oxidative stress that occurs during reperfusion and further damage an already compromised brain. We show that brief exposure (30 mins) to 100% oxygen during reperfusion worsens the histologic injury in young mice after unilateral brain hypoxia—ischemia, causes an accumulation of the oxidative metabolite nitrotyrosine, and depletes preoligodendrocyte glial progenitors present in the cortex. This damage can be reversed with administration of the antioxidant ebselen, a glutathione peroxidase mimetic. Moreover, mice recovered in 100% oxygen have a more disrupted pattern of myelination and develop a static motor deficit that mimics cerebral palsy and manifests itself by significantly worse performance on wire hang and rotorod motor testing. We conclude that exposure to 100% oxygen during reperfusion after hypoxic-ischemic brain injury increases secondary neural injury, depletes developing glial progenitors, interferes with myelination, and ultimately impairs functional recovery.
Introduction
Hypoxic-ischemic brain injury is one of the most common causes of death and long-term neurologic morbidity in both the neonatal and pediatric populations (Volpe, 2001). Resuscitation during reperfusion is important in determining the magnitude of additional injury that occurs after the initial insult. Secondary injury after ischemic stress is at least partially mediated by the presence of free radicals, which is exacerbated in the presence of high oxygen levels (Halliwell, 1992). Despite this, it is standard practice in neonates, children, and adults to resuscitate and recover patients after hypoxic-ischemic injury with 100% oxygen (Kattwinkel et al, 1999).
There are conflicting data regarding whether resuscitation with 100% oxygen after hypoxic injury is harmful in animal models (Munkeby et al, 2004; Zwemer et al, 1995). The reasons for this include the heterogeneity of models tested and an inability to establish a link between the type of resuscitation used and the functional neurologic outcome that goes beyond biochemical measurements of potentially harmful intermediates. Randomized controlled trials in human subjects are sparse, although evidence suggests improved outcomes in neonates resuscitated with room air after asphyxial birth depression (Saugstad et al, 1998). A recent meta-analysis concluded that room air resuscitation resulted in a lower mortality rate but recommended that additional studies are needed on long-term developmental outcome (Rabi et al, 2007).
Periventricular leukomalacia is the most commonly observed central nervous system abnormality in premature infants after hypoxic-ischemic injury, and although the mechanisms underlying this are still under investigation, damage to developing white matter is associated with long-term neurologic disability (Volpe, 2001). One potential mechanism is the vulnerability of myelinating progenitors to injury from oxidative stress.
Brain development is far from complete at the time of birth, as greater than two-thirds of human brain growth occurs postnatally (Dobbing and Sands, 1973). Brain growth after birth is almost exclusively glial in nature in both humans and rodents, and completion of white matter development and myelination in humans is ongoing in the first few years of life. The genesis of myelinating oligodendrocyte progenitor cells peaks around 2 weeks of age in rodents, whereas the majority of actual myelination occurs between 3 and 6 weeks (Sauvageot and Stiles, 2002). Therefore, central nervous system insults that occur at a time when there are abundant immature oligodendrocytes may be amplified because those cells are particularly sensitive to the effects of oxidative stress after hypoxic-ischemic insults.
The primary purpose of this study was to determine whether recovery in 100% oxygen after hypoxic-ischemic brain injury would exacerbate neuronal injury and impair functional recovery in young mice. Given what is currently known about the development of myelination and oligodendro-genesis in the rodent brain, we chose to subject young (postnatal day 14) mice to the Rice—Vannucci model of hypoxic-ischemic injury followed by recovery in either room air or 100% oxygen for various time points from 15 mins to 4 h (Rice et al, 1981). By using a transgenic mouse model that marks neural and glial progenitor populations with green fluorescent protein (GFP), we were able to distinguish what cell types were most vulnerable to hyperoxic recovery-mediated injury (Yu et al, 2005). We observe that a relatively brief exposure to hyperoxia after hypoxic-ischemic injury results in increased histologic injury, generation of oxidative damage, depletion of cortical oligodendrocyte progenitors, and functional motor deficits.
Materials and methods
Mice
All protocols involving the use of animals were approved by the Institutional Animal Care and Research Advisory Committee (IACRAC) at University of Texas Southwestern Medical Center at Dallas. Transgenic mice expressing GFP under control of the nestin promoter and second intron (nestin-GFP) previously generated in our lab were back-crossed into the CD1 background with wild-type CD1 mice (Charles River Laboratories, Wilmington, MA, USA) for eight generations. Offsprings were genotyped according to a previously published protocol, and nestin-GFP transgenic animals were used for all experiments (Yu et al, 2005). Pups were housed under a 12:12-h light—dark cycle, with food and water available throughout the study. A total of 242 mice were used for all of the experiments.
Hypoxic-Ischemic Injury and Recovery
Postnatal day 14 littermates were exposed to hypoxic-ischemic injury for 30 mins with 8% oxygen as an adaptation of the Rice—Vannucci model and randomized to either recovery in room air or 100% oxygen (Rice et al, 1981). Pups were anesthetized for < 10 mins with a mixture of isoflurane (4% for induction and 1.5% for maintenance), 30% oxygen, and balance nitrogen. Under sterile technique, a midline neck incision was made, and the right common carotid artery was exposed, isolated from the nerve and vein, and permanently ligated with 5–0 surgical silk. Temperature was maintained with a warming blanket at 36.5°C to 37.5°C. The incision was closed with a surgical wound clip and the pups were recovered for 1 h in their cage. The small percentage of mice suffering a stroke after ligation alone (about 5%) were euthanized and not included in the study. Animals were then placed in a constructed acrylic glass chamber with separate partitions on a warming blanket maintained at 37°C and exposed to premixed gas of 8% oxygen (balance nitrogen) at 2 L/min for 30 mins. Mice were then randomized to either return to their cage to recover in room air or to remain in the chamber to recover in 100% oxygen for defined time periods of 15 mins (
Ebselen Administration
Mice underwent carotid ligation and exposure to 8% oxygen for 30 mins as above and then randomized to receive intraperitoneal injection of ebselen (2-phenyl-1,2-benzoisoselenazole-3(2
Western Blot
Animals were divided into six groups: sham with no exposure to hyperoxia (
Histology and Immunohistochemistry
Three days after injury, animals were anesthetized with a ketamine/xylazine mixture and perfused with 20 mL of PBS followed by 20 mL of 4% PFA (paraformaldehyde) in PBS. Brains were dissected and postfixed overnight at 4°C in 4% PFA. Whole brains were blocked in 3% agarose and cut into 50 μm coronal sections on a vibratome (Leica VT 1000S).
Sections were stained with cresyl violet (Sigma-Aldrich, St Louis, MO, USA) to assess neuronal morphology after injury. Brains were scored independently by two blinded investigators using a previously described scoring system and discrepancies were resolved by taking a mean of the two assigned scores (Sheldon et al, 1998). Eight regions of the brains were scored: the anterior, middle, and posterior cortex, CA1, CA2, CA3, and dentate gyrus of the hippocampus, and caudate putamen, with the contralateral side serving as a reference for uninjured tissue. Each region was given a score from 0 to 3: 0 = no detectable neuronal cell loss; 1 = small focal areas of neuronal cell loss; 2 = columnar damage in the cortex involving predominantly layers II to IV or moderate cell loss in the hippocampus; and 3 = cystic infarction. The score for each region was summed for a final score ranging from 0 to 24. Animals that died before randomization were excluded from the study; animals that died after randomization (and before perfusion at 3 days) were included in their assigned group and given a maximal injury score of 24 if, upon post-mortem examination, they showed gross evidence of ipsilateral cerebral infarction.
For immunostaining, free floating 50 μm vibratome sections were permeabilized with 0.3% Triton X-100 PBS, washed three times, and blocked with 5% normal donkey serum for 2 h. Slices were incubated overnight at 4°C with primary antibodies: rat anti-PDGFRα (1:250; BD Pharmingen, Franklin Lakes, NJ, USA), rabbit anti-GFP (1:500; Molecular Probes, Carlsbad, CA, USA), rabbit anti-cleaved caspase 3 (1:100; Cell Signaling Technologies, Danvers, MA, USA), and rabbit anti-nitrotyrosine (1:250; Millipore). Colabeling experiments were performed with anti-PDGFRα and anti-nitrotyrosine. Samples were washed again with PBS three times and incubated in secondary antibodies: Cy2 donkey anti-rabbit, Cy3 donkey anti-mouse, and Cy5 donkey anti-rat (1:200; Jackson ImmunoResearch, West Grove, PA, USA) for 2 h at room temperature. Slides were cover-slipped with Fluoromount-G (Southern Biotech, Birmingham, AL, USA) and visualized and photographed using confocal microscopy (Zeiss, Oberkochen, Germany). For the combination of Fluoro-Jade C staining and immunocytochemistry, sections were immunostained as above with anti-cleaved caspase 3, washed with distilled water, and incubated in 0.06% potassium permanganate solution for 5 mins. They were then stained with 0.0004% Fluoro-Jade C (Millipore) for 15 mins, air-dried, and photographed using confocal microscopy. Representative midbrain sections were chosen randomly from ebselen-treated and control mice (100% oxygen only), and the Fluoro-Jade C-positive cells were counted in the CA3 region of the hippocampus.
Staining with anti-myelin basic protein (MBP) was performed in a similar manner, with free-floating slices permeabilized with 0.3% Triton X-100 PBS, washed three times, and blocked with 5% normal goat serum at room temperature for 2 h. Slices were then incubated overnight at 4°C in the primary antibody mouse anti-MBP (1:10; Millipore). Slices were washed and incubated with the secondary antibody biotinylated goat anti-mouse (1:500; Vector, Burlingame, CA, USA) for 2 h at room temperature. Samples were washed with PBS three times, incubated in 0.3% hydrogen peroxide in methanol for 30 mins, and then incubated in Avidin: Biotinylated Enzyme Complex (ABC) solution (Vector) for 2 h. After washing with PBS, slices were added to a well-containing diaminobenzidine (Vector) for 90 secs, washed again, and plated to slides. The slides were then placed in 70%, 95%, and 100% ethanol baths, cover-slipped with Permount (Fisher, Pittsburgh, PA, USA), and visualized with light microscopy (Olympus BX50) and photographed with a color CCD camera using ACT-1 software.
Fluorescent-Activated Cell Sorting
Two litters of nestin-GFP mice with identical birth dates were randomized to 1 of 3 groups: sham (
Rotorod and Wire Hang Analysis
Postnatal day 14 mice underwent randomization into 1 of 3 groups: sham injury (
Physiologic Description of the Model
The respiratory rates of postnatal day 14 sham and ligated mice were monitored before, during, and after hypoxia, with both normoxia and hyperoxic recovery (
Statistics
All statistical analysis was performed using SPSS 14.0. When data were not normally distributed, nonparametric tests (Mann—Whitney
Results
Exposure to Brief Periods of Hyperoxia after Hypoxic-Ischemic Injury Worsens Histologic Injury
To establish whether exposure to 100% oxygen after injury results in exacerbation of cerebral injury, mice underwent hypoxic-ischemic injury for 30 mins and were recovered in either room air or 100% oxygen. Mice were then histologically examined with Nissl (cresyl violet) staining 72 h after injury using a validated scoring system (Sheldon et al, 1998). Figure 1 shows a representative section from the normoxia group with a mild injury, detectable only in the CA1 layer of the hippocampus (Figures 1A to 1E), whereas hyperoxia-exposed mice sustained significantly more injury, represented in Figure 1 as a large unilateral cortical infarct with significant injury in the CA1, CA2, and CA3 areas of the hippocampus (Figures 1F to 1J). Exposure to normoxia (
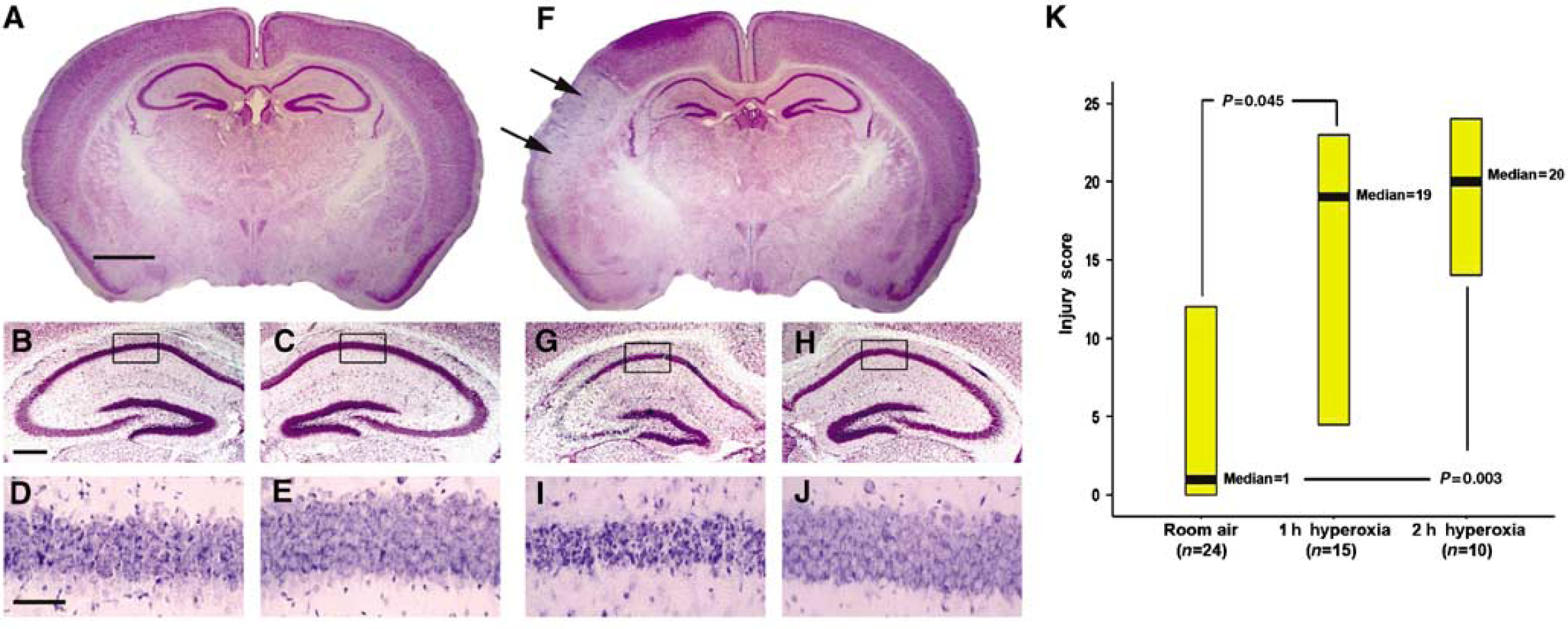
Injury scoring and histologic representation of median scores. (
Oxidant Tissue Injury is Increased in the Hyperoxia-Exposed Brain after Hypoxic-Ischemic Injury
To determine whether hyperoxia during reperfusion results in increased oxidative injury to neuronal tissue, we examined ipsilateral-injured hemispheres from normoxia-exposed and hyperoxia-exposed mice for evidence of protein nitrosylation, an established method for measuring oxidative injury (Tan et al, 1998). Nitrotyrosine is formed when tyrosine is nitrated by peroxynitrite, a toxic-free radical formed when superoxide and nitric oxide combine. Exposure for 1, 2, or 4 h consistently shows the formation of high-molecular-weight protein nitrosylation products from increased formation of the nitric oxide-derived species (Figure 2A). When quantified using densitometry, hyperoxic recovery for either 1 h (
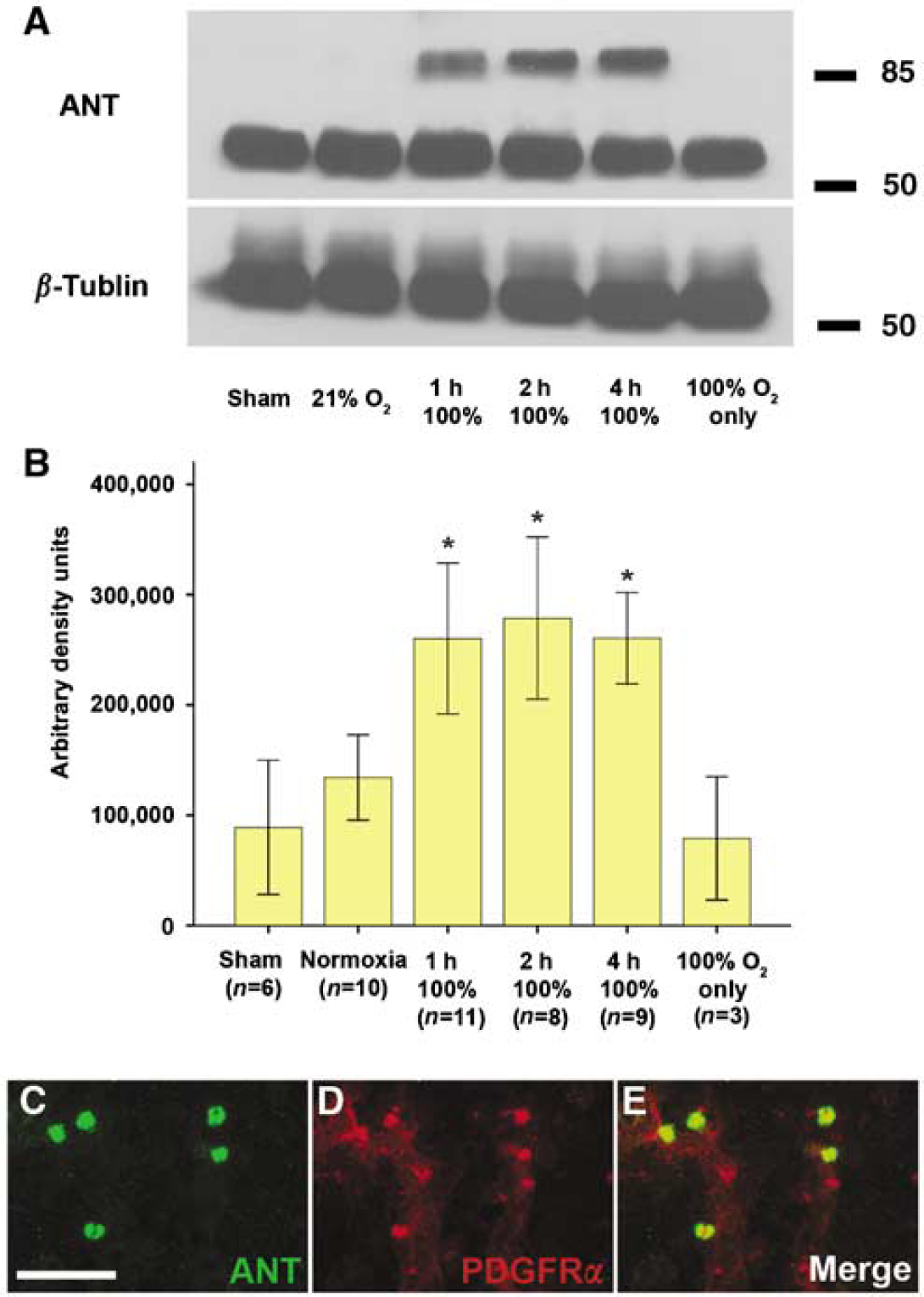
Increasing accumulation of the oxidative metabolite nitrotyrosine and localization to glial progenitors. (
Exposure to as Little as 30 mins of Hyperoxia after Injury Worsens Histologic Injury, and Injury can be Reversed with the Antioxidant Glutathione Peroxidase Mimetic Ebselen
After identifying that exposure to either 1 or 2 h of 100% oxygen during recovery increases injury susceptibility, we sought to determine the boundaries of toxicity by exposing injured mice to various times of 100% oxygen after hypoxia—ischemia. We found a consistent increase in histologic injury in mice exposed to hyperoxic recovery between 30 mins and 4 h compared with normoxic recovery. However, there was no difference with increasing duration of hyperoxic recovery, suggesting that longer exposure to 100% oxygen does not produce further injury (Figure 3A). Interestingly, mice exposed to only 15 mins of hyperoxic recovery (
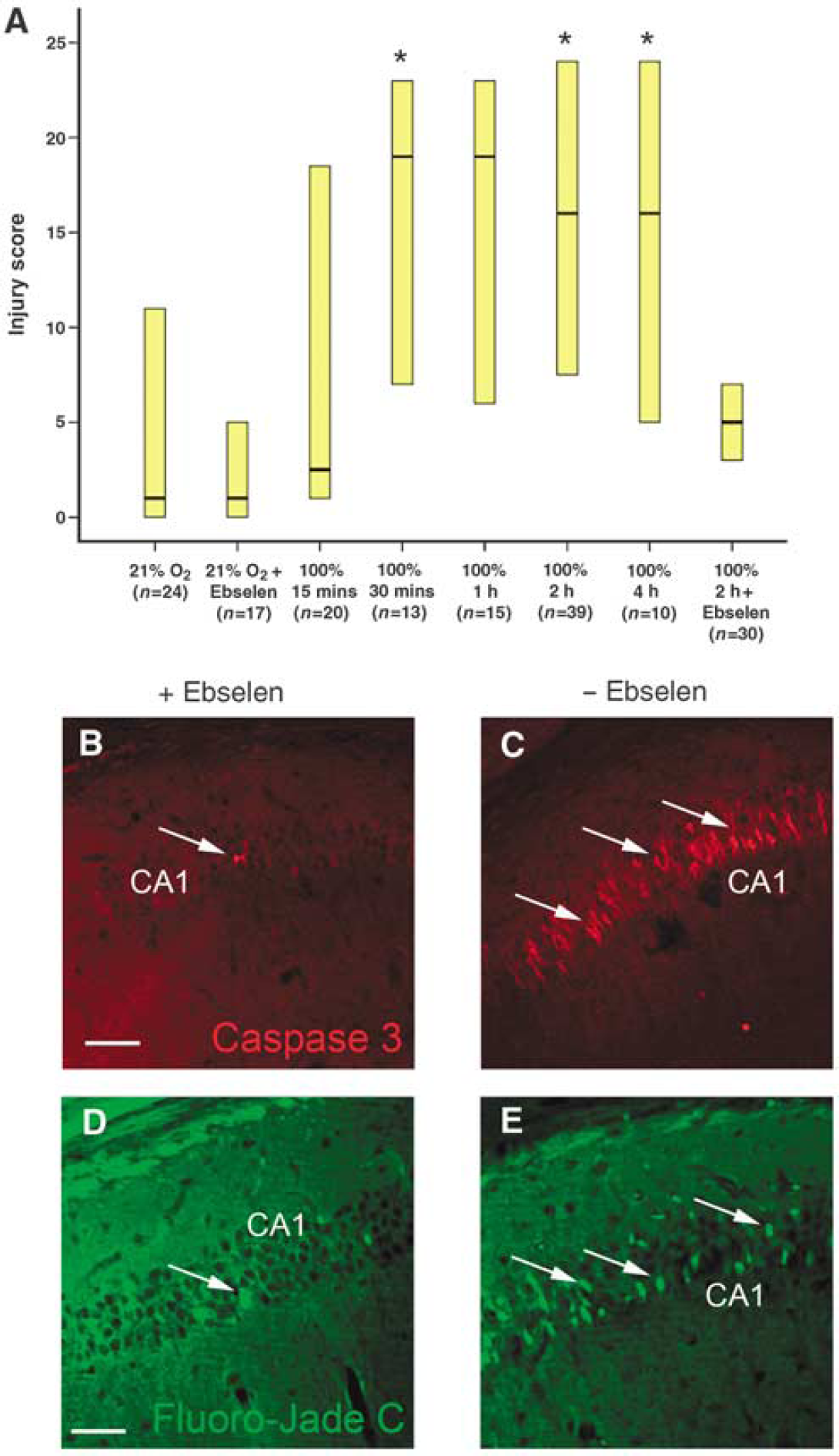
Exposure to as little as 30 mins of hyperoxia after injury worsens histologic injury, and injury can be reversed with the glutathione peroxidase mimetic ebselen. (
Our previous data suggest that brief hyperoxic recovery after hypoxia—ischemia increases neuronal injury and is associated with increases in free radical oxidative damage in early glial progenitors. To determine whether the increased injury observed with hyperoxic recovery is mediated through increasing cellular oxidative stress, we randomized mice after hypoxic-ischemic injury to immediately receive either intraperitoneal ebselen, a glutathione peroxidase mimetic, or saline, with both groups receiving subsequent exposure to 2 h of hyperoxia. Brains were examined 72 h after injury using the previously described scoring system. Mice that received saline (
To confirm that ebselen provided protection from neuronal injury, we performed immunostaining against cleaved caspase-3, a marker of apoptotic cell death, and Fluoro-Jade C, a stain for degenerating neurons, on sections obtained from ebselen-treated mice and saline-treated controls. Fluoro-Jade C staining was significantly greater in the neuronal pyramidal layers of the CA3 region of the hippocampus of saline-treated mice (
Glial but not Neuronal Progenitors are Selectively Vulnerable to Hyperoxic Exposure during Reperfusion after Hypoxia—Ischemia
Many cell types within the brain participate in the injury response, including neuronal and glial progenitor cells, astrocytes, and microglia (Urrea et al, 2007). The developing brain is unique, in that it comprises large populations of as yet undifferentiated neural and glial progenitor populations. It is currently unclear how different progenitor cell types differ in their adaptive response to injury. Nestin is expressed broadly in the developing nervous system and in many cell types, including reactive glia, muscle, and endothelial cells (Kawaguchi et al, 2001). As the brain matures, nestin expression in neural tissue is mainly apparent in neural progenitors and is restricted to two neurogenic regions, the subventricular zone and the dentate gyrus in nestin-GFP transgenic mice (Yu et al, 2005). We observe that by postnatal day 14, GFP-expressing neural progenitors have localized to the subgranular layer in the hippocampus and subventricular zone of the lateral ventricle. However, young mice in our nestin-GFP transgenic line show GFP expression in not only neural progenitors in the subgranular layer of the dentate gyrus and subventricular zone but also in immature glial progenitors expressing PDGFRα in the cortex (Figures 4A to 4F). This allows us to reliably distinguish the effects of hyperoxic recovery on GFP-expressing neural progenitors in the hippocampus from GFP-expressing glial populations in the cortex.
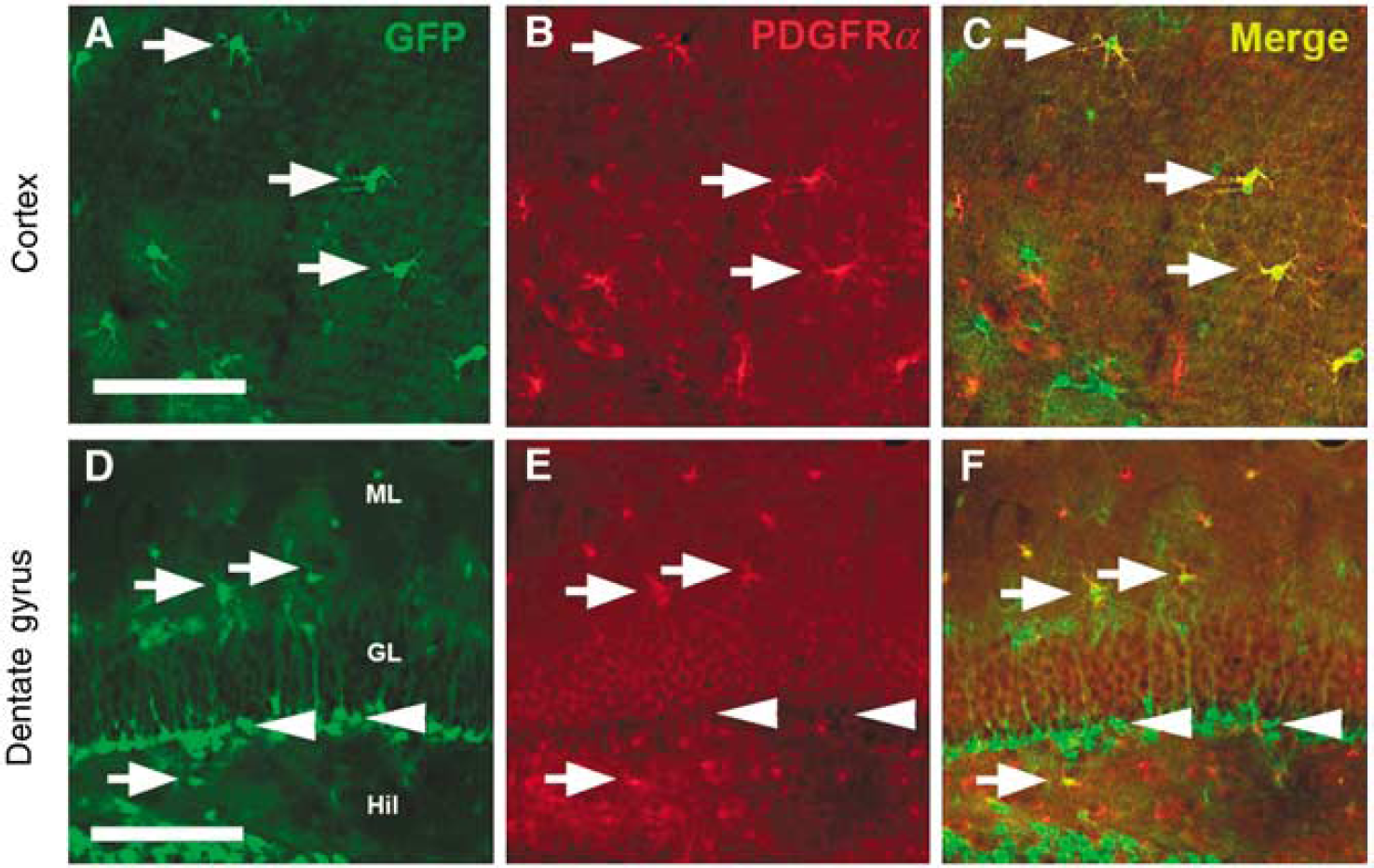
Glial progenitors are located in the cortex and pluripotent progenitors are found in the hippocampus in the immature mouse brain. (
We used flow cytometry and endogenous GFP to mark neuronal and glial progenitors, CD11b antibody to mark microglia (Wirenfeldt et al, 2005), and CD81 antibody to mark both microglia and astrocytes (Dijkstra et al, 2000). Neural progenitor cells (GFP+, CD11b-) or (GFP+, CD81+) from the hippocampus were depleted after hypoxia—ischemia injury in both normoxic and hyperoxic-recovered mice (
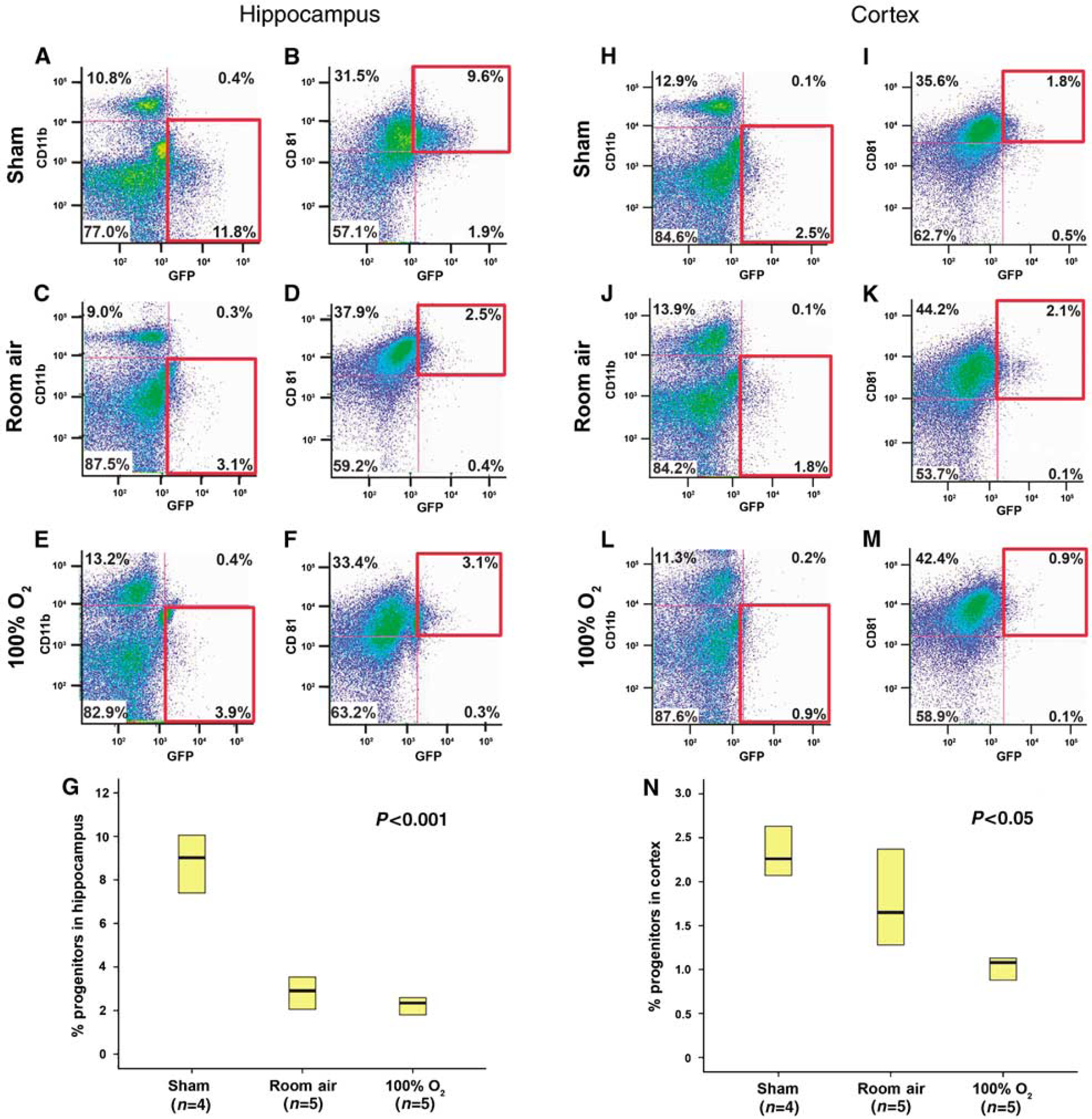
Flow cytometry shows that glial progenitors in the cortex are depleted after injury and recovery with hyperoxia. Images of hippocampal dissections (
Myelination of White Matter Tracts is Disrupted and Disorganized after Hyperoxic Recovery in Injured Mice
At postnatal day 14, although oligodendrocyte progenitors are abundant, there is relatively little mature myelination that has occurred in the rodent brain, and myelination increases over the next 2 to 4 weeks to reach adult levels (Sauvageot and Stiles, 2002). We sought to determine whether the increase in oxidative injury and decrease in glial progenitors that we observed after exposure to hyperoxia at postnatal day 14 result in dysfunctional myelin production. Mice that were injured at postnatal day 14 were killed 4 weeks later and examined for mature myelination with MBP immunohistochemistry. Figures 6A to 6C show that although myelination does not appear grossly disrupted in hyperoxic-recovered mice, when examined at higher power, the ipsilateral (injured) myelin staining pattern is reduced and disorganized compared with the contralateral (uninjured) side, suggesting that injury to early progenitors leads to long-term defects in myelination. This reduced MBP staining is most prominent in the cortical-thalamic tracts, where cortical motor neurons project to the spinal cord.
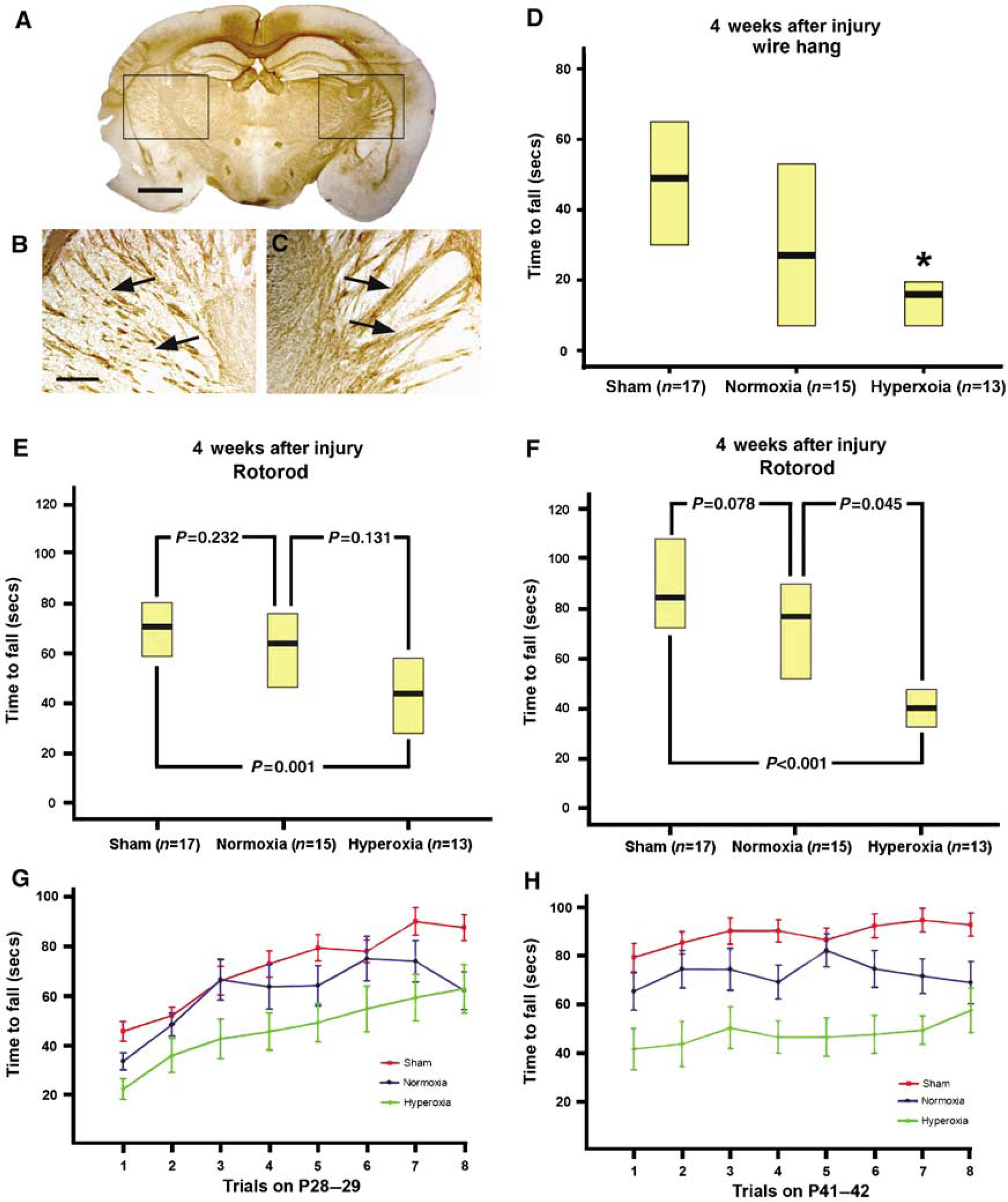
Injured mice have a more disrupted myelination pattern with myelin basic protein staining and perform significantly worse on behavior testing with rotorod and wire hang. Low-magnification views of an injured mouse recovered in hyperoxia do not reveal marked differences between the ipsilateral (injured) and the contralateral (uninjured) sides (
Exposure to Hyperoxia during Recovery Leads to Increased Functional Motor Deficits and a Static Motor Lesion
As hyperoxic-recovered mice show increased histologic injury, depletion of myelin progenitors, and impaired myelination of cortical-spinal neurons, we next tested whether recovery in 100% oxygen, would affect functional recovery after hypoxic-ischemic injury. Strength testing using the wire hang test was performed at postnatal day 42 and revealed that the hyperoxic-recovered mice performed significantly worse (median time to fall 16 secs, range = 4 to 20; 25th% to 75th%) than the sham-injured mice (49 secs, range = 29 to 69) (Figure 6D). However, there was no difference between normoxic recovery and hyperoxic recovery groups, or between the sham-operated mice and normoxic-recovered mice.
Rotorod testing was performed at postnatal days 28 to 29 and 42 to 43 (2 and 4 weeks after injury). At postnatal days 28 to 29, we observed a significant decrease in latency to fall among the injured mice recovered in 100% oxygen (47 secs, mean = 32 to 62) compared with sham-injured controls (73 secs, mean = 59 to 80;
Physiologic Description of the Model
Sham and ligated mice had similar respiratory rates before hypoxia exposure (137 ± 5 versus 134 ± 4 breaths per min). Both groups had a marked increase in respiratory rate when exposed to hypoxia (192 ± 8 and 196 ± 5 breaths per min), which was significantly different when compared with preexposure respiratory rates (
After 30 mins of hypoxia, mice (
Discussion
Hypoxic-ischemic brain injury is an important cause of neurodevelopmental disabilities in childhood, as the immature brain is both vulnerable to secondary injury and may be limited in its ability to undergo self-repair. Oxidative stress is one important factor that contributes to secondary neural injury, and despite many years of study in the context of brain injury, there are currently no clinically accepted ways of attenuating its effects. One common tenet in clinical resuscitation research after hypoxic-ischemic insults includes not only reestablishment of normoxia but also administration of hyperoxic gas, presumably to ensure a vigorous resuscitation (Niermeyer et al, 2000). In fact, many investigators advocate for global hyperoxia to the brain after injury in the hope of optimally oxygenating marginalized brain tissue (Sunami et al, 2000). This study suggests that such a practice may not be beneficial and may, in fact, result in increased injury and worse functional outcome.
Recovery in hyperoxia after hypoxic-ischemic injury has deleterious effects on cerebral metabolism and inflammation, although long-term developmental and functional outcomes are unknown (Dohlen et al, 2005). In this study, we show that young mice sustain significant increases in histologic brain injury after unilateral hypoxic-ischemic brain injury when recovered in 100% oxygen compared with room air. We observed substantial increases in neuronal injury after as little as 30 mins of hyperoxic recovery, which could easily encompass a reasonable resuscitation period in humans. Minimal exposure to hyperoxia during resuscitation is not integrated into current models of resuscitation, and our results suggest that limiting exposure to hyperoxic recovery is crucial in preventing further secondary injury.
We show that hyperoxic recovery increases the oxidative stress and free radical production resulting in nitrosylation of proteins in injured brain tissue. Furthermore, oligodendrocyte progenitor cells, which are known to be sensitive to hypoxia—ischemia and free radical injury, appear to be most susceptible to accumulating oxidative damage (Back et al, 2002; Tan et al, 1998). Reactive oxygen species are generated in a variety of ways after oxidative stress, depending on the stage of ischemia and state of reperfusion (Abramov et al, 2007). The production of reactive oxygen species appears to be a dynamic process that is driven by several factors, and defense against free radical damage is mediated by two major groups: antioxidant enzymes (superoxide dismutases, glutathione peroxidases, and catalase) and low-molecular-weight antioxidants (ascorbate, glutathione, vitamin E, and coenzyme O) (Bayir et al, 2006).
Oxidative stress upregulates antioxidant enzymes in adult rodents after various forms of neuronal injury, including traumatic brain injury, ischemic injury, and hypoxic preconditioning (Arthur et al, 2004; Goss et al, 1997; Guegan et al, 1998). Glutathione peroxidase, which detoxifies peroxynitrite, is thought to be crucial in limiting oxidative injury (Sheldon et al, 2004). Importantly, the immature brain contains significantly less glutathione peroxidase activity than the mature brain and lacks the compensatory increase seen in adult animals after neural injury (Bayir et al, 2006; Sheldon et al, 2004). The immature brain also has significantly less superoxide dismutase activity than the adult brain but increased levels of catalase (Bayir et al, 2006). This suggests that the immature developing brain, unlike the adult brain, may be uniquely vulnerable to increases in oxidative injury during resuscitation. Ebselen is a selenium-containing organic antioxidant that mimics the action of glutathione peroxidase and has been shown to improve outcomes in various models of ischemic injury in animals and humans (Takasago et al, 1997; Yamaguchi et al, 1998). We show here that the administration of ebselen immediately after a hypoxic-ischemic insult markedly attenuates the increased injury induced during hyperoxic recovery. This supports our hypothesis that hyperoxic exposure mediates increasing neural injury through mechanisms related to increased oxidative stress and toxicity.
Stem and progenitor cells in the developing brain are abundant and continually add functional neurons and other cell types to the brain throughout life (Miles and Kernie, 2006). In humans, the newborn and immediate neonatal periods are critical for normal oligodendrocyte formation and ultimate myelination, because this is the stage at which oligodendrocyte progenitors are most abundant (Back et al, 2002). Mechanisms underlying recovery may include not only progenitor-mediated replacement of lost cells but also changes in dendritic arborization, spine density, and synaptogenesis (Miles and Kernie, 2006). Developing progenitor cells are believed to be more vulnerable to hypoxic-ischemic injury and oligodendrocyte progenitor cells, in particular, are more sensitive to oxidative stress than mature oligodendrocytes (Back et al, 2002). The data presented here show that glial progenitors accumulate nitrotyrosine and are preferentially depleted after hyperoxic recovery, which is consistent with
A disruption in myelination, whether from cerebral anoxia, cerebral hemorrhage, or infection, can result in static motor lesions and underlie cerebral palsy in humans (Hoon, 2005). Here, we show that hyperoxic recovery results in disrupted myelination, and even moderately injured mice show irregular and decreased myelin deposition, particularly in the cortical-thalamic motor tracts. In addition, we show the developmental significance of this impaired myelination where exposure to hyperoxic recovery causes a static motor deficit similar to that seen in cerebral palsy. It may also be possible that injury to projecting cortical neurons and axons inhibited proper myelination in these areas. Although based on previous data that glial progenitors are depleted and accumulate oxidant injury after injury, we believe that observed defects in myelination are due, at least in part, to selective destruction of oligodendrocyte progenitors, which is exacerbated by hyperoxic recovery.
In conclusion, we establish that exposure to hyperoxia for even brief periods of time after hypoxic-ischemic brain injury results in an increased histologic injury that is accompanied by increased oxidative stress, depletion of oligodendrocyte progenitors, and impaired functional recovery from injury. Hypoxia—ischemia damages some cells irreversibly, and although other cells are destined for survival, there apparently exists a range of injured and vulnerable cells that may or may not survive, depending on the amount of secondary injury that ensues. Increasing oxidative stress with excessive exposure to oxygen favors cell death. Further studies are needed to better characterize the mechanisms underlying this sensitivity to injury and continued efforts are needed to determine the best possible concentration of oxygen required for optimal resuscitation while limiting secondary injury.
Footnotes
Acknowledgements
We thank Matthew Solove, Gui Zhang, and Ben Orr for technical assistance; Craig Powell for critical review; and Lonnie Roy for statistical assistance.
The authors state no duality of interest.