
Abstract
The effect of N-methyl-
Transient global cerebral ischemia causes selective cell death in vulnerable regions. It produces cell injury and death in the hippocampus of gerbils, rats, primates, and humans, with prominent injury to hippocampal CA1 pyramidal neurons and dentate hilar neurons (Hsu and Buzsaki, 1993; Gonzalez et al., 1991; Ito et al., 1975; Kirino, 1982; Westerberg et al., 1989; Herguido et al., 1999). This ischemic hippocampal injury results in anterograde and retrograde recent memory loss in animals and man (Squire and Zola-Morgan, 1996; Zola-Morgan et al., 1992). Neuronal damage that follows ischemia appears to involve overstimulation of glutamate receptors, with the influx of Na+ and Ca2+ ions through the channels gated by these receptors (Gill and Woodruff, 1990; Westerberg et al., 1989). N-methyl-
Recently, global ischemia has been found to stimulate the birth of new neurons in the adult brain (Liu et al., 1998). Most neurons of the mammalian adult nervous system are terminally differentiated (Gage et al., 1998; Gage, 1998). However, some cells continue to divide throughout life in the adult subventricular zone (SVZ) and subgranular zone (SGZ) of the dentate gyrus. Dividing stem and progenitor cells have been described in the SGZ of rats, mice, primates, and humans (Altman and Das, 1965; Gage et al., 1998; McKay, 1997). Subgranular zone stem cells give rise to progenitor cells that proliferate, migrate into the granule cell layer, and differentiate into mature granule cell neurons. Many factors affect dentate neurogenesis, including age, enriched environments, and stress (Gage et al., 1998; Cameron et al., 1998). Not only does the NMDAR modulate neurogenesis during brain development (Gould et al., 1994), but NMDAR blockade also increases the number of granular neuron precursors in adult brain (Cameron et al., 1995; Gould et al., 1997). Because glutamate receptors mediate ischemic injury and ischemia induces neurogenesis, the authors examined the role NMDA and AMPA/KA receptors in ischemia-induced neurogenesis.
A significant proportion of the cells born in the SGZ after ischemia mature into granule cell neurons (Liu et al., 1998), some of which extend axons to the CA3 region of hippocampus (Stanfield and Trice, 1988; Markakis and Gage, 1999). To study these synaptic responses, the authors determined whether synapsin-I, which is induced after ischemia (Marti et al., 1999), was induced during the period of neurogenesis and whether synapsin induction was modulated by NMDA or AMPA/kainate glutamate receptors. The results indicate that NMDA and AMPA/kainate receptor antagonists prevent the dentate neurogenesis and the dentate granule cell neuron synaptic responses that ordinarily occur After global ischemia. This data could suggest that NMDA and AMPA/kainate receptors mediate neurogenesis and synaptogenesis after global ischemia or that prevention of cell death with glutamate receptor antagonists prevents neurogenesis and synaptogenesis after global ischemia, or both. Because neurogenesis occurs after blockade of hippocampal glutamate receptors in normal animals, the authors hypothesize that global ischemia leads to chronic down-regulation of hippocampal glutamate receptors that stimulates neurogenesis and synaptogenesis. Consequently, the authors propose that glutamate receptor antagonists prevent neurogenesis and synaptogenesis after global ischemia by preventing hippocampal neuronal cell death and the chronic down-regulation of glutamate receptors.
MATERIALS AND METHODS
Animals
All studies were performed with adult male Mongolian gerbils (11- to 13-weeks-old; Simmenson, Gilroy, CA, U.S.A.). Animal studies were performed according to National Institutes of Health guidelines under an approved local protocol from the San Francisco VA Medical Center. Gerbils were housed singly upon arrival to the authors' animal care facility and were acclimated to the new environment for at least one week before surgery. The colony was maintained on a 12-hour light-dark period at 23°C. Food and water were available ad libitum.
Transient global ischemia
Animals were anesthetized with 3% isoflurane in 30% O2:70% N2. After bilateral neck incisions, both common carotid arteries were exposed and occluded with aneurysm clips for 10 minutes. Clips then were removed to restore cerebral blood flow. Animal temperatures were maintained at 37.5°C ± 0.5°C with a heating blanket connected with a rectal thermometer until animals recovered from surgery. Sham-operated animals were treated similarly, except that the carotids were not occluded after the neck incisions.
Glutamate receptor antagonist administration
Two different glutamate receptor antagonists were used: MK-801 (3 mg/kg; RBI labs; an NMDAR antagonist) and NBQX (30 mg/kg; RBI Labs; an AMPA/kainate R antagonist). NBQX blocks GluR1–4 of the AMPA receptors, and GluR5–6 of the kainate receptors (Chittajallu et al., 1999). MK-801 was administered intraperitoneally three times—the first injection was 30 minutes before surgery, the second was 6 hours after ischemia, and the third was 24 hours After ischemia (Gill and Woodruff, 1990). NBQX also was administered three times—the first injection was 10 minutes before the carotid clamps were placed, the second was 6 hours after ischemia, and the third was 36 hours after ischemia. These schedules were chosen because they have been reported to protect CA1 pyramidal neurons after global ischemia (Sheardown et al., 1990; Diemer et al., 1992). Control animals were injected on the same schedules, except that saline (sterile 0.9% NaCl) was injected. Nine days after the ischemia, BrdU (50 mg/kg) was administered intraperitoneally daily for 4 days. BrdU was administered on days 9 to 12 because this is the period of maximal cell proliferation after global ischemia in the gerbil brain (Liu et al., 1998). Animals were killed 15 days After the ischemia.
A second group of gerbils was submitted to the same global ischemic injury described for the first group. However, the drugs were not injected until day 7 after ischemia when MK801 (3 mg/kg, IP), NBQX (30 mg/kg, IP), or saline (0.9%, same volume IP) were injected on the same schedule (time 0, 6 and 24, or 36 hours) as the first group. The delay of 7 days was used because this is just before the period of neurogenesis that occurs after global ischemia (Liu et al., 1998). Nine days After the ischemia, BrdU (50 mg/kg) was administrated IP daily for 4 days (days 9 to 12 after ischemia) as in all of the groups.
A third group of gerbils served as drug-injected controls—that is, they were subjected to sham operations without ischemia and had glutamate antagonists or saline injected on the same schedule as the first group. Nine days after the sham surgery, BrdU (50 mg/kg) was administered intraperitoneally daily for 4 days (9 to 12 days after sham surgery). Animals were killed 15 days after sham surgery (control MK-801, control NBQX, saline control).
Microinjection of NMDA and AMPA/kainate receptor antagonists into CA1
Gerbils were anesthetized with 3% isoflurane in 30% O2:70% N2 and placed in a rodent stereotaxic. The skin over the skull was incised and bilateral burr holes were drilled manually. Needles were inserted through the burr holes and bilateral microinjections of either MK801 (0.5 μL in each side, 3 μg/μL solution) or NBQX (0.5 μL in each side, 30 μg/μL solution) were performed using a Hamilton syringe (1 μL, 30 g needle; Hamilton, Reno, NV, U.S.A.). The target for the injections was the CA1 area of the dorsal hippocampus at coordinates A: 2.8; L: 1.9; V: 1.1 Even though the targeted region was CA1, it is likely that the injections spread to dorsal and ventral hippocampus, including CA1, CA3, the dentate gyrus, and the SGZ.
BrdU labeling
The thymidine analog BrdU (5-bromo-2′-deoxyuridine-5′-monophosphate) was administered intraperitoneally (50 mg/kg, Sigma, St. Louis, MO, U.S.A.) once daily for 4 consecutive days during the peak of cell proliferation, 9 to 12 days after ischemia (Liu et al., 1998). BrdU appeared to only label dividing cells, because there was never any BrdU incorporation into CA1 neurons or any other dying cell that might be undergoing some degree of DNA repair (Liu et al., 1998). All animals were killed at 15 days after ischemia. Nonischemic animals were injected with BrdU for 4 consecutive days on days 9 to 12 after sham surgeries and then killed 3 days later on day 15.
Immunohistochemistry
Gerbils were anesthetized with ketamine (80 mg/kg; Parke-Davis, Morris Plains, NJ. U.S.A.) and xylazine (20 mg/kg; Buther, Columbus, OH, U.S.A.). They were perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde in 0.1 mM phosphate-buffered saline (PBS) (pH: 7.4). Brains were removed, postfixed for 6 hours in 4% paraformaldehyde—PBS, and placed overnight in 20% sucrose. Coronal frozen sections (45-μm-thick) were cut on a microtome and stored in PBS with azide (0.1%). To detect BrdU-labeled nuclei using immunocytochemistry, DNA was denatured to expose the antigen by treatment with 2N HCl for 30 minutes at 37°C. After rinsing twice in 0.1 mM PBS, sections then were incubated overnight at 4°C with the primary antibody to BrdU (1/500, Boehringer Mannheim) in PBS with 1% normal serum, 0.1 % bovine serum albumin, and 0.3% triton X-100. NeuN (1/500, Sigma) immunocytochemistry was used to evaluate the presence or absence of neuronal nuclei (Liu et al., 1998), and synapsin-I (1/800, Stressgen, Victoria, BC, Canada) immunohistochemistry was performed to evaluate the presynaptic protein response to ischemic injury. The same protocol used for BrdU was used for these antibodies, except that the DNA denaturation procedure was deleted.
Western blotting
Total protein was extracted from the hippocampal CA3 subregion and Western blot analysis was performed as described previously (Melloni et al., 1993). Briefly, gerbil brains from the different groups were frozen at −80°C. Slices (300-μm-thick) were cut on a cryostat, followed by microdisection of CA3 under a microscope. Four brains for each condition were used. Tissue was homogenized in 1 mL of Laemmli buffer. Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to polyvinylidenefluoride membranes, and blots were incubated in blocking solution containing 3% bovine serum albumin in 0.1 mol/L PBS, 0.1% Tween 20, and 3% normal serum. Primary antibody against syanpsin-I (1/500, Stressgen) was diluted in this same buffer and hybridized to blots overnight. Bands were visualized using ECL according to the manufacturer's specifications (Amersham, Arlington Heights, IL, U.S.A.).
Cell counting
The number of BrdU immunoreactive nuclei in each dentate gyrus (DG) was counted on 6 to 9 coronal hippocampal sections (45-μm-thick) per animal. Sections were spaced 180 μm apart and spanned the entire dorsal hippocampus. Each microscope image was digitized. BrdU-labeled nuclei were counted on a digital analysis system using National Institutes of Health v1.6 software (NIH 1.6 image program). The one focal plane per section that yielded the most nuclei was used to avoid oversampling. The density of BrdU immunoreactive cells in each section was calculated by dividing the number of BrdU-positive nuclei by the area of the DG (in mm2). These values were averaged to obtain a mean density value of BrdU-positive cells per mm2 for each subject. Differences between the mean values from each treatment group were analyzed using Krustal—Wallis analysis of variance followed by Neuman—Keuls test (SigmaStat: Jandel Scientific, San Rafael, CA, U.S.A.).
RESULTS
CA1 neurons are protected by systemic and local hippocampal injections of MK-801
Control, saline-injected animals (Fig. 1A) and control, MK-801-injected animals (Fig. 1B) (injected IP 30 minutes before, 6 and 24 hours after ischemia) showed similar numbers of NeuN-immunostained neurons in CA1 and DG. Bilateral carotid artery occlusions (10 minutes in duration) resulted in severe loss of NeuN-stained CA1 neurons in hippocampus 15 days After ischemia (Fig. 1C and 1H). Systemic injections of MK801 (injected IP 30 minutes before, 6 and 24 hours after ischemia) prevented the CA1 neuronal cell death 15 days after ischemia (Fig. 1D). Injection of MK801 at 7 days after ischemia, 2 days before the increased neurogenesis in the dentate gyrus (Liu et al., 1998), did not prevent the loss of the CA1 pyramidal neurons at 15 days after ischemia (Fig. 1E). These results confirm many previous studies that demonstrate that a majority of the CA1 neurons die and disappear sometime between 2 and 5 days after global ischemia (using 10-minute carotid occlusions in the gerbil model), although cell death can continue for 2 weeks or more in some models (Colbourne et al., 1999). Animals microinjected with saline in the CA1 hippocampal subfield showed the typical loss of CA1 neurons 15 days after ischemia (Fig. 1F). Remarkably, animals microinjected just one time in CA1 with MK-801 at 30 minutes before ischemia demonstrated nearly complete preservation and neuroprotection of CA1 neurons 15 days After ischemia (Fig. 1G).
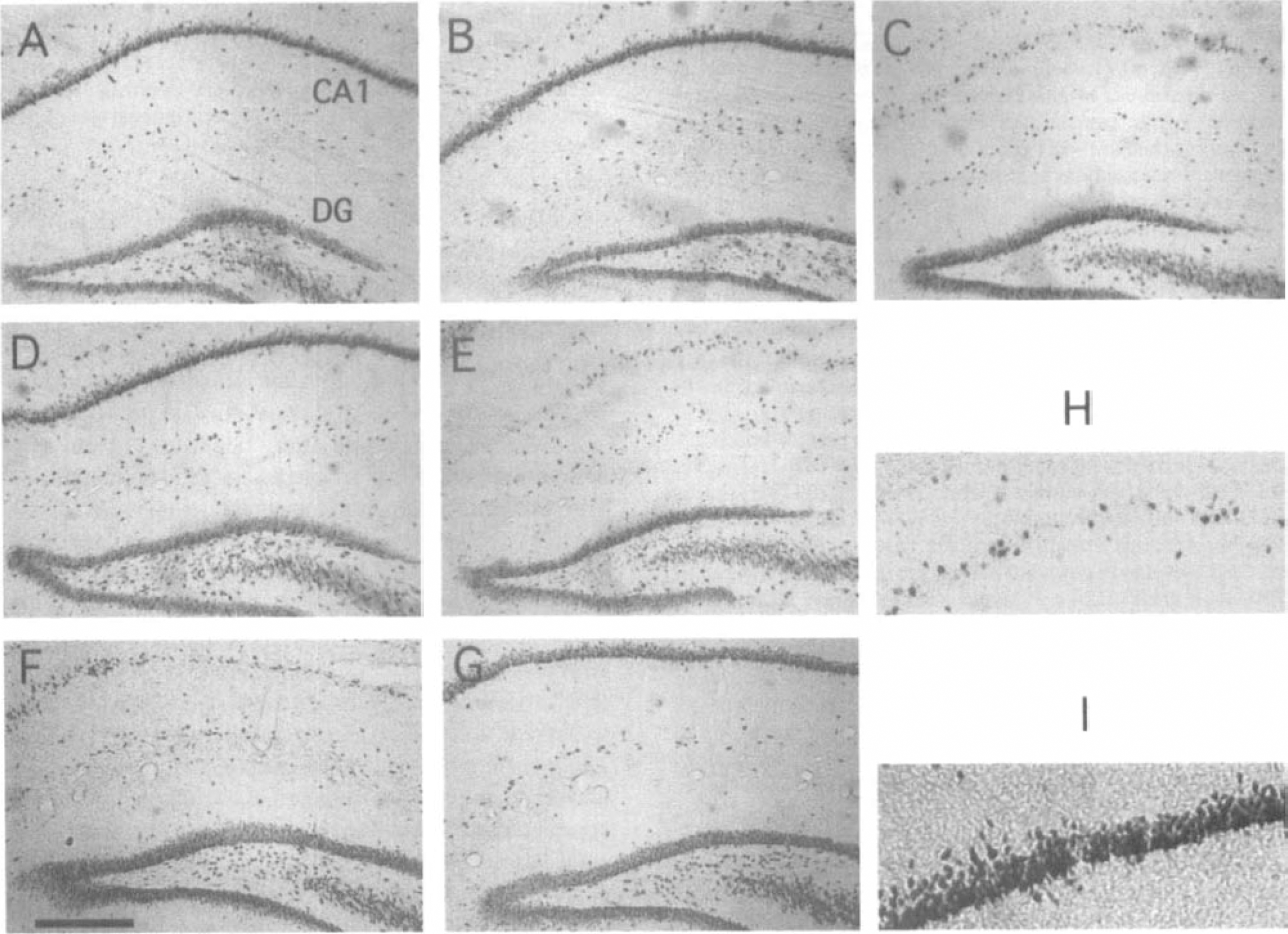
MK-801 protects CA1 pyramidal cells against global ischemia. Coronal sections (45-μm-thick) of rat hippocampus show NeuN-immunostained neuronal nuclei. Control animals
MK801 blocks the cell proliferation that occurs in dentate gyrus
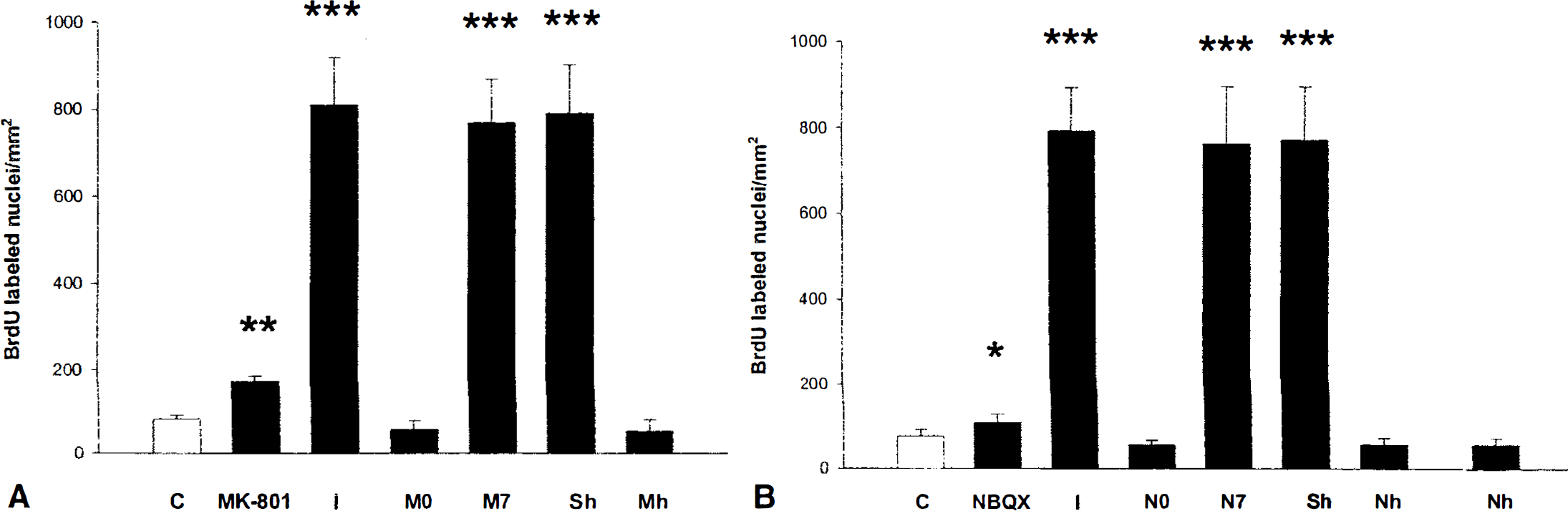
To evaluate cell proliferation in the DG, BrdU was injected between 9 and 12 days after ischemia (when there is a maximal granular cell proliferation) or for 4 consecutive days in control, nonischemic animals. Animals were killed 15 days after surgery, 3 days after the last BrdU injection. There was a basal level of BrdU incorporation into newborn cells in the SGZ of the DG and in the SVZ (not shown) in the control animals (Fig. 2A). This is similar to the findings obtained using 3H thymidine in previous studies (Cameron et al., 1995; Kaplan and Hinds, 1977; Stanfield and Trice, 1988). MK-801 injected systemically (IP) into control animals significantly increased the number of BrdU-labeled cells (Fig. 2B and 5A); these results agree with previous studies (Cameron et al., 1995). At 15 days after global ischemia, BrdU incorporation into cells in the dentate was significantly increased (Figs. 2C and 5A), in a pattern previously described for newborn cells in DG after ischemia in gerbils (Liu et al., 1998). Systemic injections of MK801 (30 minutes before ischemia, and 6 and 24 hours after ischemia) completely blocked BrdU incorporation into DG cells (Fig. 2D), with the number of BrDU-labeled cells similar to those in control animals (Fig. 5A, C vs. MO). Moreover, systemic injections of MK-801 given after the period of CA1 neuronal loss, but before the neurogenesis increase (7 days after ischemia), had no effect on the marked increase of BrdU into cells in the DG at 9 to 12 days after ischemia (Figs. 2E and 5A). The numbers of BrdU-labeled cells were similar in the ischemia group and the ischemia group that received MK801 systemically 7 days after ischemia (Fig. 5A, I vs. M7).
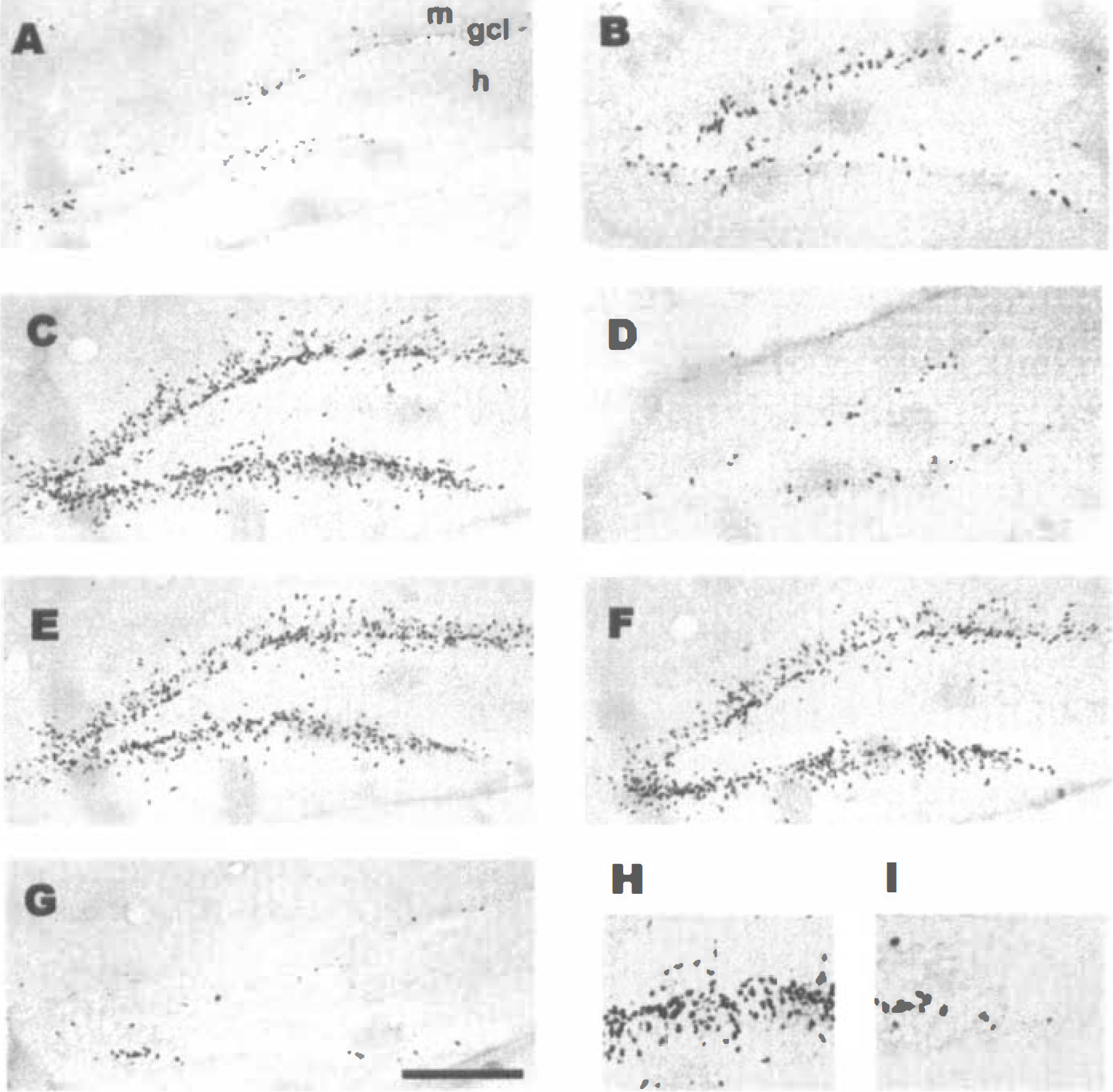
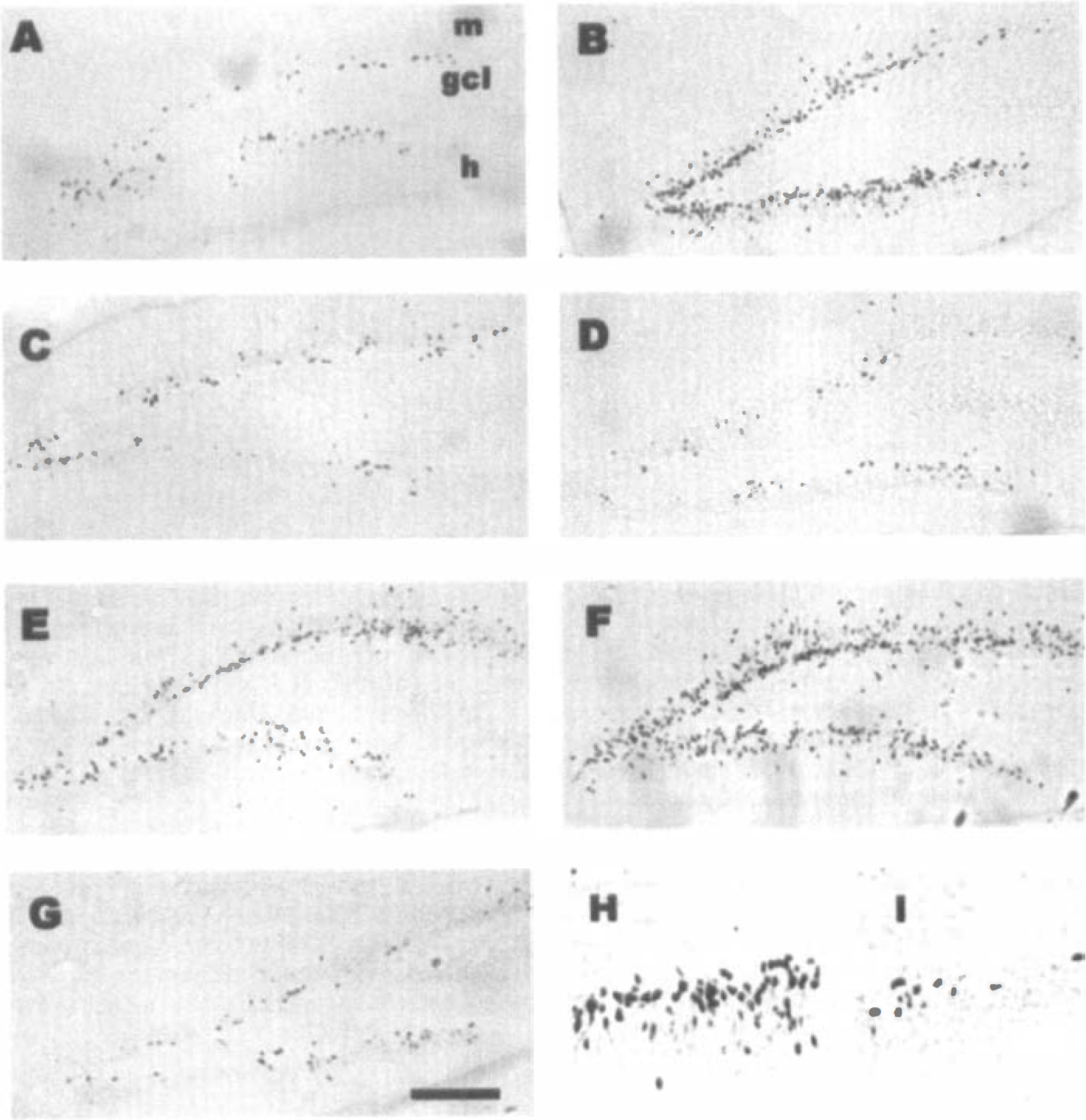
MK-801 prevents the neurogenesis that occurs in dentate gyrus after ischemia. BrdU (50 mg/kg, IP) was administrated on days 9 to 12 after globalischemia (10 minutes) or for 4 consecutive days after sham surgery in controls. All subjects were killed 15 days after ischemia. Panels
MK-801 and NBQX prevent ischemia-induced neurogenesis. Gerbils were subjected to 10 minutes of global ischemia, injected daily with BrdU for 4 days between 9 and 12 days after ischemia, and killed at 15 days after ischemia (3 days after the last injection of BrdU).
To evaluate where MK801 acts, local injections were made into hippocampus. Saline microinjected into hippocampus at 30 minutes before ischemia (Fig. 2F) did not affect the marked increase of BrdU-labeled cells in the DG (Figs. 2F and 5A-Sh) compared with ischemic, noninjected animals (Figs. 2C and 5A to 5I). However, microinjections of MK-801 into hippocampus (Figs. 2G and 5A-Mh) at 30 minutes before ischemia produced a significant decrease of BrdU labeling at 15 days after ischemia compared with saline-injected animals (P < 0.001, in comparison with saline microinjection, Fig. 5A). Notably, the number of BrdU-labeled cells in animals injected with MK801 into hippocampus (Figs. 2G and 5A-Mn) was the same or even slightly less than in control animals (Figs. 2A and 5A to 5C).
CA1 neurons are protected by systemic and local hippocampal injections of NBQX
Control, saline-injected animals (Fig. 3A) and control, NBQX-injected animals (Fig. 3B) (injected IP 30 minutes before, and 6 and 36 hours after ischemia) showed similar numbers of NeuN-immunostained neurons in CA1 and DG. Bilateral carotid artery occlusions (10 minutes in duration) resulted in severe loss of NeuN-stained CA1 neurons in hippocampus 15 days after ischemia (Fig. 3C). Systemic injections of NBQX (injected IP 30 minutes before. and 6 and 36 hours after ischemia) prevented the CA1 neuronal cell death 15 days after ischemia (Fig. 3D). The NBQX neuroprotection of CA1 pyramidal cells was similar to that observed by others (Sheardown et al., 1990; Diemer et al., 1992; Gorter et al., 1997). Injection of NBQX at 7 days after ischemia. 2 days before the increased neurogenesis in the DG (Liu et al., 1998). did not prevent the loss of the CA1 pyramidal neurons at 15 days after ischemia (Fig. 3E). Animals microinjected with saline in the CA1 hippocampal subfield showed the typical loss of CA1 neurons 15 days after ischemia (Fig. 1F). However, animals microinjected with just one dose of NBQX in CA1 at 30 minutes before ischemia demonstrated nearly complete preservation and neuroprotection of CA1 neurons (Fig. 3F).
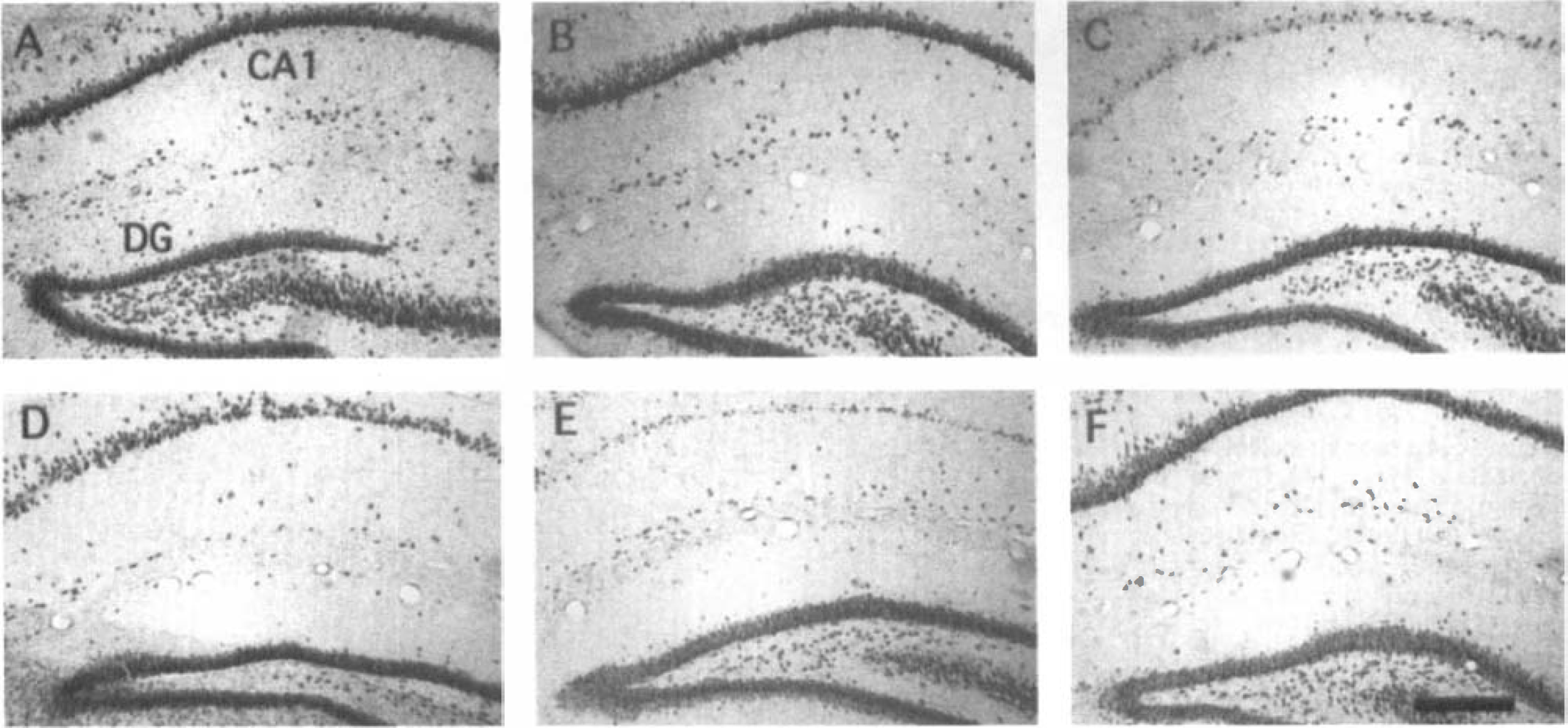
NBQX protects CA1 pyramidal cells against global ischemia. Coronal sections of rat hippocampus show NeuN-immunostained neuronal nuclei. Control animals
NBQX blocks the cell proliferation and neurogenesis that occurs in dentate gyrus
To repeat the evaluation of cell proliferation and neurogenesis in the DG. BrdU was injected between 9 and 12 days after ischemia or for 4 consecutive days in control, nonischemic animals. Animals were killed 15 days after surgery, 3 days after the last BrdU injection. There was a basal level of BrdU incorporation into newborn cells in the SGZ of the DG and in the SVZ (not shown) of the control animals (Fig. 4A). NBQX injected systemically (IP) into control animals produced a modest increase in the number of BrdU-labeled cells (Figs. 4C and 5B). At 15 days after global ischemia, BrdU incorporation into cells in the dentate was significantly increased (Figs. 4B, 4H, and 5B). Systemic injections of NBQX (30 minutes before, and 6 and 36 hours after ischemia) completely blocked BrdU incorporation into DG cells (Fig. 4D and 4I), with the numbers of BrdU-labeled cells similar to those in control animals (Fig. 5B). Moreover, systemic injections of NBQX given after the period of CA1 neuronal loss, but before the neurogenesis increase (7 days after ischemia), had little effect on the marked increase of BrdU into cells in the DG at 9 to 12 days after ischemia (Figs. 4E and 5B-N7). The number of BrdU-labeled cells was similar in the ischemia group (I) and the ischemia group that received NBQX systemically (N7) at 7 days after ischemia (Fig. 5B).
NBQX prevents the neurogenesis that occurs in dentate gyrus after ischemia. BrdU (50 mg/kg, IP) was administrated on days 9 to 12 after global ischemia (10 minutes) or for 4 consecutive days in controls. All subjects were killed 15 days after ischemia. Panels
To evaluate where NBQX acts, local injections were made into hippocampus. Saline microinjected into CA1 at 30 minutes before ischemia (Fig. 4F) did not affect the marked increase of BrdU incorporation into DG (Figs. 4F and 5B-S) compared with ischemic, noninjected animals (Figs. 4B and 5B to 5I). However, microinjections of NBQX into CA1 of hippocampus (Fig. 4G and 5B-Nh) at 30 minutes before ischemia produced a significant decrease of BrdU labeling at 15 days after ischemia (P < 0.001, in comparison with saline microinjection, Fig. 5B). Notably, BrdU labeling in animals injected with NBQX into hippocampus of ischemic animals (Figs. 4G and 5B) was the same or even slightly lower than in control animals (Figs. 4A and 5B).
Glutamate receptor antagonists block the increased CA3 synapsin-I expression
Synapsin-I immunoreactivity was observed as a fine reticular staining in the neuropil of the plexiform layers of the hippocampus (Figs. 6 and 7). Granule cell mossy fiber nerve terminals in the stratum lucidum layer of CA3 were particularly well stained compared with other portions of the hippocampus in control animals (Figs. 6A and 7A). Western blots confirmed the presence of synapsin-I in CA3 of hippocampus of control animals (Fig. 6H, lane B and Fig. 7H, lane B). In addition, administration of either MK-801 (Fig. 6H, lane C) or NBQX (Fig. 7H, lane C) produced a small decrease of synapsin-I protein in CA3 of nonischemic controls.
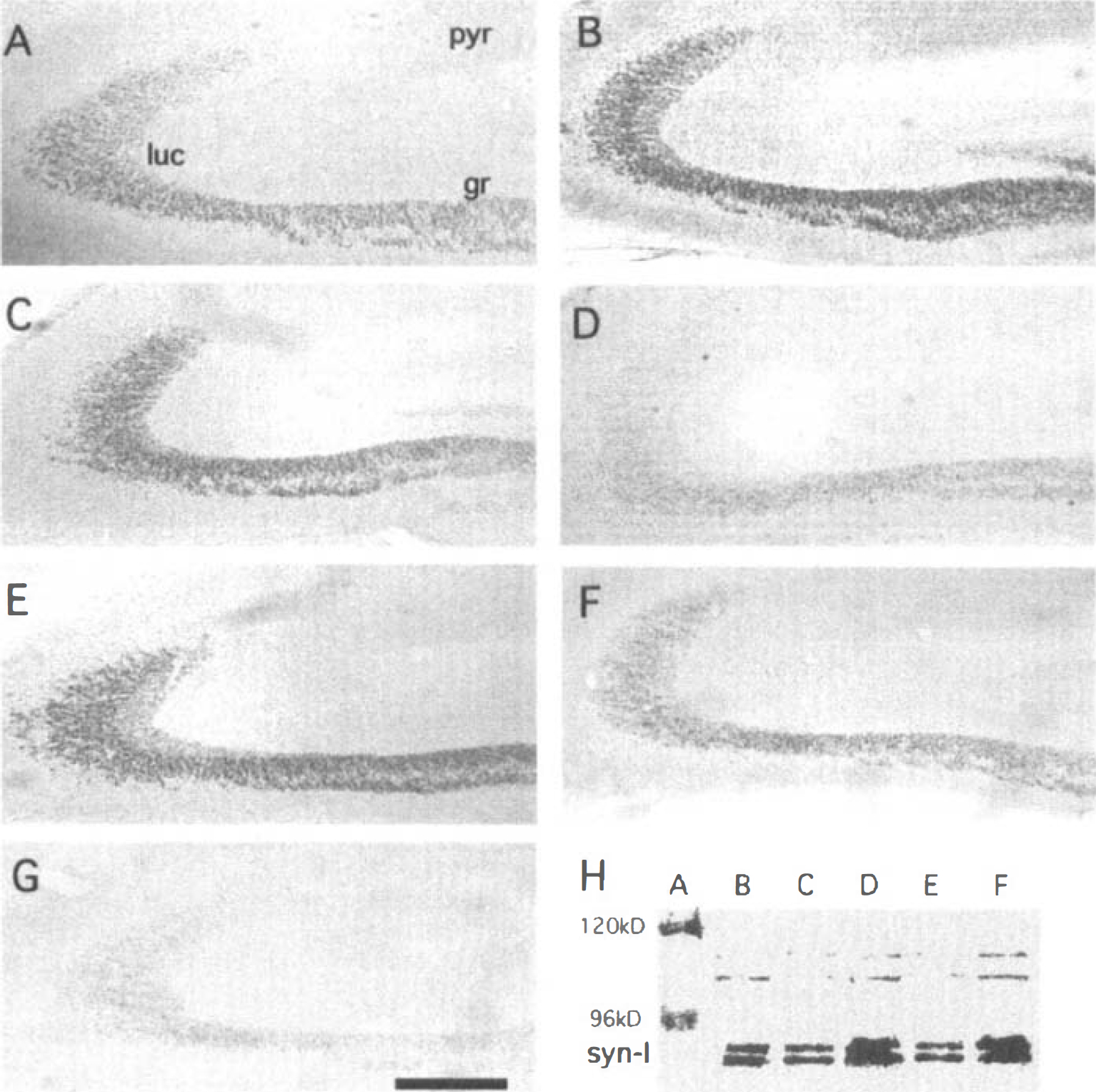
Induction of synapsin-I protein in CA3 of hippocampus after global ischemia is blocked by MK-801. Panels
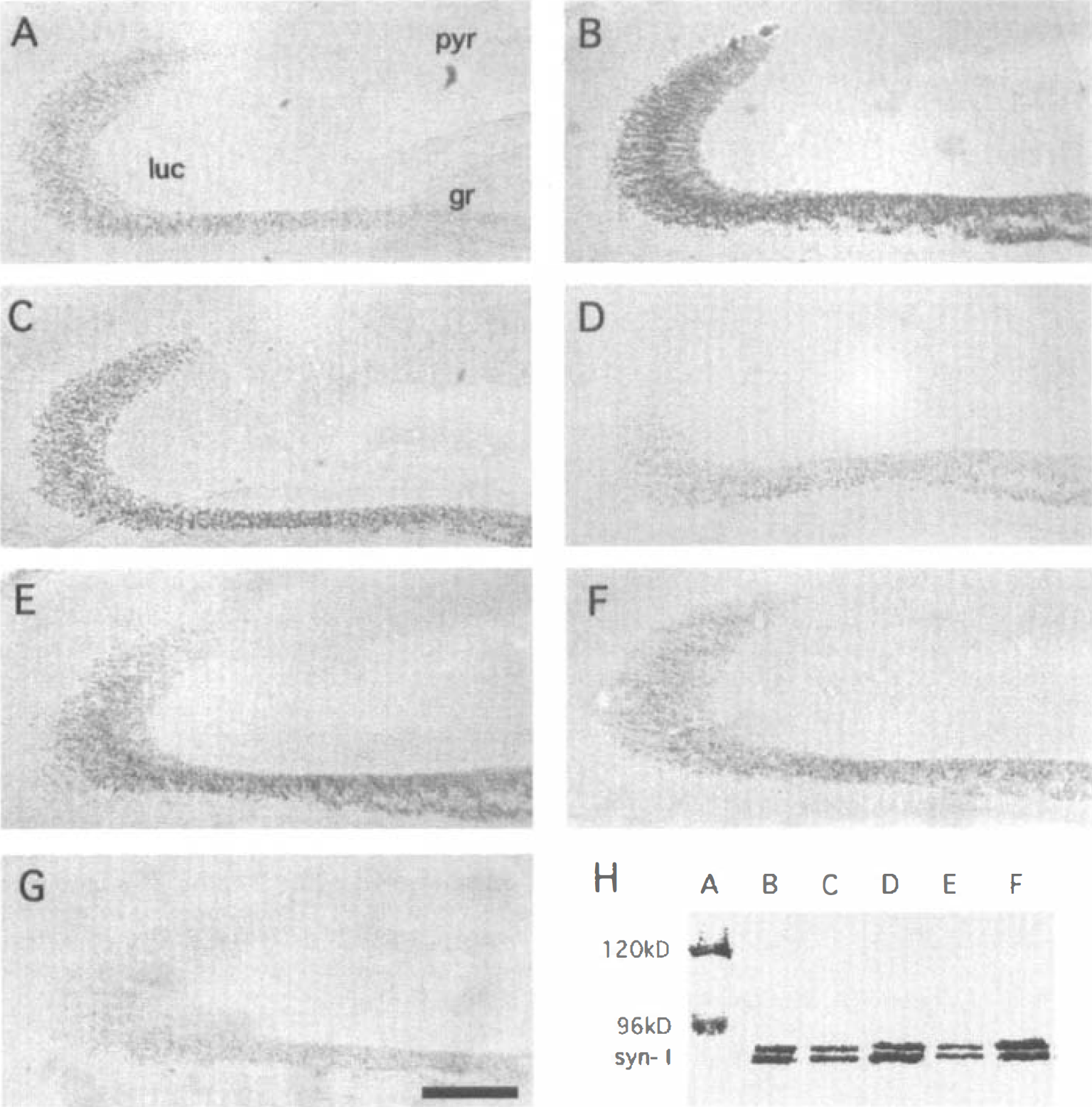
Induction of synapsin-I protein in CA3 of hippocampus after global ischemia is blocked by NBQX. Panels
At 7 days after global ischemia (Figs. 6B and 7B) and 15 days after ischemia (Figs. 6C, and 7C), there was a marked increase in synapsin-I immunoreactivity in stratum lucidum of CA3, and a marked increase of synapsin protein in CA3 on Western blots (Figs. 6H, lane D and Fig. 7H, lane D).
Systemic administration of MK801 (30 minutes before, and 6 and 24 hours after ischemia) markedly decreased the induction of synapsin-I in CA3 at 15 days after ischemia as assessed using immunocytochemistry (Fig. 6D) and Western blotting (Fig. 6H, lane E). Systemic administration of MK-801 at 7 days after ischemia had little effect on the marked induction of synapsin-I in CA3 (Fig. 6E and 6H) when compared with ischemia alone either by immunocytochemistry (compare Fig. 6E with Fig. 6B and 6C) or by Western blotting (compare Fig. 6H, lanes D and F).
Systemic administration of NBQX (10 minutes before, and 6 and 36 hours after ischemia) markedly decreased the induction of synapsin-I in CA3 at 15 days after ischemia as assessed using immunocytochemistry (Fig. 7D) and Western blotting (Fig. 7H, lane E). Systemic administration of NBQX at 7 days after ischemia had little effect on the marked induction of synapsin-I in CA3 (Fig. 7E and 7H) when compared with ischemia alone either by immunocytochemistry (compare Fig. 7E with Fig. 7B and 7C) or by Western blotting (compare Fig. 7H, lanes D and F).
The next experiments determined whether MK-801 and NBQX act on hippocampus. Saline injections into hippocampus slightly decreased synapsin-I staining in CA3 of hippocampus (Figs. 6F and 7F) when compared with ischemia (Figs. 6C and 7C), but the staining was still greater than in controls (Fig. 7 A). Remarkably, single injections into hippocampus of either MK-801 (Fig. 6G) or NBQX (Fig. 7G) 30 minutes before ischemia completely blocked the increased expression of synapsin-I protein in CA3 of hippocampus on immunocytochemically-stained sections (Figs. 6G and 7G) and Western blots (Fig. 6H, lane E; Fig. 7H, lane E).
Notably, systemic injections of MK-801 slightly decreased synapsin-I in CA3 compared with control animals (Fig. 6H, lane C compared with lane B). In addition, systemic injections of NBQX also slightly decreased synapsin-I in CA3 compared with control animals (Fig. 7H, lane C compared with lane B). These data suggest that baseline levels of synapsin in CA3 may be regulated by the activity of NMDA and AMPA/kainate glutamate receptors even in normal control animals.
DISCUSSION
Results show that systemic and hippocampal administration of MK801 and NBQX prevent CA1 pyramidal neuronal cell death, dentate cell proliferation, and the induction of synapsin-I protein that occur in hippocampus after global ischemia. The results are consistent with the possibilities that NMDA and AMPA/kainate receptors modulate neurogenesis, or that injuries that decrease dentate glutamate receptor activation stimulate dentate stem cell proliferation, or both. It is postulated that preventing CA1 cell death after ischemia with MK-801 and NBQX may prevent glutamate receptor down-regulation in CA1 and dentate (Gorter et al, 1997; Aronica et al, 1998), thereby preventing hippocampal neurogenesis.
Systemic administration of an NMDAR antagonist, MK-801, to control animals stimulated cell proliferation in DG in the current study. This supports other studies that show that MK-801 stimulates dentate precursor cell proliferation, with BrdU-labeled newborn cells developing into neurons and some into astrocytes (Gould et al., 1994, 1997; Cameron et al., 1995, 1998). Notably, in studies by Gould et al., (1977), MK-801 led to changes in cell proliferation within hours of drug administration, whereas the current study demonstrated similar changes many days later. These studies suggest that even temporary blockade of NMDA receptors may alter dentate cell proliferation for several days. The current study also shows that the AMPA/KAR antagonist, NBQX, increases proliferation of dentate progenitor cells in normal brain, and suggests that in the normal, intact brain decreased glutamate activation of either NMDAR or AMPA/KAR increases dentate cell proliferation. This is consistent with cell culture studies that show that cell depolarization generally decreases DNA synthesis (Cui and Bulleit, 1998; LoTurco et al., 1995).
Therefore, it was surprising that administration of either MK-801 or NBQX blocked the neurogenesis that occurs after global ischemia. Ischemia should acutely stimulate NMDAR and AMPA/KAR because there is glutamate release during ischemia (Meldrum, 1995; Peruche and Krieglstein, 1993) and glutamate receptor antagonists prevent injury (Buchan et al., 1991b; Diemer et al., 1992; Gill and Woodruf, 1990; Lippert et al., 1994; Sheardown et al., 1990; Simon et al., 1984). The pilocarpine-induced seizures that stimulate neurogenesis (Parent et al., 1997) also would be expected to increase glutamate release and acutely activate NMDA and AMPA/kainate glutamate receptors. Although ischemia and seizure activation of glutamate receptors may increase neurogenesis, it is difficult to understand why glutamate receptor antagonists also induce neurogenesis in control animals. It seems likely, therefore, that ischemia and seizure-induced neurogenesis occurs through some other mechanism other than stimulating glutamate receptors.
Ischemia, seizures, and glutamate antagonists may all increase neurogenesis because there is decreased activation or down-regulation of hippocampal glutamate receptors after hippocampal injuries of any type (Gorter et al, 1997); that is, both NMDA and AMPA/kainate receptors may continuously modulate neurogenesis in the dentate. After global ischemia (Liu et al., 1998) there is death of CA1 pyramidal neurons, dentate hilar neurons, and some CA3 neurons and dentate granule cell neurons (Fukuda et al., 1999; Hsu and Buzsaki, 1993; Li et al., 1997; Onodera et al., 1990, 1993; Sugimoto et al., 1993). Although ischemia acutely activates glutamate receptors, this cell death chronically decreases cell firing in all hippocampal circuits, decreasing the activity of dentate granule cell neurons and hippocampal CA3 and CA1 neurons (Aoyagi et al., 1998; Gao et al., 1998; Howard et al., 1998; Doherty and Dingledine, 1998). In addition, there is down-regulation of NMDA and AMPA/kainate receptors after ischemia in CA1 and to some degree in dentate (Pellegrini-Giampietro et al., 1992, 1997; Onodera et al., 1989; Zhang et al., 1997). Although dentate granule cell activation mediates ischemic CA1 and hilar cell loss (Johansen et al., 1986), it is possible that decreased activation or down-regulation of dentate glutamate receptors is the stimulus for increased neurogenesis after ischemic and other types of chronic hippocampal injury.
Neurogenesis also occurs after ischemia-induced ischemic tolerance in which there is no death of CA1 neurons (Liu et al., 1998). However, there is death of dentate hilar neurons, even in the ischemia-induced tolerance model (Johansen et al., 1987, 1993; Sugimoto et al., 1993; Benveniste and Diemer, 1988), that provide significant input to dentate granule cells (Sugimoto et al., 1993; Johansen, 1993). This could decrease dentate glutamate receptor activation, through hilar interneurons, and trigger dentate neurogenesis in ischemic tolerant animals. Notably, NMDA but not AMPA receptors appear to mediate ischemia-induced tolerance (Bond et al., 1999), whereas both glutamate receptors modulate neurogenesis in normal and ischemic hippocampus.
Seizure-induced neurogenesis also could be related to cell death and glutamate receptor down-regulation, because the generalized seizures produced by pilocarpine produce death of CA3 neurons (Parent et al., 1997). Seizure-induced neurogenesis caused by kainate (Gray and Sundstrom, 1998) is associated with CA3 (and CA1) death that likely leads to chronic decreased activation of hippocampal glutamate receptors. Entorhinal cortical lesions (Cameron et al., 1995) and granule cell lesions that stimulate neurogenesis (Gould and Tanapat, 1997) should decrease dentate glutamate receptor activation. MK-801 or NBQX given systematically would obviously decrease stimulation of hippocampal glutamate receptors (Gould et al., 1994; Cameron et al., 1995). Administration of MK-801 or NBQX before ischemia in the current study would prevent the death of neurons that occurs after ischemia, would maintain normal hippocampal activity, and therefore would prevent ischemia-induced decreases of glutamate receptor activation and increases of dentate neurogenesis.
Pharmacologic blockade of NMDAR-dependent processes with MK-801 decreases long-term stabilization of hippocampal activity (Zhang et al., 1997; Kentros et al., 1998). Blockade of AMPA and Kainate receptors with NBQX also affects neurotransmission and plasticity in hippocampus (Chittajallu et al., 1999; Vignes and Collingridge, 1997). Long-term stabilization of firing fields in the hippocampus is required for normal glutamatergic function and related glutamatergic-dependent processes (Kentros et al., 1998; Bertram and Zhang, 1999). Modifications of glutamatergic conductance produced by glutamate antagonists or lesions of entorhinal cortex, amygdala, dentate, CA3, or CA1 could provoke long-term changes in glutamatergic neurotransmission and glutamate receptors in the hippocampus with subsequent changes of the rates of dentate neurogenesis. This contrasts with the SVZs in which ischemia and glutamate receptor antagonists (data not shown) do not appear to modulate neurogenesis (Liu et al., 1998; Doetsch et al., 1999a,b).
Considerable evidence indicates that Ca2+ permeable AMPAR and NMDAR are critical mediators of global ischemia-induced death of CA1 neurons (Simon et al., 1984; Sheardown et al., 1990; Diemer et al., 1993; Meldrum, 1995; Colbourne et al., 1999; Hicks et al., 1999). Some previous studies showed CA1 protection with systemic drugs (Gill and Woodruff, 1990; Warner et al., 1991) and others did not (Buchan et al., 1991a,b). The current study supports some protective effects of MK-801 against global ischemic injury when given directly into hippocampus (Simon et al., 1984).
Blockade of AMPA/KAR also protects CA1 pyramidal neurons against injury produced by global ischemia (Sheardown et al., 1990; Diemer et al., 1992; Buchan et al., 1991a,b). The current study supports a primary role for hippocampal AMPA/KAR because both systemic and intrahippocampal NBQX protected against CA1 cell death after global ischemia. Recent studies indicate possible mechanisms of injury. NR2A and NR2B subunits of NMDAR, and GluR2 and GluR4 subunits of AMPAR decrease at early times after reperfusion in CA1 pyramidal cells perhaps producing changes of ion flux that mediate selective neuronal cell death after global ischemia (Pellegrini-Giampietro et al., 1992; Gorter et al., 1997; Ying et al., 1997; Zhang et al., 1997).
Results confirm that synapsin-l increases in the mossy fiber layer of the CA3 region of hippocampus after global ischemia (Marti et al., 1999) and show that NMDAR and AMPA/KAR antagonists prevent synapsin-I induction. Stimuli for the CA3 synapsin up-regulation are uncertain, but could be similar to those that stimulate neurogenesis after ischemia.
Synapsin-I is a presynaptic protein involved in neurotransmitter release and regulation of axonal elongation and new synapse formation (Greengard et al., 1993; Ferreira et al., 1996), synapsin-I phosphorylation releasing presynaptic vesicles from the reserve pool (Turner et al., 1999). Synapsin-I up-regulation in the CA3 region of the hippocampus After global ischemia suggests that it is induced in the presynaptic terminals of mossy fibers from dentate granule cell neurons that synapse on CA3 pyramidal neurons. This suggests altered synaptic function or formation of new synapses, or both, by granule cell mossy fibers on CA3 pyramidal neurons. The stimulus for increased synapsin-I expression in dentate granule cell neuron mossy fibers could be death of some granule cell neurons with sprouting of their neighbors, or death of CA3 target neurons with synaptic reorganization of mossy fiber afferents onto surviving CA3 neurons, or both (Fukuda et al., 1999; Hsu and Buzsaki, 1993; Li et al., 1997; Onodera et al., 1990, 1993). Thus, MK-801 and NBQX may prevent synapsin-I induction simply by preventing ischemia-induced cell death.
The synapsin-I induction probably occurs mainly in granule cell neurons that project to CA3. Because cell proliferation does not begin until 7 days after ischemia (Liu et al., 1998), and because newborn granule cells with neuronal phenotypes are not detected until 1 to 3 weeks after ischemia (Liu et al., 1998), the synapsin-I induction at 7 and 15 days is probably not due to synaptogenesis of neurons born after ischemia. At later times it is possible that newborn granule cell neurons contribute to mossy fiber synaptogenesis in CA3 because newborn neurons extend axons to CA3 (Stanfield and Trice, 1988; Markakis and Gage, 1999). The suppression of neurogenesis and synaptogenesis by glutamate receptor activation and its enhancement by NMDAR and AMPA/KAR inactivation, may provide a means whereby natural alterations in the degree of excitatory input control the numbers of newborn neurons and the sprouting of existing neurons.
Footnotes
Acknowledgment
The authors thank Dr. Jialing Liu for help in the early portions of this study.