
Abstract
Lipid peroxidation and the cytotoxic by-product 4-hydroxynonenal (4-HNE) have been implicated in neuronal perikaryal damage. This study sought to determine whether 4-HNE was involved in white matter damage in vivo and in vitro. Immunohistochemical studies detected an increase in cellular and axonal 4-HNE within the ischemic region in the rat after a 24-hour period of permanent middle cerebral artery occlusion. Exogenous 4-HNE (3.2 nmol) was stereotaxically injected into the subcortical white matter of rats that were killed 24 hours later. Damaged axons detected by accumulation of β-amyloid precursor protein (β-APP) were observed transversing medially and laterally away from the injection site after intracerebral injection of 4-HNE. In contrast, in the vehicle-treated animals, axonal damage was restricted to an area immediately surrounding the injection site. Exogenous 4-HNE produced oligodendrocyte cell death in culture in a time-dependent and a concentration-dependent manner. After 4 hours, the highest concentration of 4-HNE (50 μmol/L) produced 100% oligodendrocyte cell death. Data indicate that lipid peroxidation and production of 4-HNE occurs in white matter after cerebral ischemia and the lipid peroxidation by-product 4-HNE is toxic to axons and oligodendrocytes.
The plethora of reactive oxygen and nitrogen species that are generated after ischemic or traumatic brain injury interact with multiple essential cellular components leading to tissue damage. Free radical attack on lipid membranes initiates a self-propagating process of lipid peroxidation (Radi et al., 1991; Darely-Usmar et al., 1992; Rubbo et al., 1994), which results in the generation of lipid peroxidation by-products including 2-hydroperoxynonenal, 4,5-hydroxydecanal, and 4-hydroxynonenal (4-HNE) (Comporti 1985; Comproti 1989). There has been intense interest in the role of 4-HNE in acute brain injury and chronic neurodegenerative diseases because this lipid peroxidation product is highly cytotoxic due to its interactions with a host of different cellular components and proteins (Esterbaur et al., 1991). For example, 4-HNE covalently binds to cytoskeletal proteins (Montine et al., 1996; Mattson et al., 1997) and thus has been implicated in disruption of cytoskeletal structure, an important pathologic mechanism in cerebral ischemia and traumatic brain injury (Posmantur et al., 1994; Dewar and Dawson 1995; Saatman et al.,1996; Yam et al., 1997; 1998; McCracken et al., 1999). Other proteins to which 4-HNE conjugates include neurotransmitter, glucose, and ion transporters (Siems et al., 1996; Springer et al., 1997; Keller et al., 1997; Blanc et al., 1998); 4-HNE also inhibits specific components of brain mitochondrial respiration (Picklo et al., 1999). This multiplicity of actions of 4-HNE suggests that damage caused by 4-HNE after brain injury could occur not just in neuronal perikarya, but in all cellular compartments (neurons, glia, and axons). The induction of apoptosis (Kurman et al., 1997) and cell death (Montine et al., 1996; Pedersen et al., 1999) in neuronal cultures indicates the vulnerability of neuronal perikarya to 4-HNE. The disruption of synaptosomal function (Keller et al., 1997) after 4-HNE treatment in vitro suggests the vulnerability of synaptic terminals to this lipid peroxidation product. However, the susceptibility of axons and oligodendrocytes to 4-HNE-induced damage is not currently known.
Axons and oligodendrocytes are the major components of cerebral white matter. The authors have highlighted the importance of white matter damage in ischemic brain damage (Dewar et al., 1999). Damage to white matter tracts is also a significant pathologic consequence of traumatic brain injury (Gentry et al., 1988; Maxwell et al., 1993; Saatman et al,. 1996; McCracken et al., 1999). The mechanisms leading to white matter damage after acute brain injury are poorly understood in contrast to the comprehensive mechanistic insight that relates to neuronal perikarya. In vitro studies have implicated influx of calcium into the axoplasm as a critical pathogenic event in cytoskeletal breakdown (Stys, 1998). In cultured neurons, intracellular levels of calcium are elevated in response to 4-HNE treatment (Siems et al., 1996). Accumulation of 4-HNE in fibers of the corticospinal tract after in vivo spinal cord injury (Springer et al., 1997) raises the possibility that 4-HNE toxicity may play a role in this type of white matter damage. Elevated levels of 4-HNE have been detected in the brain after transient ischemic (Urabe et al., 1998; Horsburgh et al., 2000) or traumatic injury (Zhang et al., 1999), although immunohistochemical labeling of 4-HNE-modified proteins in these studies focused only on neuronal or microglial cells. It is currently not known whether 4-HNE accumulates in white matter after an ischemic insult.
The toxic effects of 4-HNE on neurons have been investigated predominantly in vitro in cell culture systems, although one study has demonstrated damage to neuronal perikarya after injection of 4-HNE directly into the basal forebrain of rats (Bruce-Keller et al., 1998). The goals of the current study were threefold: first, to determine if endogenous 4-HNE accumulates in white matter after an ischemic injury; second, to determine if intracerebral injection of exogenous 4-HNE damages axons in vivo; and third, to determine if exogenous 4-HNE is toxic to oligodendrocytes in vitro.
MATERIALS AND METHODS
Focal cerebral ischemia in the rat
Tissue sections used for 4-HNE immunostaining were obtained from rats subjected to 24 hours of permanent middle cerebral artery (MCA) occlusion (Longa et al., 1989). Male Sprague Dawley rats weighing 270 to 320 g were anesthetized with 5% halothane and 70% N2O:30% O2 (n = 6). The authors previously reported the quantification of white matter pathology in these animals (Valeriani et al., 2000). Paraffin sections (5 μm) from six rats at the level of the anterior hypothalamus were used. All animals had a previously defined ischemic lesion in grey matter and extensive axonal damage in the internal capsule, as reflected by accumulation of amyloid precursor protein (Valeriani et al., 2000).
Intracerebral injection of 4-HNE
Adult male Sprague Dawley rats weighing 290 to 330 g were anesthetized with 5% halothane and 70% N2O:30% O2. Animals then were placed in a David Kopf stereotaxic frame (Clark Electromedical, U.K.), a face mask was fitted over the snout, and the halothane was reduced to 1% to 2% for the duration of surgery. An incision was made in the scalp and the skull was exposed. The subcortical white matter was stereotaxically injected with 0.5 μL of either 4-HNE (64 mmol/L; Cayman chemicals), vehicle (ethanol), or artificial cerebrospinal fluid (aCSF) at coordinates 0.26 mm from bregma, 2 mm lateral from the midline, and 3 mm dorsal from the surface of the brain at a rate of 0.1 μL/min. Anesthesia was discontinued and the animals were allowed to recover for 24 hours. Animals were reanesthetised with 5% halothane, 70% N2O:30% O2, and transcardially perfusion-fixed with 0.9% saline and 4% paraformaldehyde. Rats were decapitated and the heads post-fixed in 4% paraformaldehyde for 48 hours before being processed and paraffin embedded. Five-micrometer sections were cut for histology and immunohistochemistry.
Cell culture
Mixed glial cells were prepared from postnatal (0 to 3 days) Sprague Dawley rats. Rats were decapitated and the hippocampus and meninges carefully removed in culture media—hanks Balanced Salt solution without calcium and Magnesium (Life Technologies). The neopallia was finely chopped and enzymatically dissociated using 2000 U/mL collagenase (Sigma), 0.25% trypsin (Sigma), and 0.02% ethylenediaminetetraacetic acid (EDTA; Sigma). Cell suspension was triturated and then centrifuged at 1000 g for 7 minutes (Hettich zentrifugen, Universal 16/16R). Cell density was determined by trypan blue exclusion using an inverted phase contrast microscope (Lecia DMIL). Cells then were plated at 6 × 106 per 75 cm3 flask in culture media (20% fetal calf serum in Dulbecco's Modified Medium (GIBCO). Culture media were changed every 2 to 3 days until 80% confluent or 12 days maximum. To obtain oligodendrocyte cultures, a modified method of McCarthy and DeVellis (1980) shaking protocol was used. After overnight shaking and centrifugation, cell density was determined as above and plated at 2 × 104 onto poly-L-lysine coverslips. Culture meida were changed every 2 to 3 days in Sato's media (0.029 μL BSA pathocyte4 in PBS; 1.61 mg/mL putrescine/H2O; 0.4 mg/mL thyroxine T4/EtOH; 0.34 mg/mL tri-iodo-thryonine T3/EtOH; 0.062 mg/mL progesterone/EtOH; and 0.387 mg/mL sodium selenite/EtOH [Sigma]) until 7 days old. Oligodendrocytes and astrocytes were immunolabeled with the galactocerebroside or glial fibrillary acidic protein, respectively, to determine the purity of oligodendrocyte cultures (∼95% pure).
Statistical analysis
To determine if there was a significant difference between the lesion volume of grey and white matter in the 4-HNE-treated group compared with the vehicle-treated groups, a one way analysis of variance was used with subsequent Student's t-test with Bonferroni corrections for the multiple comparisons. Oligodendrocyte cell death in culture after increased concentrations of exogenous 4-HNE and exposure time was assessed by analysis of variance, with subsequent Student's t-test, and Bonferroni corrections for the multiple comparisons.
RESULTS
4-HNE immunoreactivity after focal cerebral ischemia in vivo
In the hemisphere ipsilateral to the occluded MCA, there was a marked increase in the intensity of 4-HNE immunostaining in axons of the internal capsule tracts permeating the striatum and in axons of the medial forebrain bundle (Fig. 1A). Axons at the margin of the ischemic lesion were strongly 4-HNE-positive, whereas those in the core of the ischemic lesion were not as intensely stained. In the hemisphere contralateral to the occluded MCA, 4-HNE immunoreactivity was pale and diffuse both in white matter tracts permeating the striatum and the internal capsule (Fig. 1B). The pattern and distribution of 4-HNE immunostaining was reminiscent of the axonal bulbs and swellings that are labeled with the axonally transported protein APP (Fig. 1C). Amyloid precursor protein immunoreactivity was not present in the contralateral hemisphere (Fig. 1D). Increased 4-HNE immunoreactivity in neuronal perikarya was also present at the boundary between normal and ischemic tissue in the ipsilateral cortex (Fig. 1E), but neuronal perikaryal staining was pale and diffuse in the contralateral cortex (Fig. 1F).
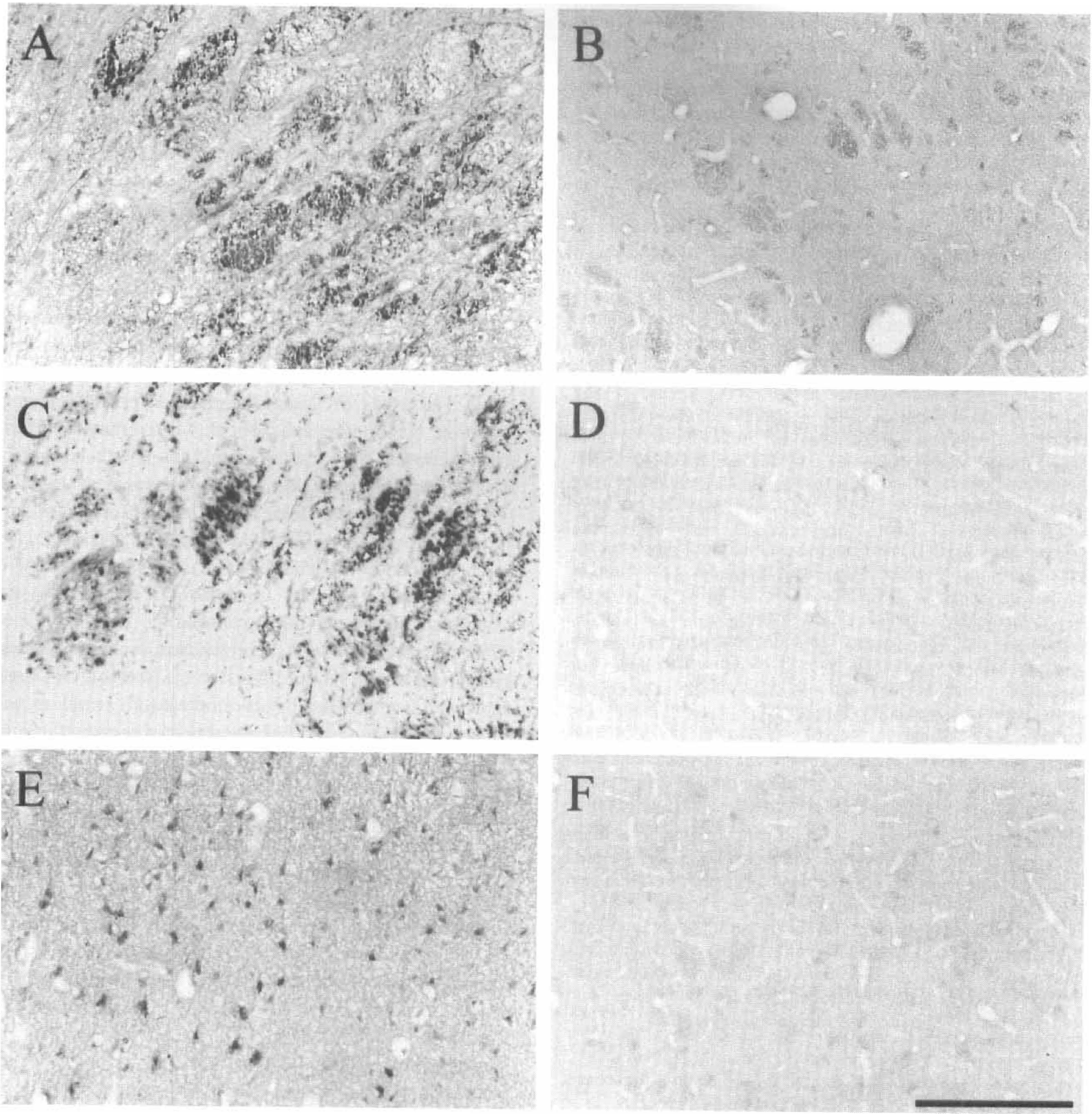
4-Hydroxynonenal (4-HNE) immunoreactivity after permanent focal cerebral ischemia. In the hemisphere ipsilateral to the occluded middle cerebral artery (MCA), 4-HNE immunoreactive axons were prominent in the myelinated fiber tracts of the caudate nucleus and internal capsule
Intracerebral injection of 4-HNE
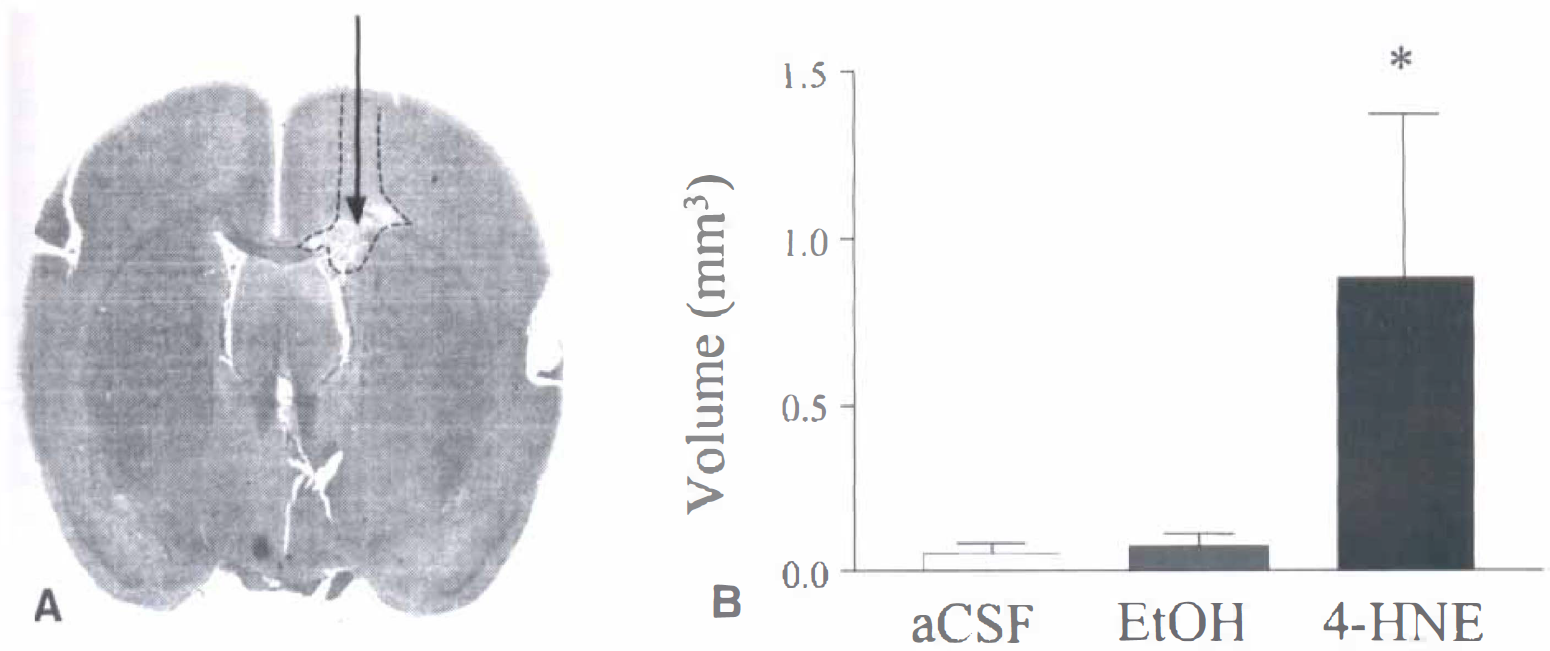
Intracerebral injection of 4hydroxynonenal (4-HNE). The digitized image
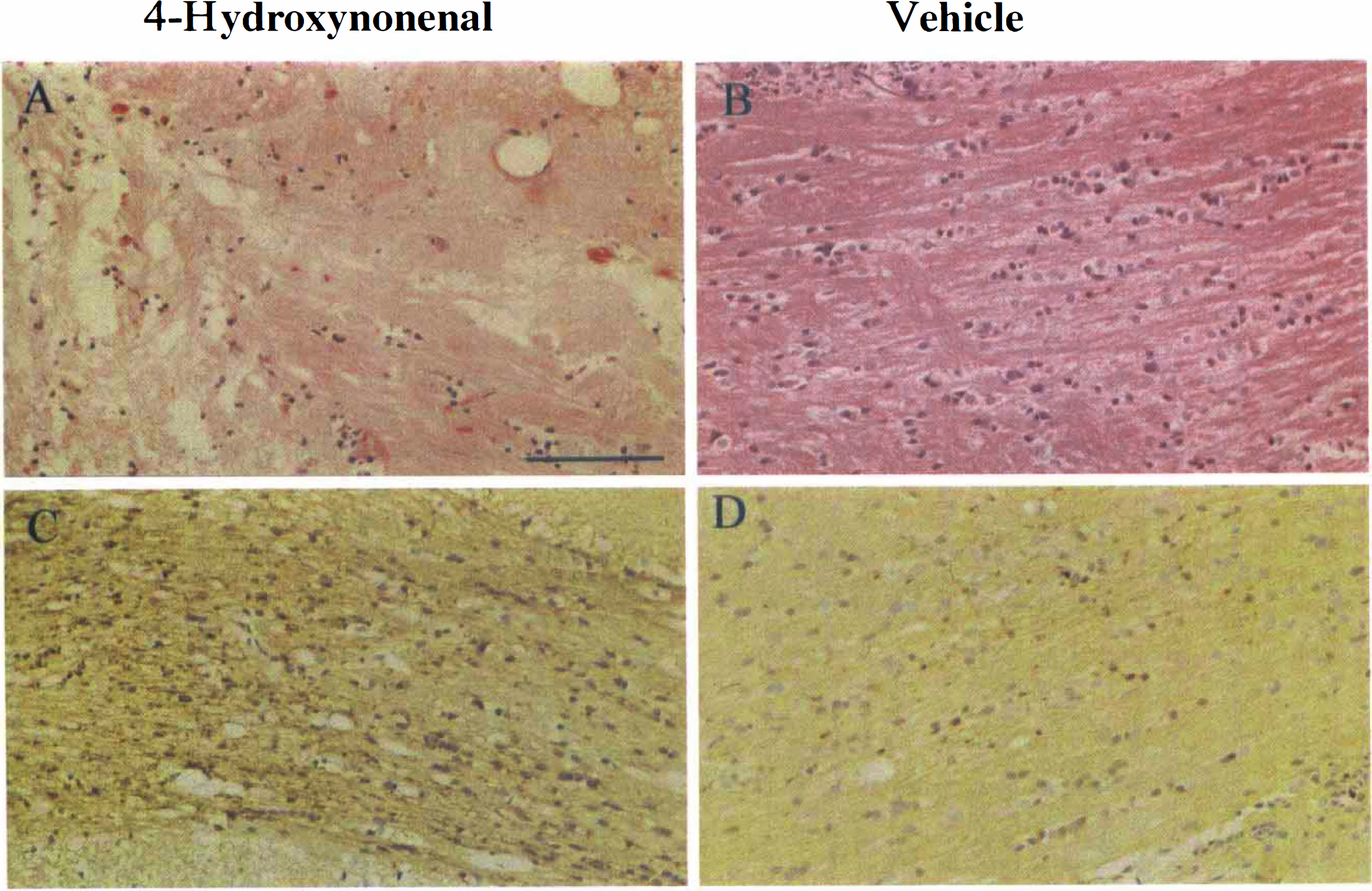
Histology and immunohistochemical staining after intracerebral injection of 4-HNE and vehicle. In hematoxylin and eosin stained sections
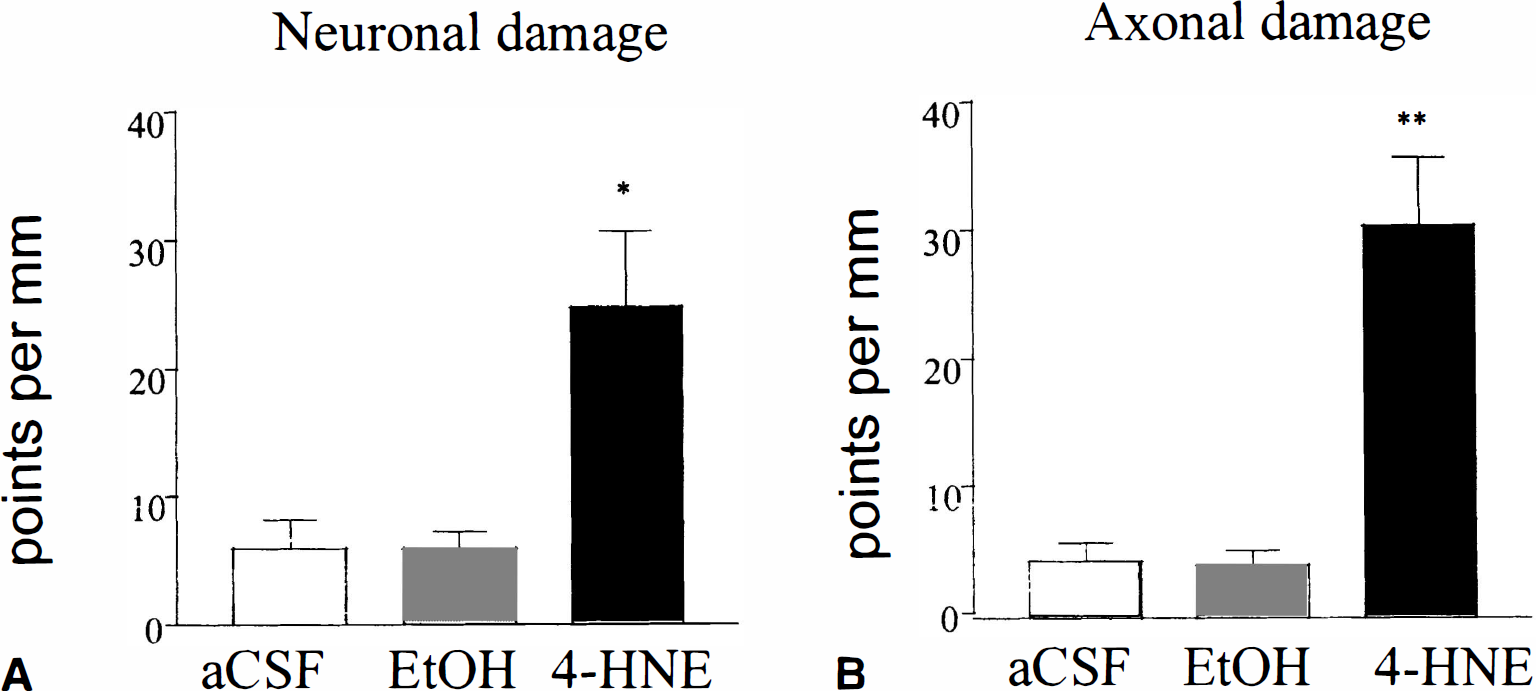
Extent of neuronal and axonal damage. The extent of neuronal necrosis in cortical grey matter was significantly greater in the 4-HNE–treated group compared with the vehicle-treated groups
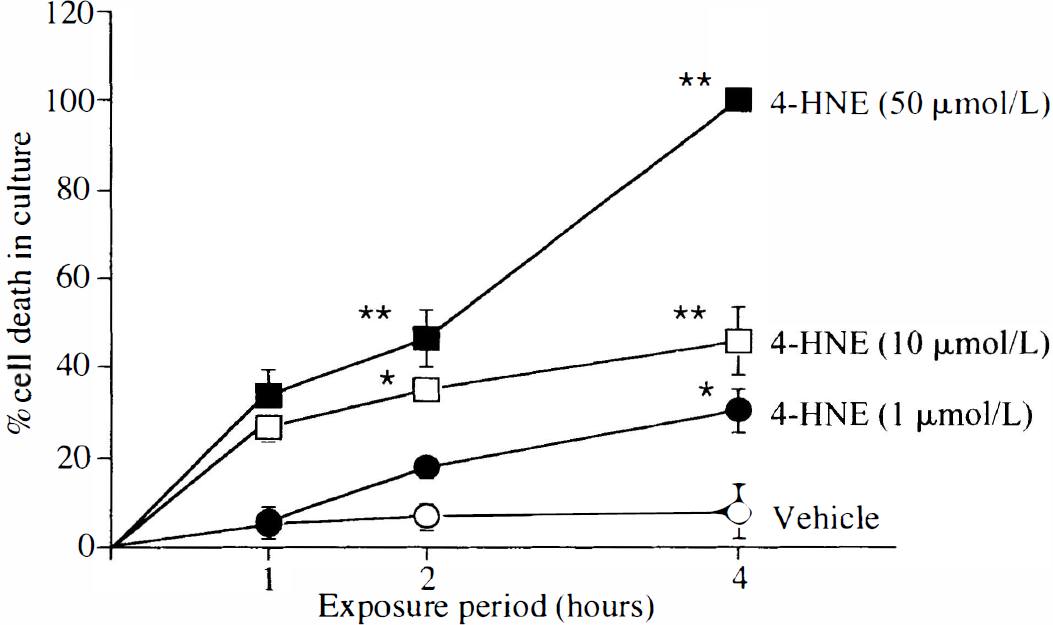
Exogenous 4-HNE increased oligodendrocyte cell death in a time-and concentration-dependent manner. The quantitative data demonstrates an increase in the percentage of oligodendrocyte cell death in culture after exposure to 1, 10, or 50 μmol/L 4-HNE over 1, 2, or 4 hours. Data are expressed as mean ± SD. * P < 0.01 and ** P < 0.001 for comparison with vehicle at the same time point.
DISCUSSION
The current study demonstrates that axons contained increased levels of 4-HNE immunoreactivity after focal cerebral ischemia in vivo, and that intracerebral injection of 4-HNE in vivo caused axonal damage. In addition, the study demonstrates that oligodendrocytes in vitro were vulnerable to 4-HNE-induced toxicity. These findings suggest that lipid peroxidation and the generation of 4-HNE may play a role in the pathophysiology of white matter damage after an ischemic insult.
Currently, the mechanisms leading to ischemic white matter damage in vivo are poorly understood in comparison with those in neuronal perikarya (Flamms et al., 1978; Braughler et al., 1989; Chan 1994). Lipid peroxidation is a major contributor to ischemic damage in neurons and this process results in the generation of aldehyde by-products, including 4-HNE. The presence of 4-HNE therefore indicates that lipid peroxidation has taken place. 4-HNE immunoreactivity was detected in neuronal perikarya and microglia after focal ischemia (Urabe et al., 1998), and increased levels of 4-HNE have been detected in tissue homogenates after transient forebrain ischemia in the rat. In the current study, increased 4-HNE immunostaining was observed not only in neuronal perikarya within the ischemic hemisphere but also within axons. To determine whether 4-HNE has a primary role in white matter damage a 10-fold lower concentration of substances known to damage neuronal perikarya (Bruce-Keller et al., 1998) was stereotaxically injected into the subcortical white matter of rats, and the extent of axonal damage was assessed by the accumulation of APP. The authors, as well as others, have previously used APP immunoreactivity as a sensitive marker of axonal damage after focal ischemia and trauma (Gentleman et al., 1993; Yam et al., 1997, 1998; McCracken et al., 1999; Valeriani et al., 2000; Graham et al., 2000). Amyloid precursor protein is transported anterogradely and retrogradely within axons by fast axonal transport and accumulates at sites where this process is disrupted (Koo et al., 1990). Microtubules are an essential component of fast axonal transport and it has been suggested that 4-HNE causes damage in neuronal perikarya by binding to microtubular and microtubule-associated proteins (Montine et al., 1996; Mattson et al., 1997; Neely et al., 1999). Conformational changes in the structure of β-tubulin, a major component of microtubules, were induced by exposure of synaptosomes to 4-HNE (Subramanian et al., 1997). Previously, the authors reported alterations in axonal β-tubulin immunostaining in response to focal ischemia in the rat (Dewar and Dawson, 1997). It is possible that similar changes could occur in axons exposed to 4-HNE with dissolution of microtubule structure, disruption of axonal transport, and APP accumulation.
Neuronal perikarya damage was anatomically circumscribed in the cerebral cortex above the site of 4-HNE injection site in subcortical white matter. The reasons for this could include tracking of 4-HNE up the needle track, postinjection diffusion of 4-HNE, or other pathogenic substances produced from damaged axons into the cortex. However, damaged axons were also prominent in areas of subcortical white matter distant, both medially and laterally, from the 4-HNE injection site where the overlying cortex appeared morphologically normal. 4-HNE may track preferentially along myelinated fibers away from its initial site of delivery and cause axonal pathology until concentrations within the tissue fall below the threshold for damage. In addition to the direct attack on microtubule structure, 4-HNE can also interact with other proteins that are pertinent to axonal damage. For example, 4-HNE promotes the cross-linking of the glutamate transporter, GLT-1 (Blanc et al., 1998), and inhibition of this transporter by dihydrokainate reduced anoxia-induced damage in spinal cord white matter in vitro (Li et al., 1999). The integrity of Na+-K+-ATPase at the node of Ranvier is essential for the maintenance of axonal ionic homeostasis, and 4-HNE can inhibit this transporter either directly by binding to the protein (Siems et al., 1996; Mark et al., 1997; Mattson et al., 1998) or by reducing ATP levels through its ability to cause mitochondrial dysfunction (Keller et al., 1997; Picklo et al., 1999). The resulting dysregulation of axoplasmicCa2+ may initiate a cascade of events including Ca2+-activated breakdown of the cytoskeleton, disruption of axonal transport, and finally axonal damage.
After intracerebral injection of 4-HNE, glial cells within the subcortical white matter had a shrunken eosinophilic appearance indicative of necrosis. A proportion of these glial cells was putatively oligodendrocytes, although the authors' histologic and immunohistochemical markers could not confirm this. To determine if oligodendrocytes themselves were susceptible to 4-HNE toxicity and not damaged as a secondary consequence of axonal damage, the authors exposed cultured oligodendrocytes to 4-HNE. The data demonstrate that oligodendrocytes were vulnerable to 4-HNE-induced cytotoxicity. A 4-hour exposure to 4-HNE (10 μmol/L) caused approximately 40% cell death in the oligodendrocyte cultures. This is comparable to results obtained from cultures hippocampal neurons in which a 5-hour exposure to 10 μmol/L 4-HNE caused a similar amount of cell death (Mark et al., 1997). Although these data suggest that neurons and oligodendrocytes are equally vulnerable to attack by 4-HNE, it still remains to be established whether oligodendrocytes and neurons are exposed to similar concentrations of 4-HNE during cerebral ischemia in vivo.
In summary, exogenous 4-HNE was toxic to axons and neurons in vivo and to oligodendrocytes in culture in vitro. In addition, endogenous levels of 4-HNE were increased in axons, neurons, and glia after acute brain injury induced by ischemia. Therefore, the results suggest that 4-HNE–induced damage may constitute a common pathologic mechanism in grey and white matter after acute brain injury.