
Abstract
Numerous studies indicate a role for oxidative stress in the neuronal degeneration and cell death that occur during ischemia–reperfusion injury. Recent data suggest that inhibition of the proteasome may be a means by which oxidative stress mediates neuronal cell death. In the current study, the authors demonstrate that there is a time-dependent decrease in proteasome activity, which is not associated with decreased expression of proteasome subunits, after cerebral ischemia–reperfusion injury. To determine the role of oxidative stress in mediating proteasome inhibition, ischemia–reperfusion studies were conducted in mice that either overexpressed the antioxidant enzyme glutathione peroxidase [GPX 1(+)], or were devoid of glutathione peroxidase activity (GPX −/−). After ischemia–reperfusion, GPX 1(+) mice displayed decreased infarct size, attenuated neurologic impairment, and reduced levels of proteasome inhibition compared with either GPX −/− or wild type mice. In addition, GPX 1(+) mice displayed lower levels of 4-hydroxynonenal-modified proteasome subunits after ischemia–reperfusion injury. Together, these data indicate that proteasome inhibition occurs during cerebral ischemia–reperfusion injury and is mediated, at least in part, by oxidative stress.
Increasing evidence suggests a role for oxidative stress in mediating neuron death and neuron degeneration in numerous disorders, including cerebral ischemia–reperfusion injury (IRI). For example, increased levels of oxidative damage occur during IRI (Granger et al., 1986; Chan, 1996) and before neuron death (Keller et al., 1998). Increasing the levels of antioxidant enzymes (Yoshida et al., 1996; Keller et al., 1998; Chan et al., 1998), or application of antioxidants (Imaizumi et al., 1990), attenuates neuron damage and neuron death after IRI. Conversely, lowering the antioxidant capacity increases neuron death after IRI (Kawase et al., 1999). Recent studies indicate that oxidative stress may mediate neuron death by inhibiting essential enzymatic pathways or through the generation of toxic oxidation products (Granger et al., 1986; Girotti, 1998). In particular, the lipid peroxidation product 4-hydroxynonenal (HNE) has been demonstrated to be generated after IRI (Yoshino et al., 1997), whereas application of HNE is sufficient to induce neuron death (Kruman et al., 1997).
The proteasome is a large ∼700 kDa complex, which is responsible for the majority of overall protein degradation (Rock et al., 1994), and has been best characterized for its role in the ubiquitin-ATP dependent proteasome pathway (Hershko and Ciechanover, 1992; Alves-Rodriguez et al., 1998). In particular, the proteasome is responsible for the removal of most oxidized, aggregated, and damaged proteins (Murakami et al., 1990; Hershko and Ciechanover, 1992; Tanaka, 1998). The proteasome is a barrel-like structure composed of 4 rings, with each ring containing 7 α- oragr;-or β-subunits (Tanaka, 1998). The two inner rings of the proteasome contain the β-subunits, which possess the proteolytic activity, and the two outer rings are composed of the α-subunits, which are believed to aid in stabilizing the proteasome complex (Goldberg et al., 1997; Tanaka, 1998). Proteasome activity is essential for neuron survival, with inhibition of the proteasome sufficient to induce caspase activation and neuron death (Qiu et al., 2000).
Recent data indicate that oxidative stress may inhibit proteasome activity during IRI. Elevations in ubiquitinated proteins occur during IRI (Alves-Rodriguez et al., 1998), which is consistent with the presence of inhibited proteasome activity. The proteasome can be inhibited by exposure to oxidative stress in vitro and in vivo (Reinheckel et al., 1998; Glockzin et al., 1999; Okada et al., 1999; Keller et al., 2000a). In particular, the lipid peroxidation product HNE has been demonstrated to inhibit proteasome activity (Friguet and Szweda, 1997; Okada et al., 1999). The focus of the current study was to characterize IRI-induced alterations in proteasome activity and to determine the involvement of oxidative stress.
MATERIALS AND METHODS
Focal cerebral ischemia
Glutathione peroxidase transgenic mice [GPX1(+)] and GPX deficient mice (GPX−/−) were generated as described previously (Cheng et al., 1997; Beck et al., 1998). Briefly, GPX1(+) mice were derived from B6C3 (C57B1 × C3H) hybrid mice (Taconic, Germantown, NY, U.S.A.). A 5.3-kb SacI fragment containing the entire mouse GPX1 gene was used for microinjection into fertilized mouse eggs from B6C3 mice. The GPX−/− mice were generated by Dr. Ye-Shih Ho (Ho et al., 1997). Twelve animals each of wild type, GPX1+, and GPX −/− mice were used in the ischemic studies for behavior, infarct volume, and physiologic parameters. Five mice of each type were used for each time point in proteasome assays. Arterial blood gas samples (50 μL) were analyzed for pH, arterial oxygen pressure, and partial pressure of carbon dioxide using a blood gas/pH analyzer (Corning 178; CIBA-Corning Diagnostics, Medford, MA, U.S.A.). Samples were taken immediately before and 10 minutes after middle cerebral artery occlusion (MCAO), and 10 minutes after the commencement of reperfusion. Rectal and temporalis muscle temperature was maintained at 37 ± 0.5°C with a temperature control unit (FHC, Brunswick, ME, U.S.A.). Cerebral blood flow was determined in anesthetized animals under resting conditions, 30 minutes into ischemia, and 10 minutes after reperfusion. Measurements were made using a laser–Doppler flowmeter (Laserflo BP; Vasomedic, Westbury, NY, U.S.A.). Two flexible probe tips were secured 2 mm posterior and 3 mm lateral to bregma, and 2 mm posterior (periinfarct region) and 6 mm lateral to bregma on the ischemic hemisphere. Neurologic deficits were assessed 22 hours after reperfusion based on a scale from 0 (no deficits) to 3 (severe deficits) as described previously (Huang et al., 1994).
The protocol for performing focal cerebral ischemia using a MCAO reperfusion method was similar to that used in the authors' previous studies (Keller et al., 1998), which were adapted from a previously described method (Huang et al., 1994). Briefly, mice were anesthetized with 350 mg/kg chloral hydrate. In all mice, the left common carotid artery was exposed, and the occipital, superior thyroidal, maxillary, and lingal branches of the external carotid artery were coagulated; the pterygopalatine artery was ligated. An incision was made in the external carotid artery, and a polylysine-coated suture was advanced into the MCA. The thread was left in place for 60 minutes and then removed to allow reperfusion. After surgery, mice were returned to their cages and given ad libitum access to food and water. At the times indicated, mice were killed by anesthesia overdose, and perfused with sterile normal saline. For quantifying infarct area, coronal brain sections (1-mm-thick) were stained with 1% triphenyltetrazolium chloride (TTC). Images of stained sections were analyzed using a Hitachi KPD50 color digital camera (Medical Video System, Winston-Salem, NC, U.S.A.), and infarct size volume was calculated using a Macintosh G3 computer and image analysis software (Scion Image NIH version 1.59; Frederick, MD, U.S.A.) as described previously (Keller et al., 1998). Infarct volume was calculated using the following formula: [(contralateral volume − ipsilateral undamaged volume) × 100%/contralateral volume] to eliminate effects of edema (Bonventre et al., 1997). For analysis of proteasome activity, saline perfused brains were collected on ice and processed as described below.
Analysis of proteasome activity
Brains were analyzed for chymotrypsin activity, postglutamyl-peptidase, and trypsin activity using N-LLVY-MCA, N-LLE-β-napthylamine, and N-LLR-MCA substrates, respectively. Proteasome activity was determined in tissue homogenates by measuring the rate of substrate cleavage as described previously (Rock et al., 1994; Keller et al., 2000a, 2000b; Qiu et al., 2000). Briefly, tissue samples were collected on ice in proteasome buffer (10 mmol/L Tris-HCl (pH 7.8), 1 mmol/L EDTA, 0.5 mmol/L dithiothreitol, 2 mmol/L ATP, and 5 mmol/L MgCl2). Protein determinations were made on the resulting lysate (Pierce, Rockford, IL, U.S.A.), and aliquots were prepared (250 μL/250 μL) for determination of proteasome proteolytic activity. The aliquots were incubated for 30 minutes at 37°C with 2.5 μL of the indicated proteasome substrate (5 mmol/L stock) and the activity determined as the increase in the fluorescence of the reaction products. The fluorescence of trypsin and chymotrypsin activity was monitored at 340 nm excitation and 440 nm emission, whereas postglutamyl-peptidase activity was determined at 335 nm excitation and 410 nm emission, as described previously (Rock et al., 1994; Keller et al., 2000a, 2000b; Qiu et al., 2000). Background fluorescence was determined by incubating lysates with the proteasome inhibitor MG 115 (50 μmol/L) for 30 minutes before the addition of proteasome substrate. To ensure that the changes were because of proteasome activity, initial experiments were completed in which the proteasome was immunoprecipitated from lysates. Samples from which the proteasome had been immunoprecipitated displayed no increase in fluorescence (data not shown).
Western blot and Northern blot analysis
Immunoprecipitations were conducted as described previously (Keller et al., 1997). For Western blot analysis, tissue was harvested into 1 mL of cold PBS, and sonicated in homogenization buffer. A total of 50 μg of total protein was separated on a 7.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, transferred to nitrocellulose, and processed as described previously (Keller et al., 1998). Homogenates were analyzed for proteasome subunit expression using anti-α and anti-β (Calbiochem) antibodies. The levels of GPX mRNA were determined by Northern blot analysis as described previously (Cheng et al., 1997).
Statistical analysis
Statistical analysis of proteasome activity and proteasome expression was determined by analysis of variance with Scheffe's post hoc test. Differences between the wild type, transgenic, and knock-out mice were compared by paired and unpaired two-tailed Student's t-test, Scheffe's test (physiologic parameters), Bonferrini test (cerebral blood flow), or Mann-Whitney test (neurologic assessment). For all statistical analysis, 0.05 was set as the maximum amount of variance allowed to be considered statistically significant.
RESULTS
Ischemia–reperfusion injury causes inhibition of the proteasome
To determine the effect of IRI on proteasome activity, studies were conducted in mice using the MCAO model of focal IRI. No alterations in proteasome activity were observed during the first 45 minutes of ischemia (data not shown). However, after reperfusion there was a time-dependent decrease in proteasome activity within the ipsilateral hemisphere (Table 1). In contrast to the ipsilateral hemisphere, proteasome inhibition was not evident in the contralateral hemisphere (Table 1). The loss of proteasome activity was greatest in the ipsilateral hippocampus (Table 1), although significant decreases in proteasome activity occurred in the ipsilateral cortex. Interestingly, a transitory increase in proteasome activity was observed in the contralateral hippocampus and cortex after IRI (Table 1). The IRI-induced inhibition of proteasome activity resulted in inhibition of all three proteolytic activities (Fig. 1), with all three activities affected similarly. To determine if impairment of proteasome activity was because of decreased levels of α- oragr;-oragr;-or β-proteasome subunits, the authors conducted Western blot analysis. At no time point after IRI was there observed to be altered levels of either α- or β-subunits (Fig. 2).
Proteasome activity is decreased after ischemia–reperfusion injury
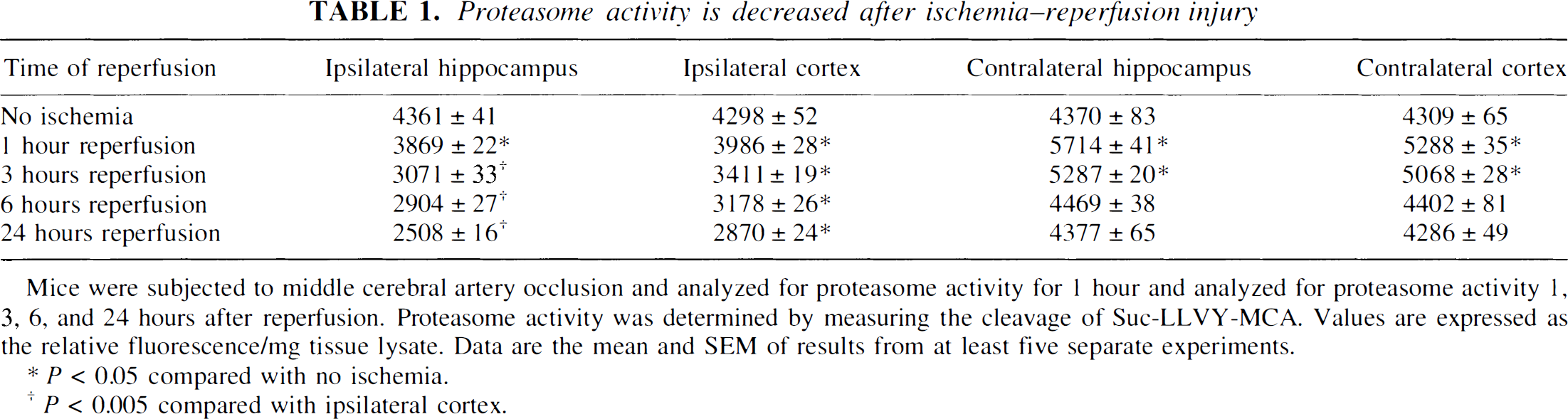
Mice were subjected to middle cerebral artery occlusion and analyzed for proteasome activity for 1 hour and analyzed for proteasome activity 1, 3, 6, and 24 hours after reperfusion. Proteasome activity was determined by measuring the cleavage of Suc-LLVY-MCA. Values are expressed as the relative fluorescence/mg tissue lysate. Data are the mean and SEM of results from at least five separate experiments.
P < 0.05 compared with no ischemia.
P < 0.005 compared with ipsilateral cortex.
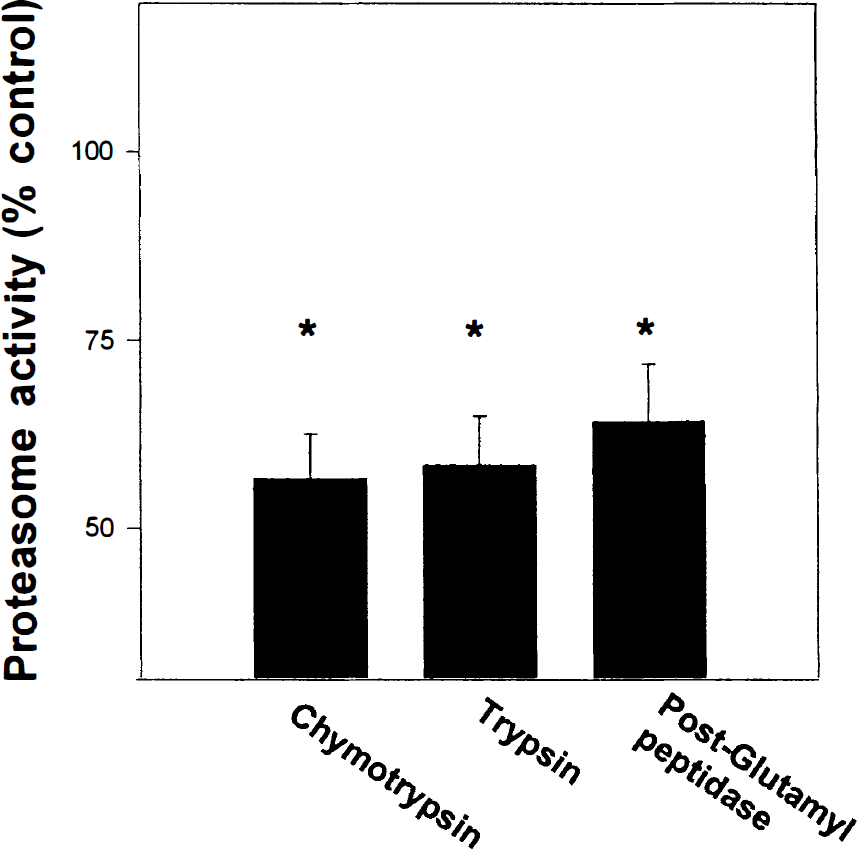
All three proteasome activities are decreased after ischemia–reperfusion injury. The chymotrypsin, trypsin, and postglutamyl peptidase activities of the proteasome were analyzed from the ipsilateral hippocampus after 6 hours reperfusion. The fluorescence values for nonischemic hippocampal tissue ranged from 4325 to 4575 per mg, and ischemia–reperfusion values ranged from 2635 to 2890 per mg. Data are the mean and SEM from five separate experiments. * P < 0.05 compared with values for no ischemia.
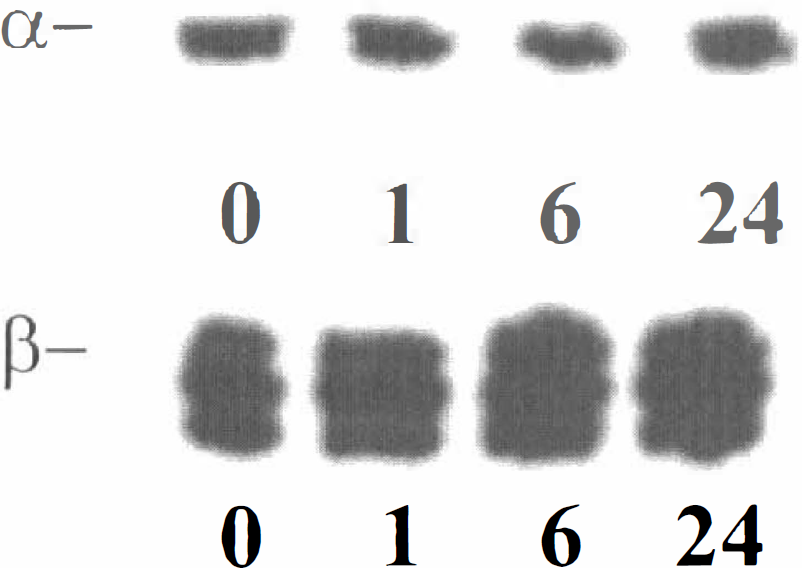
Levels of α- andagr;-andagr;-and β-proteasome subunits do not decrease during ischemia–reperfusion injury. Tissue lysates from the ipsilateral hippocampus were analyzed for α- and β-immunoreactivity 0, 1, 6, and 24 hours after reperfusion. Data are representative of results from five separate experiments.
The lack of changes in proteasome expression suggested that proteasome activity may be impaired by posttranslational modifications. To determine if oxidative stress-induced modification of the proteasome occurs during IRI, the authors conducted immunoprecipitation experiments using an antibody that recognizes HNE modified proteins (Montine et al., 1997). As early as 1 hour after reperfusion, increased HNE-immunoprecipitatable β-proteasome subunits were observed (Fig. 3). Similar results were obtained when anti-β-immunoprecipitates were analyzed for HNE immunoreactivity (data not shown).
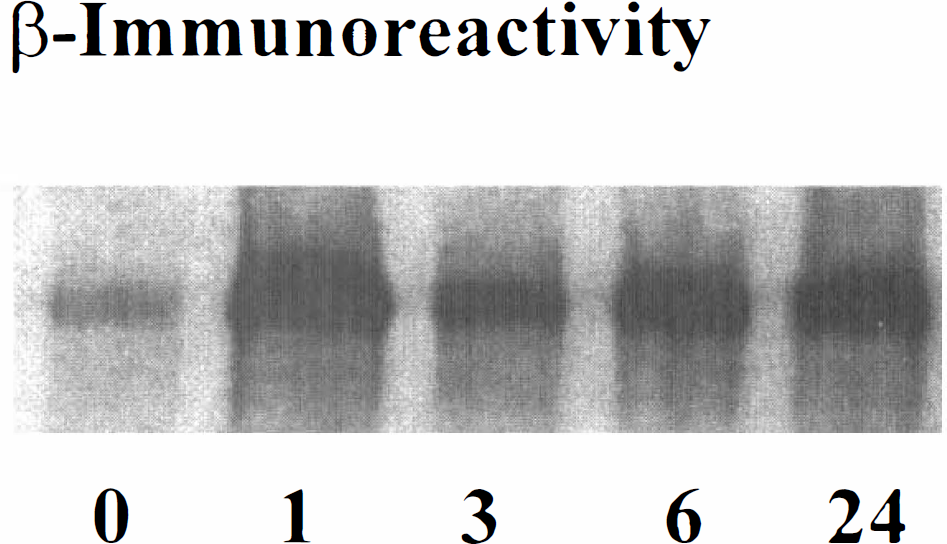
Levels of HNE-immunoprecipitatable proteasome subunits increases after ischemia–reperfusion injury. Tissue from the ipsilateral hippocampus was collected 0, 1, 3, 6, and 24 hours after reperfusion, immunoprecipitated with an antibody to HNE modified proteins, and the resulting immunoprecipitate analyzed for β-immunoreactivity. Data representative of results from three separate experiments.
Increased expression of glutathione peroxidase attenuates IRI-induced neuronal injury and proteasome inhibition
To further elucidate the involvement of oxidative stress in IRI-induced impairment of proteasome activity, experiments were conducted in mice expressing increased levels of the antioxidant enzyme glutathione peroxidase [GPX1(+)] or mice devoid of glutathione peroxidase activity (GPX1 −/−). Glutathione peroxidase transgenic animals displayed a threefold increase in GPX1 mRNA levels compared with wild type animals, whereas GPX1 −/− mice displayed no detectable GPX1 mRNA (Fig. 4A). Glutathione peroxidase activity was increased 2.1-to 5.5-fold in the brains of GPX1(+) mice, as compared with wild type mice, whereas glutathione peroxidase activity was undetectable in GPX −/− mouse brain (data not shown). After 24 hours reperfusion, the infarct size was quantified, with GPX1(+) mice displaying decreased infarct size compared with wild type mice (Fig. 4B). In contrast, GPX −/− mice exhibited increased infarct area after 24 hours reperfusion, compared with either GPX1(+) or wild type mice (Fig. 4B). Neurologic assessment revealed preserved neurologic function in GPX1(+) mice, whereas GPX −/− mice exhibited exacerbated neurologic impairment compared with wild type mice (Fig. 4C), after IRI. No significant difference in physiologic parameters (cerebral blood flow, blood pO2, pCO2, pH) was evident between wild type, GPX1(+), and GPX −/− mice (Table 2).
Physiologic parameters of wild type, GPX −/−, and GPX1 (+) mice at baseline, during ischemia, and after reperfusion
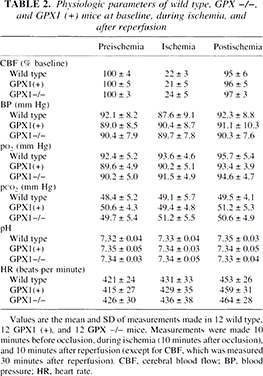
Values are the mean and SD of measurements made in 12 wild type,12 GPX1 (+), and 12 GPX −/− mice. Measurements were made 10 minutes before occlusion, during ischemia (10 minutes after occlusion), and 10 minutes after reperfusion (except for CBF, which was measured 30 minutes after reperfusion). CBF, cerebral blood flow; BP, blood pressure; HR, heart rate.
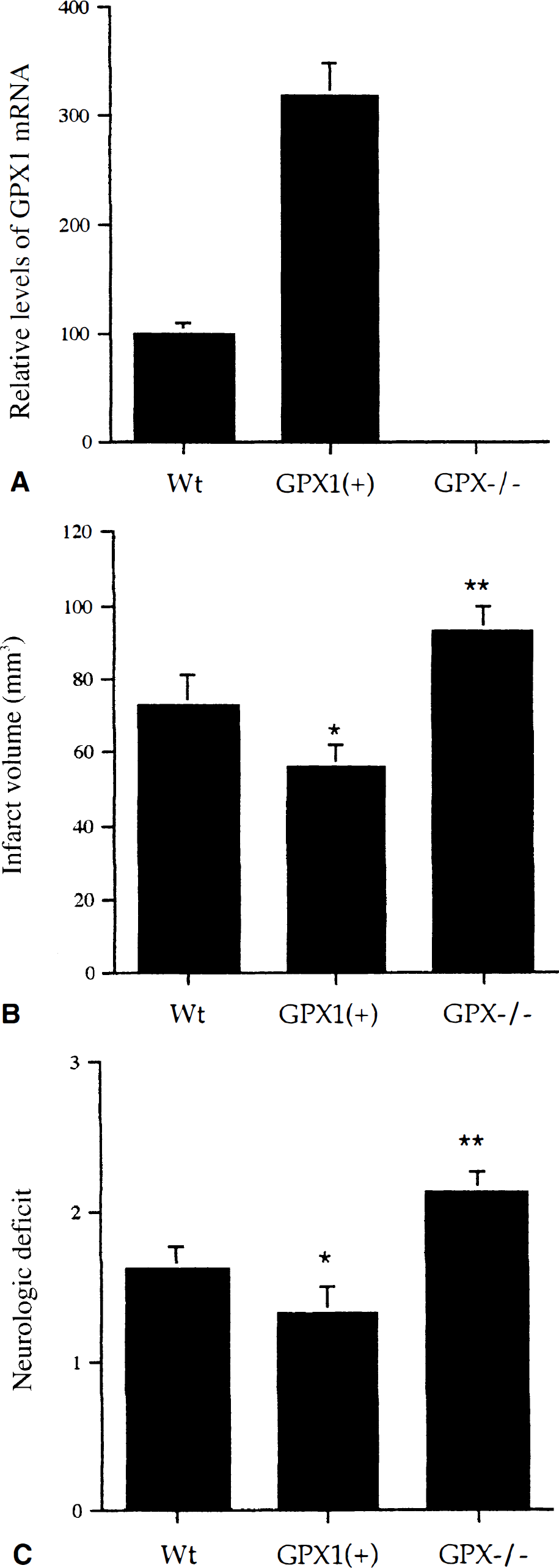
Ischemia–reperfusion injury is attenuated in mice expressing increased levels of glutathione peroxidase.
To determine if the neuroprotective effects of elevated glutathione peroxidase activity were associated with maintained proteasome function, the authors analyzed proteasome activity in GPX1(+), GPX −/−, and wild type mice 6 hours after reperfusion. Although no significant difference in basal levels of proteasome activity and proteasome expression were observed between mice (data not shown), GPX1(+) mice exhibited less severe proteasome inhibition after IRI compared with wild type mice (Table 3). In contrast, GPX1 −/− mice displayed a significantly greater degree of proteasome inhibition as compared with either GPX1(+) or wild type mice (Table 3). Consistent with the involvement of oxidative stress, after 6 hours reperfusion GPX1(+) mice displayed lower, and GPX −/− mice displayed higher, levels of HNE-immunoprecipitatable proteasome subunits compared with wild type mice (Fig. 5A). This coincided with a generalized attenuation of HNE production in the GPX1(+) mice, and an increase in HNE production in GPX −/− mice, after IRI (Fig. 5B).
Elevated levels of glutathione peroxidase attenuate ischemia–reperfusion-induced impairment of proteasome activity
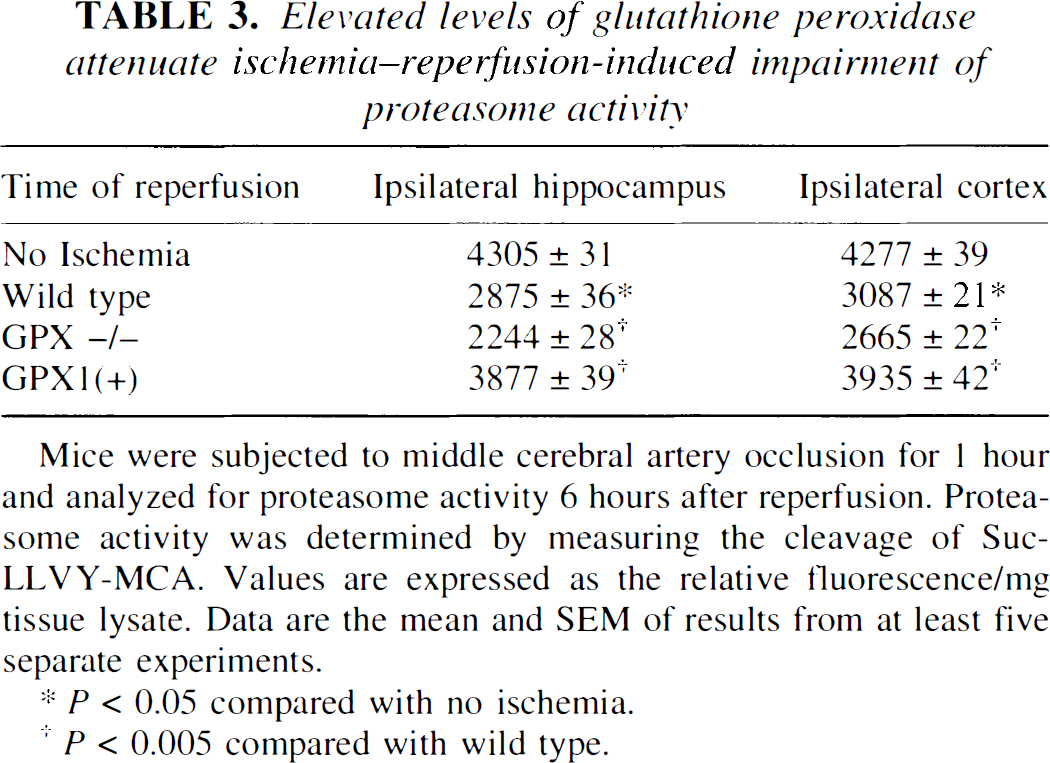
Mice were subjected to middle cerebral artery occlusion for 1 hour and analyzed for proteasome activity 6 hours after reperfusion. Proteasome activity was determined by measuring the cleavage of Suc-LLVY-MCA. Values are expressed as the relative fluorescence/mg tissue lysate. Data are the mean and SEM of results from at least five separate experiments.
P < 0.05 compared with no ischemia.
P < 0.005 compared with wild type.
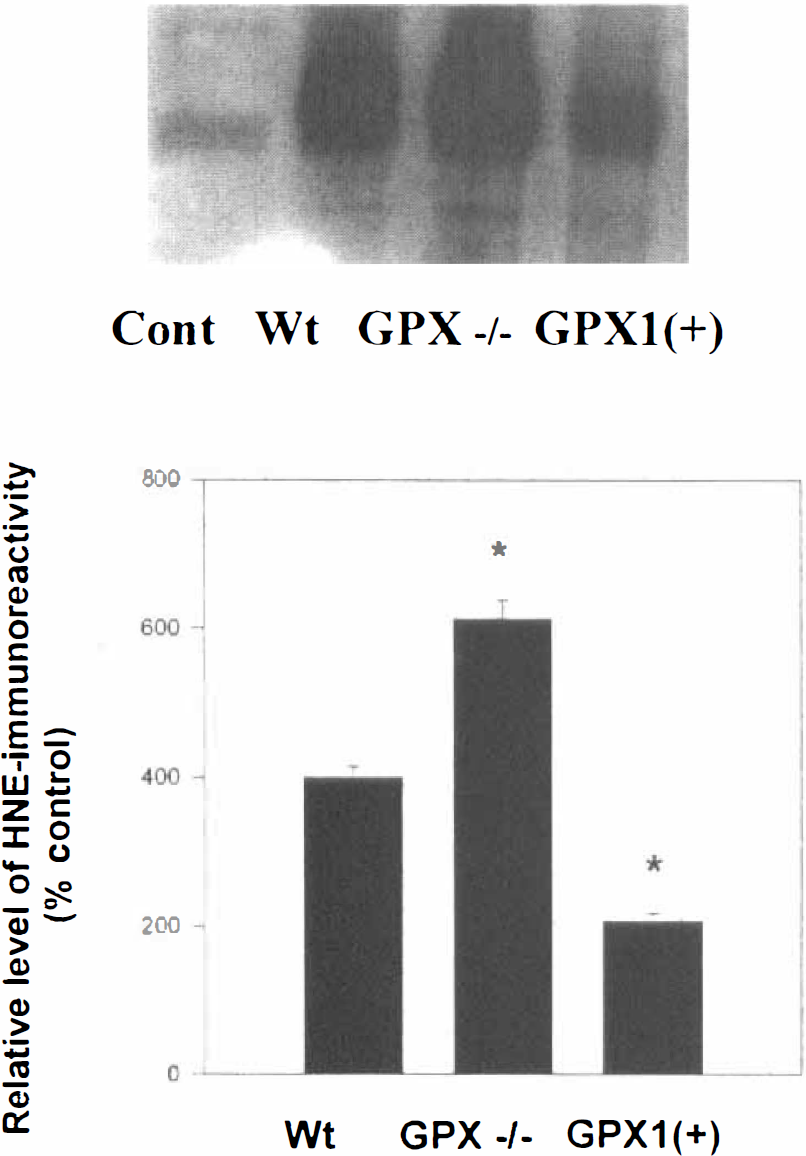
Levels of HNE-immunoprecipitatable proteasome subunits are decreased in mice expressing increased levels of glutathione peroxidase.
DISCUSSION
The current study demonstrates that proteasome activity is decreased after IRI and may therefore contribute to neuronal injury during IRI. For example, inhibition of the proteasome is sufficient to induce neuron death (Qiu et al., 2000). In addition to directly inducing neuron death, subtoxic levels of proteasome inhibition would be expected to increase neuronal viability. Proteasome activity is required for most forms of NF kB activation (Hershko and Ciechanover, 1992), a transcription factor which has been demonstrated to have neuroprotective properties in vitro and in vivo (Mattson, 1998). Because the proteasome is responsible for the removal of damaged and oxidized proteins (Grune et al., 1997), impaired proteasome activity may contribute to oxidative stress during IRI. It is interesting to speculate that the increase in proteasome activity in the contralateral hemisphere, which did not undergo significant increases in neuron damage after IRI in the current study, may represent a neuroprotective activation of proteasome activity. Alternatively, low levels of oxidative stress have been demonstrated to stimulate proteasome activity (Grune et al., 1997) and may account for increases in proteasome activity within the contralateral hemisphere.
All three proteasome activities were inhibited by cerebral IRI. The chymotrypsin-, trypsin-, and postglutamyl peptidaselike activities of the proteasome occur at distinctive sites within the proteasome (Goldberg et al., 1997; Tanaka, 1998). These data suggest that IRI may cause a severe alteration in proteasome stability and proteasome structure. The proteasome degrades substrates in a processive fashion (Goldberg et al., 1997) that is believed to be particularly important for preventing the accumulation of partially degraded proteins and the partial degradation of a substrate, which may allow for its continued functioning. For example, by degrading a regulatory portion, but not the active enzymatic portion of a proteasome substrate, the activity of that substrate could continue to aberrantly function. Alternatively, proteasome inhibition could increase the half-life of proapoptotic proteins within neurons, increasing the toxicity of their activation. It is interesting to point out that pharmacologic inhibition of the proteasome is sufficient to induce caspase activation, which may be due in part to the decreased catabolism of activated caspases (data not shown).
Data from the current study suggest that oxidative stress contributes to IRI-induced proteasome inhibition. Increased expression of the antioxidant enzyme glutathione peroxidase decreases IRI-induced proteasome inhibition, whereas decreased levels of glutathione peroxidase exacerbates IRI-induced proteasome inhibition. In particular, data from the current study indicate a role for lipid peroxidation and the lipid peroxidation product HNE in IRI-induced proteasome inhibition. Lipid peroxidation modification of the proteasome coincides with the loss of activity, whereas decreased HNE conjugation coincides with maintained proteasome activity. Once formed, HNE can form Michael adducts with cysteine, histidine, and lysine residues, and interact with proteins through Schiff-base reactions (Esterbauer et al., 1991). Numerous metabolic transporters and enzymes have been demonstrated to be impaired after exposure to HNE (Esterbauer et al., 1991), whereas application of HNE or HNE-modified proteins can inhibit proteasome activity in vitro (Friguet and Szweda, 1997; Keller et al., 2000a). It is unclear in the current study if IRI-induced proteasome inhibition is because of HNE directly modifying proteasome subunits, or whether HNE-modified proteins are becoming associated with the proteasome and thus impairing its activity. In addition to lipid peroxidation, proteasome activity can be impaired by nitric oxide and nitrosylated glutathione (Glockzin et al., 1999), which are both increased during IRI (Chan, 1996; Keller et al., 1998).
Glutathione peroxidase is a ubiquitous antioxidant enzyme that catalyzes the breakdown of hydrogen peroxide into water (Girotti, 1998; Brigelius-Flohe, 1999). In vitro studies have demonstrated that glutathione peroxidase is a more potent antioxidant than either superoxide dismutase or catalase (Toussaint et al., 1993). The potent antioxidant effects of glutathione peroxidase can be attributed to its presence in the cytosol and mitochondria, and the fact that it is able to use lipid peroxides and hydrogen peroxide as substrates (Girotti, 1998). The current study clearly indicates that increased expression of glutathione peroxidase is associated with decreased conjugation of HNE to individual proteasome subunits. Preventing HNE formation, by the scavenging of lipid peroxides and hydrogen peroxide, is likely a primary source of glutathione peroxidase conferred protection in the current study. Consistent with such an observation, GPX1(+) mice displayed lower levels of overall HNE-modified proteins after IRI than either GPX −/− or wild type mice, suggesting that glutathione peroxidase decreases the overall generation of HNE. Similarly, the authors have demonstrated that increased expression of the antioxidant enzyme manganese superoxide dismutase decreases IRI-induced lipid peroxidation and increases neuron survival after IRI (Keller et al., 1998).
The current study indicates that proteasome inhibition may be a primary source of oxidative stress-induced toxicity during IRI. Consequently, these data suggest that pharmacologic interventions designed to preserve proteasome activity may be beneficial in attenuating neuron death in conditions associated with oxidative stress, including IRI.
Footnotes
Acknowledgements
The authors thank Dr. T. Montine for supplying the HNE antibody.