
Abstract
Many studies have reported ischemia protection using various preconditioning techniques, including single dose 3-nitropropionic acid (3-NPA), a mitochondrial toxin. However, the cellular signal transduction cascades resulting in ischemic tolerance and the mechanisms involved in neuronal survival in the tolerant state still remain unclear. The current study investigated the mRNA and protein expression of the antiapoptotic bcl-2 and the proapoptotic bax, two antagonistic members of the bcl-2 gene family, in response to a single dose of 3-NPA, to global cerebral ischemia–reperfusion, and to the combination of both 3-NPA-pretreatment and subsequent global cerebral ischemia–reperfusion. Brain homogenates of adult Wistar rats (n = 25) were analyzed for bcl-2 and bax mRNA expression using a new highly sensitive and quantitative polymerase chain reaction (PCR) technique that allows real-time fluorescence measurements of the PCR product (LightCycler; Roche Diagnostics, Mannheim, Germany). Animals for mRNA analysis received 3-NPA (20 mg/kg, intraperitoneal; “chemical preconditioning”) or vehicle (normal saline), and were either observed for 24 plus 3 hours or were subjected to 15 minutes of global cerebral ischemia 24 hours after the pretreatment and observed for 3 hours of reperfusion. Immunohistochemistry was applied to serial brain sections of additional rats (n = 68) to determine amount and localization of the respective Bcl-2 and Bax protein expression in various brain areas. One set of animals was injected with 3-NPA and observed for 3, 12, 24, and 96 hours; a second set was exposed to 15 minutes global cerebral ischemia, 3, 12, and 24 hours reperfusion; and a third set was pretreated with 3-NPA or saline 24 hours before the ischemic brain insult and observed for 96 hours of reperfusion. The authors found single dose 3-NPA treatment to be associated with an elevated bcl-2:bax ratio (increased bcl-2 expression, decreased bax expression), both on the transcriptional (mRNA) and the translational (protein) level. The differential influence of 3-NPA was maintained during early recovery from global cerebral ischemia (3 hours), when 3-NPA pretreated animals showed higher bcl-2 and lower bax mRNA levels compared with rats with saline treatment. Respective changes in protein expression were localized predominately in neurons vulnerable to ischemic damage. Compared with baseline, Bcl-2 protein was significantly higher in surviving neurons at 96 hours after the insult, whereas Bax protein remained unchanged. However, at this late time of postischemic recovery (96 hours), the protein expression pattern of surviving neurons was not different between animals with and without 3-NPA pretreatment. To the authors' knowledge, the current study is the first report on the differential expression of pro-and antiapoptotic genes after a single, nonlethal dose of 3-NPA. The current results suggest alterations in the balance between pro-and antiapoptotic proteins as a potential explanation for the reported protection provided by chemical preconditioning using 3-NPA in rats.
Tolerance to cerebral ischemia can be induced (preconditioning) by sublethal stress before the cerebral insult as, for example, short ischemic episodes (Kitagawa et al., 1990), cytokines (Barone et al., 1998), drugs (Heurteaux et al., 1995), and chemicals such as 3-nitropropionic acid (3-NPA;Riepe et al., 1997; Brambrink et al., 1998; Wiegand, 1999; Sugino et al., 1999).
Ischemic tolerance may have been of evolutionary benefit to mammals, and a common mechanism may be present (Hochachka et al., 1998). It has been shown that changes in cellular signal transduction may be involved in tolerance induction in the brain; for example, activation of ATP-dependent potassium channels (KATP-channels;Heurteaux et al., 1995) may result in neuronal hyperpolarization, reduction of postischemic reactive oxygen species (ROS), and conservation of energy rich phosphates (Riepe et al., 1997) during subsequent ischemia–reperfusion. Rapid tolerance induction may partially depend on this mechanism, independent of the stimulus used (Stagliano et al., 1999), although it may only postpone neuronal death (Pérez-Pinzón, 1997).
Sustained and long-lasting ischemic tolerance, however, requires at least one day to develop. Various stress stimuli to neuronal tissue are accompanied by increased levels of ROS, and it has recently been proposed that, independent of the stimulus, a ROS burst may trigger the induction of brain ischemic tolerance (Kato et al., 1995; Wiegand et al., 1999). This suggests an antioxidant pathway as a common mechanism, but levels of antioxidant proteins such as copper–zinc-or manganese-superoxide dismutase have not been found to increase in neurons after preconditioning (Wiegand et al., 1999).
Although the respective contribution of necrosis and apoptosis to the development of brain damage after cerebral ischemia has not yet been conclusively determined (Martin et al., 1998; Roy and Sapolsky, 1999), programmed cell death represents an important mechanism for postischemic cell death (Steller, 1995). Apoptosis has been successfully suppressed in the myocardium after preconditioning (Piot et al., 1997), therefore it can be speculated that a differential expression of cell death-regulating genes and proteins may occur during tolerance induction in the brain.
In addition to other genes, an entire family of mammalian genes, bcl-2 homologues, has been identified to be involved in the regulation of cell death and survival (Adams and Corey, 1998). Some members of this family (for example, bcl-2 and bcl-xl) prolong cell survival in neurons and in other mammalian cells by antioxidant and antiapoptotic activity (Hockenberry et al., 1993; Zhong et al., 1993). In contrast, other members of the family (for example, bax, bad, and bcl-xs) actively promote cell death by disruption of the mitochondrial membrane potential and caspase activation (Zamzami et al., 1998; Jürgensmeier et al., 1998). The balance between pro-and antiapoptotic members of this gene family has been suggested to be of pivotal importance in the regulation of cell death or survival (Oltvai et al., 1993), and increased bcl-2 levels were observed after nonchemical tolerance induction using short ischemia (Shimazaki et al., 1994).
Using the same experimental paradigm as in the current study, the authors have recently shown that partial respiratory chain inhibition in rats improved neuronal survival after global ischemia after a single dose of 3-NPA in neocortex and hippocampus by approximately 20% and 30%, respectively (Brambrink et al., 1998).
In the current study the authors investigated the following: whether 3-NPA treatment was associated with a change in bcl-2 and bax expression; the influence of ischemia–reperfusion on bcl-2 and bax levels in the current model; and whether postischemic bcl-2 and bax expression can be influenced by prior administration of a single dose 3-NPA (known to result in significant neuroprotection). For these experiments the authors used a new, highly sensitive and quantitative polymerase chain reaction (PCR) technique, which allows real-time fluorescence measurements of the PCR product (LightCycler, Wittwer et al., 1997). The authors further subjected serial brain sections to immunohistochemistry to localize the respective proteins in various brain areas and to asses changes in their differential expression.
MATERIALS AND METHODS
Experimental groups
The current study used 104 male Wistar rats (body weight = 307 ± 52 g, mean ± SD; Charles River, Kisslegg, Germany). Animals randomized for ischemia–reperfusion were fasted the night before the insult.
For the mRNA expression study (n = 25), animals were randomly divided into the following groups: rats of one cohort receiving either 3-NPA (20 mg/kg, diluted in 1.8 mL sodium chloride 0.9%, buffered (pH 7.4) with 0.2 mL NaOH (1 mol/L), intraperitoneal (IP), n = 5) or vehicle (sodium chloride, 0.9%, n = 5) were observed for 24 plus 3 hours and then killed. Another cohort was treated with either 3-NPA (20 mg/kg, IP, n = 5) or saline (n = 5), and 24 hours later the animals were subjected to transient global cerebral ischemia and killed after 3 hours of reperfusion. Sham animals for the mRNA-expression study were pretreated with saline 24 hours before the experiment, sham-operated, and killed after 3 hours of observation (mRNA-sham, n = 5).
The animals assigned to the immunohistochemistry study (n = 68) were allocated at random to four groups. One set of rats (n = 28) was injected with 3-NPA (20 mg/kg, IP, n = 20), observed for 3 (n = 5), 12 (n = 5), 24 (n = 5), or 96 hours (n = 5), and then transcardially perfusion-fixed. Eight animals were perfused without prior intervention to determine baseline immunostaining (naive control, n = 8). Animals of a second set (ischemia without pretreatment, n = 16) were subjected to transient global cerebral ischemia, observed for 3 (n = 5), 12 (n = 5), and 24 (n = 6) hours, and perfusion-fixed. A third set of animals (n = 14) was pretreated with 3-NPA (20 mg/kg, IP; chemical preconditioning, n = 7) or vehicle (sodium chloride, 0.9%; n = 7) 24 hours before the experiment. They were then subjected to transient global ischemia and perfusion-fixed after 96 hours of observation. For each respective observation period or pretreatment strategy, or both, two rats were sham-operated (immunohistochemistry-sham, n = 10).
Eleven animals were excluded from data evaluation. Eight of those 11 animals showed inappropriate postischemic recovery (low mean arterial blood pressure, no cerebral reperfusion, no EEG activity [zero line]: 1 animal for mRNA-expression experiments [vehicle group]; 7 animals for immunohistochemistry [untreated, n = 4; vehicle, n = 2; 3-NPA, n = 1]). Three animals (3-NPA, n = 2; vehicle, n = 1) did not survive the 96-hour observation period. The excluded animals were replaced according to the randomization protocol.
All animal related procedures were conducted in accordance with national and international laws and policies (Guide for the Care and Use of Laboratory Animals, National Institutes of Health, 1985) and approved by the regional ethics committee for animal research (Regional Government Rhineland-Palatinate).
Surgical preparations and transient global cerebral ischemia
Surgery. The experimental procedures on animals randomized for ischemia–reperfusion were performed as previously described in detail (Brambrink et al., 1999). In brief, the rats were anesthetized (chloral hydrate: 360 mg/kg body weight, IP), orally intubated, and mechanically ventilated (35 to 40 mm Hg Pa
The head of the animal was fixed in a stereotaxic frame for EEG and cerebral blood flow (CBF) measurements (Brambrink et al., 1999). The calvarium was exposed by a median incision and two depressions designed to hold chlorinated silver pin EEG electrodes were made (high speed dental drill, positioned over the left somatosensory cortex and frontal sinus). Over the right hemisphere approximately 24 mm2 of the skull bone was removed leaving a thin bone lamina (1.3 to 5.3 mm lateral to the right and 1.5 to 7.5 mm occipital from the bregma). A laser–Doppler flow probe (BPM 403A; TSI, St. Paul, MN, U.S.A.) controlled with a computer driven micromanipulator was used to determine regional CBF (rCBF) at 30 locations using the “scanning technique” (Soehle et al., 1998). On the contralateral side (3.3 to 5.3 mm lateral to the left and 5.0 to 7.0 mm occipital of the bregma), a smaller (4 mm2) window was established to measure local CBF (lCBF) using a stationary laser–Doppler flow probe (BPM 2; Vasamedics, St. Paul, MN, U.S.A.).
Temperature probes were placed, as described previously (Brambrink et al., 1999), in the right temporal muscle, in the left auricular tube, and at 6-cm depth in the rectum. Core temperature (thermostatically controlled heating blanket) and temporal muscle temperature (near-infrared heat radiator) were controlled to 37.5 ± 0.1°C throughout the experimental procedures.
The lower body section of the animal was placed in an air-tight chamber. This allowed the establishment of hypobaric pressures by an electronically regulated vacuum pump, thereby reducing arterial blood pressure of the animal (venous pooling) in a controlled fashion (hypobaric hypotension;Soehle et al., 1998; Brambrink et al., 1999).
Global brain ischemia and reperfusion.
After a 30-minute postsurgical stabilization period, transient global cerebral ischemia was initiated by pulling the nylon thread attached to a defined weight around the right carotid artery to occlude the vessel (arterial pressure was measured by a catheter placed in the left carotid artery); mean arterial blood pressure was simultaneously reduced to 35 mm Hg using the hypobaric hypotension technique. Brain ischemia was confirmed by continuous lCBF, rCBF, and EEG measurements. After 15 minutes of global cerebral ischemia, the thread was cut and vacuum was terminated to allow reperfusion.
Temperatures, lCBF, EEG, and mean arterial blood pressure were continuously monitored. Arterial blood gases, pH, base excess, hematocrit, hemoglobin, blood glucose (GLU), and lactate (LAC) levels were determined 7 minutes before (baseline) and at the end of ischemia (ischemia), as well as after 60 minutes of reperfusion (reperfusion).
After 90 minutes of recovery, the common carotid artery catheter was removed, incisions were closed, and the animals were weaned from the ventilator. On extubation (approximately 110 minutes of reperfusion) the rats were returned to their cage and a heating blanket was used to prevent heat loss. The rats were further evaluated for behavioral and neurologic abnormalities once daily the 2 days before, and for 4 days after the ischemic insult. Animal body weight, and 24-hour food and water consumption were monitored, and lactated ringer solution (2 mL/100 g body weight, subcutaneous) was substituted in animals with zero water intake.
Reverse transcriptase polymerase chain reaction
At predetermined times (outlined above), deeply anesthetized animals (chloral hydrate 360 mg/kg, IP) were decapitated. Sparing the cerebellum and the medulla, the brain was removed rapidly (< 60 seconds), frozen in liquid nitrogen, and stored at −80°C. Tissue samples were homogenized for each animal, and total RNA was extracted (Chomczynski and Sacci, 1987).
The total RNA of all animals from the respective experimental groups was then pooled and mRNA was isolated from approximately 200 μg total RNA using oligo-dT-cellulose (FastTrack 2.0 kit; Invitrogen, Groningen, The Netherlands). Isolation of PolyA+-RNA was followed by DNAse I treatment (300 U RNAse-free DNAse I [Roche Diagnostics, Mannheim, Germany], 50 U RNAsin [Promega, Germany]; 37°C, 30 minutes). After digestion, mRNA was purified by phenol extraction and precipitated. Final RNA concentration on photometric analysis (Beckmann DU-640, München, Germany) ranged from 234 to 592 ng/μL.
First-strand cDNA was synthesized from 2 μg pooled mRNA per experimental group using an oligo-dT primer (200 pmol), 20 pmol dNTPs, 2 μCi 33P-dCTP as trace-label, DTT (500 pmol), SuperScript II buffer, RNAsin (10 U), and 400 U SuperScript II (Life Technologies, Karlsruhe, Germany). cDNA synthesis efficiency was estimated by incorporation measurement (Packard Tri-Carb 2100 TR scintillation counter, Dreieich, Germany).
The recently introduced LightCycler sytem (Roche Diagnostics, Mannheim, Germany) allows rapid quantitative PCR analyses using real-time fluorescence detection of amplicon formation by SybrGreen DNA intercalation. For each PCR experiment, fresh two-or threefold dilution series of respective cDNA were prepared. Reverse transcription-polymerase chain reaction experiments were repeated twice for bcl-2 and three times for bax under identical conditions to verify results. cDNA amounts were normalized with three housekeeping genes (cyclophilin, gapdh, and s20) to calculate relative expression ratios for bcl-2 and bax mRNA. Primers specific for rat were designed using the DNAstar PrimerSelect program (Lasergene, Madison, WI, U.S.A.) and synthesized by ARK Scientific (Darmstadt, Germany).
For bcl-2 quantification, the following primers (rat) were used: bcl-2 antisense, 5′-TTT CAT ATT TGT TTG GGG CAG GTC-3′ and bcl-2 sense, 5′-ATG GGG TGA ACT GGG GGA GGA TTG-3′. Bcl-2 PCR was done for five consecutive twofold dilutions of cDNA (1:2, 1:4, 1:8, 1:16, 1:32) per experimental group, including one negative control (no template). The reaction was performed in a final volume of 20 μL in the LightCycler glass capillaries according to the manufacturer's recommendations. MgCl2 concentration was adjusted to 2 mmol/L. Conditions for bcl-2 amplification were as follows: 95°C for 1 second (denaturation), followed by 64°C for 10 seconds (annealing), and 72°C for 30 seconds (extension); the cycle was repeated 40 times (40 cycles) per experiment. At the end of every cycle, fluorescence of each sample was measured at 88°C to allow specific quantification of the PCR product.
Quantitative analysis of the fluorescence data was performed according to the instructions of the manufacturer. The dilution series of one cDNA-pool (no ischemia/vehicle group) were used to produce a standard curve. Specificity of the obtained product was verified by melting point analysis and agarose gel electrophoresis.
All procedures for bax PCR were performed according to the protocol used for bcl-2 with the following exceptions: 2 μL of serial threefold cDNA dilutions (1:3, 1:9, 1:27, 1:81, 1:243, 1:729, 1:2187) were used for each experimental group, MgCl2 was adjusted to 4 mmol/L, and annealing temperature was 65°C (primers [rat], bax-antisense: 5′-GGT GAG CGA GGC GGT GAG GAC T-3′; bax-sense: 5′-GAG AGG ATG GCT GGG GAG AC-3′).
Polymerase chain reaction of housekeeping genes (primers [rat]— cyclophilin-sense: 5′-ACC CCA CCG TGT TCT TCG AC-3′, cyclophilin-antisense: 5′-CAT TTG CCA TGG ACA AGA TG-3′, s20-sense: 5′- GAG GAA CAA GTC GGT CAG G-3′, s20-antisense: 5′-CTC CAC TCC AGG CTC AAT ACT-3′; gapdh-sense: 5′-CCT GGC CAA GGT CAT CCA TGA CAA CTT TGG-3′, gapdh-antisense: 5′-GCC ATG AGG TCC ACC ACC CTG TTG CTG TAG-3′) was performed with serial threefold dilutions as performed for bax PCR. Annealing and measuring were performed at 63°C/83°C, 58°C/85°C, and 61°C/80°C, respectively.
The authors' original data of PCR product quantifications for each group were normalized to the respective mean mRNA expression of the three different housekeeping genes (cyclophilin, s20, and gapdh) determined for the respective cDNA pool. The mean value (± SD) for each experimental group was derived from the normalized PCR product concentration of each dilution step. The resulting quantitative differences represent relative changes in mRNA expression among the experimental groups.
Immunohistochemistry
The animals were reanesthetized (chloral hydrate) and transcardially perfused with ice cold (4°C), freshly prepared 4% phosphate buffered paraformaldehyde (pH 7.4). The brains were removed, postfixed for 48 hours at 4°C, and subsequently embedded in paraffin.
Immunostaining was used simultaneously on a large number of brain sections. According to a randomized protocol each series included animals from all different groups and negative controls. All necessary procedures were performed during a short period of time and by the same individuals according to a rigid protocol. Tissue sections (3 μm) were deparaffinized for bcl-2 immunohistochemistry using graded xylene and alcohols, treated with 0.3% H2O2 in methanol (100%) for 25 minutes, washed in methanol (50%), exposed to sodium borohydride 0.05% in methanol (50%) for 25 minutes, washed in phosphate buffered saline (0.1 mol/L, 0.9%, pH 7.4; PBS), and microwaved (600 W, to enhance antigen exposure) twice for 5 minutes in citrate buffer (0.01 mol/L, pH 6.0) to replace citrate buffer loss. After cooling and subsequent washing steps (distilled water, PBS, PBS plus bovine serum albumin [PBS+BSA] 0.5%, pH 7.5), sections were incubated in 3% normal goat serum in PBS+BSA at 37°C for 30 minutes. The tissue was exposed overnight to rabbit polyclonal anti-bcl-2 antibody at a dilution of 1:3,000 in PBS+BSA at 4°C (bcl-2 [N-19], 200 μg/mL; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A., cat no. sc-492). The antibody recognizes the amino terminus of bcl-2 protein in rats (previously used on rat brain by Chen and Chuang, 1999; Zhu et al., 1999).
On the following day, the sections were first washed twice in PBS+BSA and then incubated with biotinylated goat anti-rabbit immunoglobulins at a dilution of 1:200 (cat. no. PK 6101; Vector Laboratories, Burlingame, CA, U.S.A.) in PBS+BSA for 60 minutes at 37°C, followed by two washing steps (PBS+BSA, PBS), and then exposed to avidin-biotin-peroxidase (30 minutes at 37°C). After washing (PBS, distilled water), the tissue sections were visualized with 3,3′-diaminobenzidine (DAB-Chromogen-Kit, code no. K 3467; DAKO, Carpinteria, U.S.A.), embedded in glycerol gelatin, and coverslipped. As a negative control, the primary antibody was omitted.
The protocol outlined above was also used for bax immunohistochemistry, with the exception of omitting sodium borohydride and using a different dilution (1:800) of the primary anti-bax antibody (rabbit polyclonal IgG to bax protein [P-19], 200 μg/mL; Santa Cruz; cat. no. sc-526; previously used on rat brain by Chen and Chuang 1999; Zhu et al., 1999).
Differences between individual staining series were very small, and the intensity of immunoreactivity to bcl-2 and bax protein was evaluated using the semiquantitative grading procedure published by Krajewski et al. (1995). The authors used light microscopy (250x) attached to a color video camera (SSC-C370P; Sony Corporation, Japan), and the PC-based Optimas imaging system (Stemmer, Martinsreid, Germany) served to store and evaluate appropriate images of sections from different regions of the sensomotoric neocortex, hippocampus (CA1, CA2, CA3, CA4), dentate gyrus, and thalamus. The three investigators were blinded to the randomization protocol of the neutrally coded tissue sections. Immunoreactivity of viable neurons was graded independently according to a score ranging from zero (0) for no staining to four (4) for strong immunoreactivity (zero [0], faint [1], mild [2], moderate [3], and strong [4]). The median for the different gradings was established as the immunoreactivity score for the respective brain region of each animal. If grading differed by more than one point among the investigators (fewer than 15% of all evaluations), sections were reevaluated and a common decision was taken. In addition, all brain sections were evaluated for morphologic evidence of neuronal cell damage (shrunken cytoplasm, cytoplasmatic vacuolization, condensed nucleus [pyknosis], and loss of nuclear detail (Martin et al., 1998).
Statistical analysis
Data are presented as mean values (± SD) for the respective group. Physiologic variables, relative mRNA expression, and immunoreactivity were assessed by one-way analysis of variance or analysis of variance on ranks where required, and the appropriate parametric post hoc test for multiple comparisons (physiologic variables, Tukey Test; mRNA expression, Student Newman–Keuls method; protein expression, Dunn's method), using SigmaStat-2.0 routines (Jandel, Erkrath, Germany). Differences were considered statistically significant at P < 0.05.
RESULTS
Physiologic variables
All physiologic variables of animals randomized for global cerebral ischemia and reperfusion or sham operation were within normal ranges at baseline (Table 1). Mean arterial blood pressure was reduced during transient cerebral ischemia (35 mm Hg), and cerebral blood flow remained at biological zero (rCBF = 2 ± 2 LD-units; lCBF measurements confirmed results, data not shown) for 15 minutes, which was significantly different from the values recorded in sham-operated animals. At the end of the insult period, arterial blood analysis of ischemic animals showed mild metabolic acidosis, reduced serum glucose, and elevated serum lactate levels, as well as a small reduction in temperature at all sites measured compared with shams. All animals included in the analysis showed robust postischemic cerebral reperfusion and simultaneous EEG-recovery. Within the first hour of reperfusion, all parameters returned to baseline values, except for a moderately reduced rCBF (hypoperfusion) and a reduced base excess. There were no statistical differences in the physiologic variables monitored between the different intervention groups (vehicle and 3-NPA) at any experimental time point (Table 1).
Physiologic variables of ischemia–reperfusion subgroups
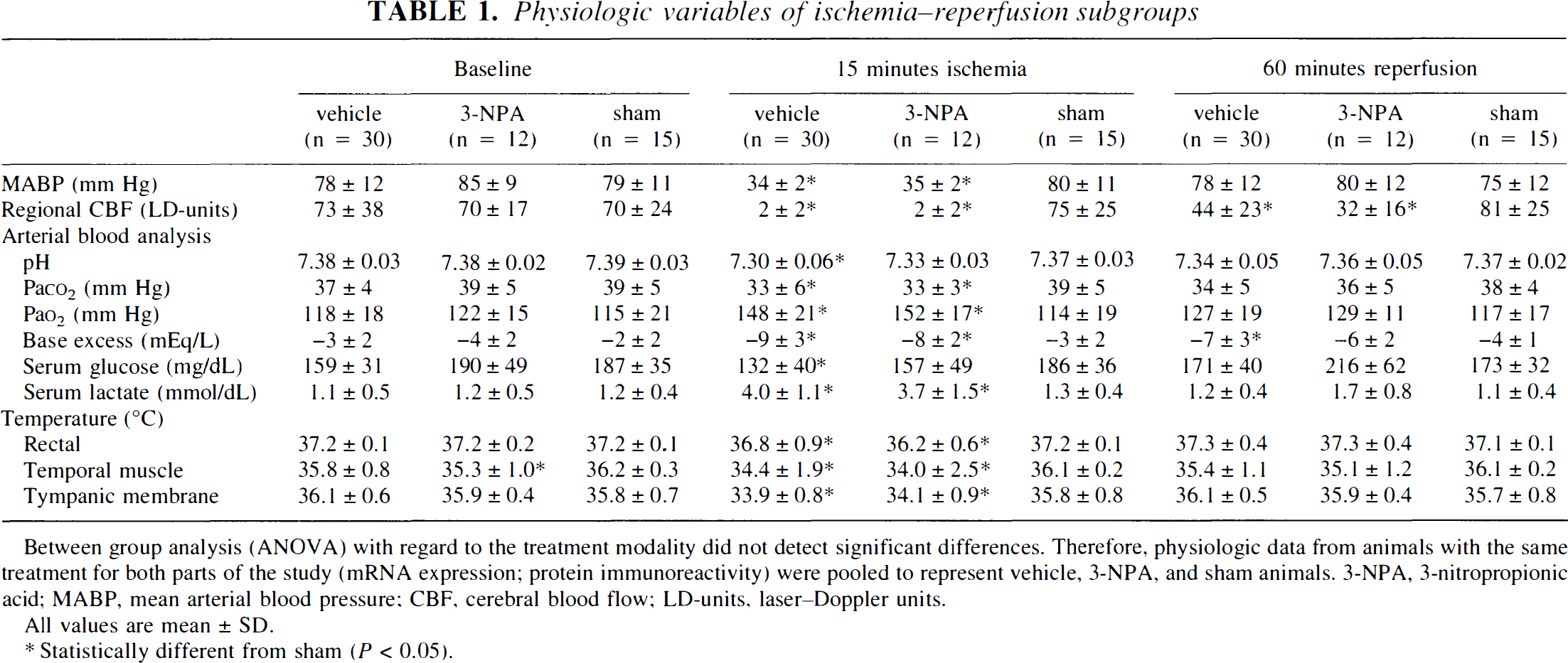
Between group analysis (ANOVA) with regard to the treatment modality did not detect significant differences. Therefore, physiologic data from animals with the same treatment for both parts of the study (mRNA expression; protein immunoreactivity) were pooled to represent vehicle, 3-NPA, and sham animals. 3-NPA, 3-nitropropionic acid; MABP, mean arterial blood pressure; CBF, cerebral blood flow; LD-units, laser–Doppler units.
All values are mean ± SD.
Statistically different from sham (P < 0.05).
Animals receiving a single dose of 3-NPA intraperitoneally developed no behavioral or neurologic abnormalities at any time during the observation period, nor was there any evidence of necrotic or apoptotic cell damage in the histomorphologic analysis.
Expression of bcl-2 and bax mRNA
Bcl-2 mRNA.
Lightcycler RT-PCR revealed a 1.5-fold increased relative expression of brain bcl-2 mRNA in animals pretreated with 3-NPA compared with those receiving normal saline (vehicle). Relative bcl-2 mRNA expression with 3-NPA pretreatment was also significantly higher in animals 3 hours after global brain ischemia compared with that of postischemic animals with saline-pretreatment, or after sham operation (Fig. 1A).
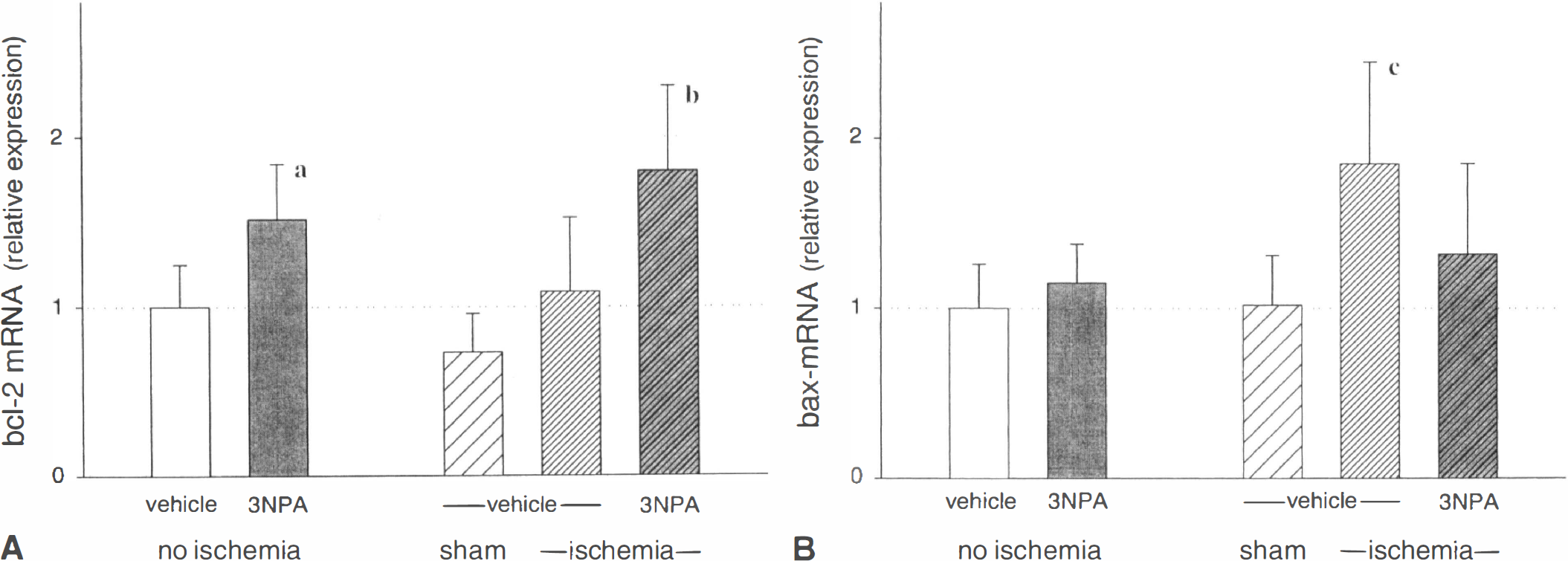
Quantitative real-time polymerase chain reaction (PCR) analysis of relative expression of bcl-2
Bax mRNA.
Relative bax mRNA expression was not changed by 3-NPA pretreatment (no ischemia animals), although a change was observed after the ischemic brain insult. Three hours after global ischemia, bax mRNA expression was almost twofold increased in animals receiving saline 24 hours before the insult compared with sham control. In contrast, when animals were pretreated with 3-NPA, postischemic bax mRNA expression was significantly reduced (Fig. 1B).
Expression of Bcl-2 and Bax protein
At baseline.
Fig. 2 shows photomicrographs of representative animals. Neuronal immunoreactivity for Bcl-2 protein was moderate in hippocampal CA3 (Fig. 2C), faint in hippocampal CA1 (Fig. 2A), CA2, and CA4, and in the neocortex (Fig. 2G), and zero in the dentate gyrus of naive control animals (Fig. 2E).
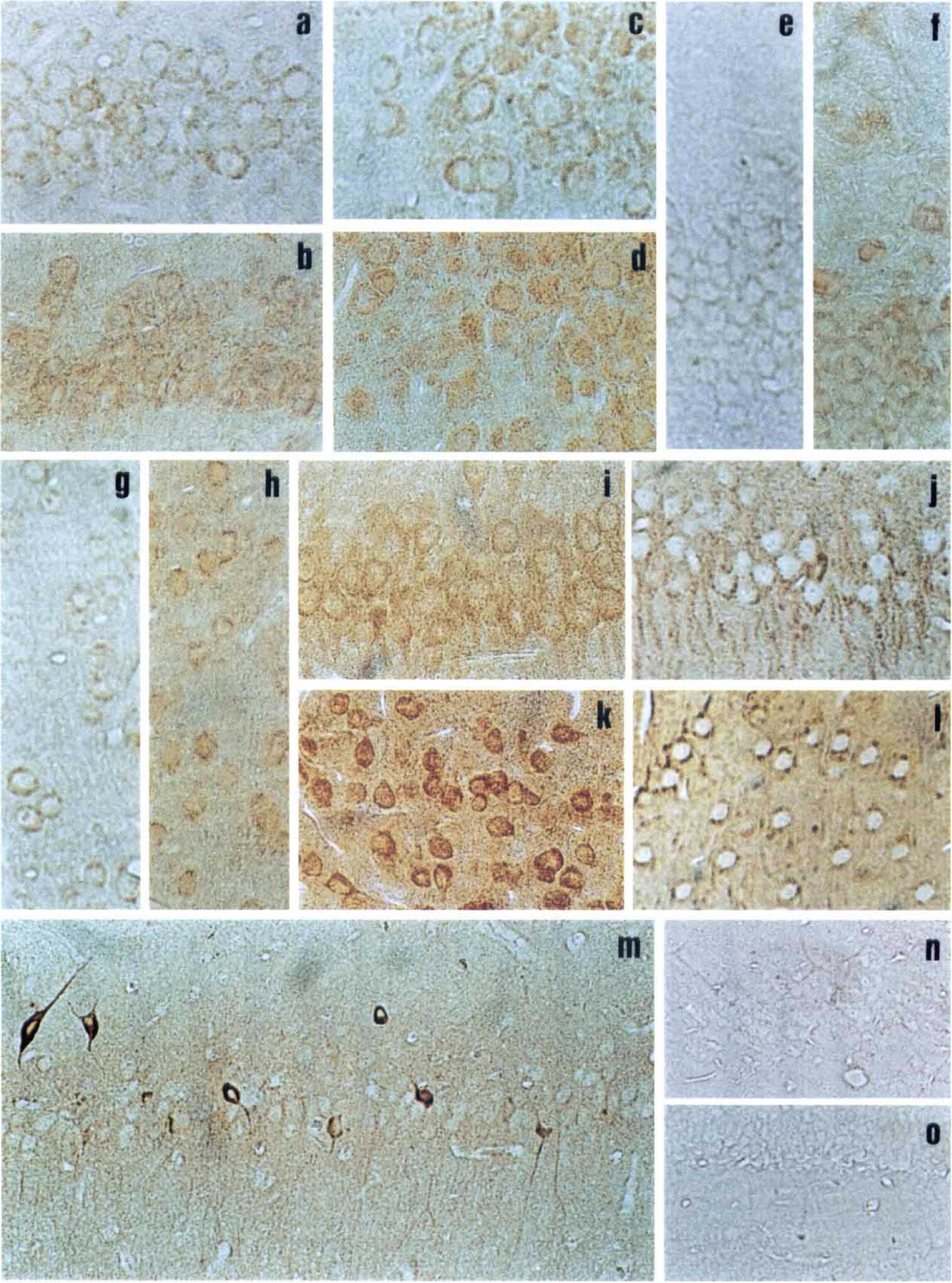
Immunohistochemical analysis of Bcl-2 and Bax proteins in selected brain areas of normal (naive controls), 3-nitropropionic acid (3-NPA) treated, and postischemic rats. Tissue sections (3 μm) were immunostained as described in Materials and Methods using antibodies specific for Bcl-2 and Bax proteins (rabbit), which were detected by diaminobenzidine (DAB) protocol. The primary antibody was omitted in negative controls.
Immunoreactivity for Bax protein at baseline was mild to moderate in all areas evaluated (Figs. 2I and 2K).
After 3-NPA administration.
Neuronal immunoreactivity for Bcl-2 protein increased over the first hours after 3-NPA-injection, reaching statistical significance at 24 hours after 3-NPA administration in CA1, CA2, and CA4 of the hippocampus, and in the neocortex compared with naive control animals (Figs. 3A, 2B, 2D, 2F, and 2H). Notably, baseline immunoreactivity for Bcl-2 in CA3 of the hippocampus was already in the moderate range (Fig. 2C). Animals with 3-NPA-pretreatment, observed for 96 hours, showed no further increase in Bcl-2 immunoreactivity and exhibited near baseline levels in some areas of the hippocampus—for example, CA1, CA2, and CA3. However, Bcl-2 immunoreactivity remained at higher levels in the CA4, dentate gyrus, and neocortex (Fig. 3A).
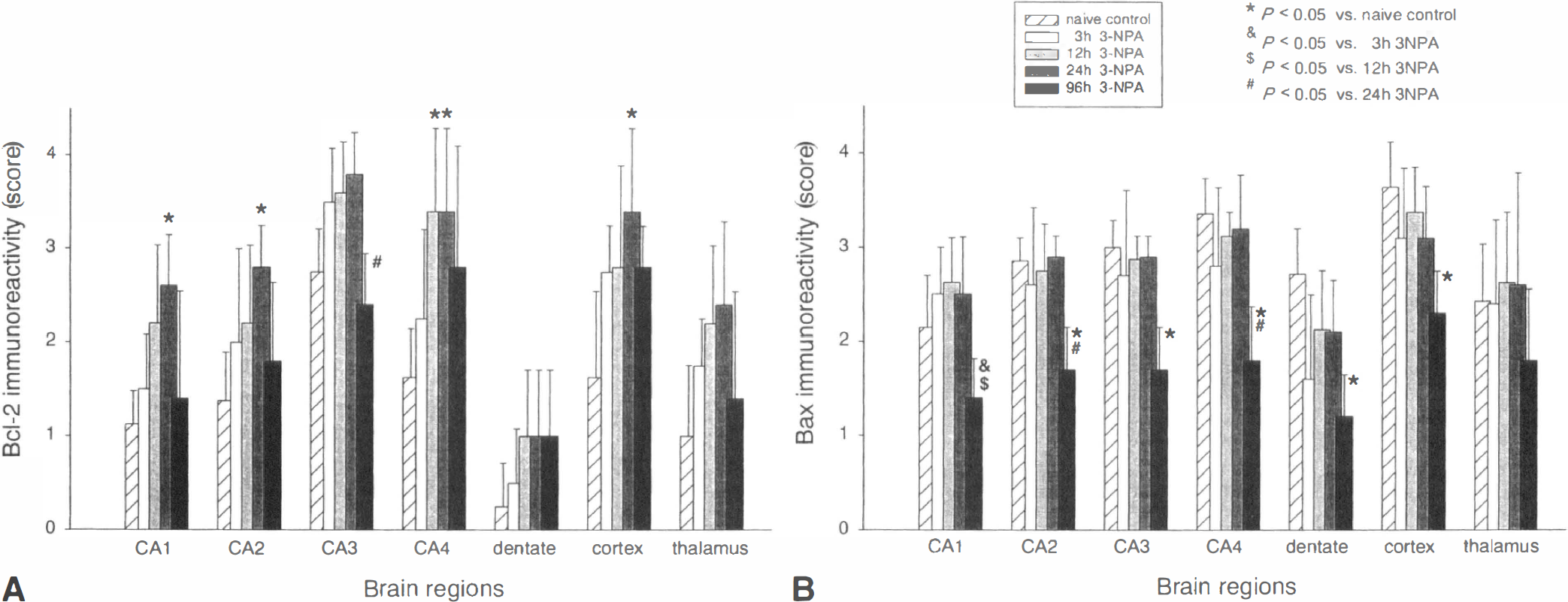
Changes of Bcl-2
In contrast, immunoreactivity for Bax protein decreased significantly from baseline (P < 0.05) at 96 hours after 3-NPA-administration in CA2, CA3, and CA4 of the hippocampus, and in the dentate gyrus and cortex (Figs. 3B, 2J, and 2L). No differences were observed at earlier time points.
After global cerebral ischemia.
Bcl-2 immunoreactivity in viable cells was not different from baseline values during early postischemic recovery (3, 12, and 24 hours). However, Bcl-2 protein expression in surviving neurons of all brain areas evaluated was significantly more pronounced after 4 days compared with naive controls (96 hours, P < 0.05; Fig. 4A).
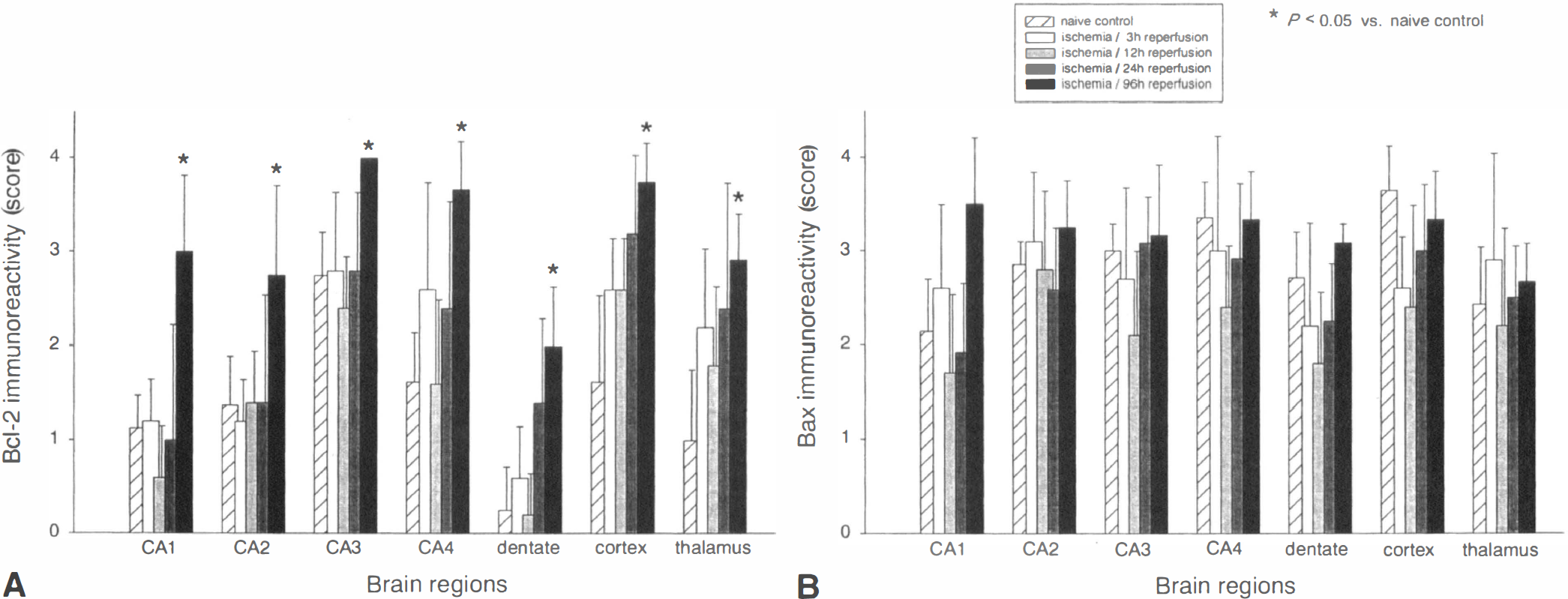
Changes in expression of Bcl-2
Cells with strong immunoreactivity for Bax protein were observed as early as 3 and 12 hours after cerebral ischemia in a small number of animals. These cells showed the typical morphology of ischemic neuronal cell death (Fig. 2M), and represented approximately 10% to 30% of hippocampal (preferentially in CA1, CA2, and CA4) and neocortical neurons at these time points. At 24 hours, only 1 out of 6 animals presented with strong Bax positive cells disseminated in the thalamus, although 96 hours after global ischemia all but 1 animal in each group had markedly Bax positive neurons in the CA1, CA2, and cortex, or thalamus, or both.
In contrast, immunoreactivity for Bax protein in viable neurons was not statistically different from baseline at all time points evaluated. However, during early recovery from ischemia (especially at 12 hours) there was a trend towards lower Bax protein expression in viable neurons of all brain regions, except CA2 and the thalamus (Fig. 4B).
After global cerebral ischemia with and without 3-NPA pretreatment.
At 96 hours of recovery from global cerebral ischemia, Bcl-2 or Bax immunoreactivity of viable neurons was not different in any brain region for animals pretreated with 3-NPA or vehicle before the ischemic insult (data not shown).
Negative controls.
Incubation of rat brain sections in the absence of primary bcl-2 or bax antibodies, but with secondary anti-rabbit immunoglobulins, avidine-biotin-peroxidase, and 3,3′-diaminobenzidine produced no immunolabeling in any staining procedure (Figs. 2N and 2O). These negative controls demonstrate that the respective cell body labeling for Bcl-2 and Bax was not a nonspecific observation resulting, for example, from tissue injury.
DISCUSSION
The results presented here suggest changes in the balance between pro-and antiapoptotic proteins as one potential explanation for the observed protection provided by chemical preconditioning using 3-NPA in rats. Furthermore, the authors describe the application of a novel and highly sensitive real-time PCR technique for the quantification of differential gene expression in cerebral ischemia and tolerance.
Although ischemia protection was observed after various preconditioning techniques, the cellular signal transduction cascades resulting in ischemic tolerance, and the mechanisms involved in neuronal survival in the tolerant state remain unclear. The current study investigated the expression of bcl-2 and bax, two antagonistic members of the bcl-2 gene family, in response to a single dose 3-NPA, to global cerebral ischemia, and to the combination of 3-NPA-pretreatment and subsequent global cerebral ischemia. The authors observed a differential expression of proapoptotic bax and antiapoptotic bcl-2 after 3-NPA-treatment in brain areas vulnerable to ischemic damage on the transcriptional (Fig. 1A) and translational level (Fig. 2A).
Effects of single dose 3-NPA on neuronal bcl-2:bax ratio in nonischemic animals
Plant and fungus-produced 3-NPA acts as an irreversible inhibitor of the respiratory chain complex II (Coles et al., 1979), thus representing a potent mitochondrial toxin. Repeated doses result in selective striatal damage in animals (Beal et al., 1993; Kim et al., 2000) and humans (Ludolph et al., 1991). In contrast, a single injection of 3-NPA (20 mg/kg, IP), which results in approximately 20% to 30% inhibition of succinate dehydrogenase (authors' unpublished observation, Wiegand et al., 1999), induces tolerance to cerebral ischemia (chemical preconditioning;Riepe et al., 1997), and results insignificant neuroprotection after transient global cerebral ischemia in rats (Brambrink et al., 1998) and gerbils (Sugino et al., 1999), and after permanent and transient focal ischemia in rats (Wiegand et al., 1999).
After a single dose of 3-NPA in rats, the authors observed an elevated bcl-2:bax ratio at the transcriptional (RT-PCR, whole brain) and translational level (immunohistochemistry, brain slices). The current data show that the increase in Bcl-2 protein expression occurred in neurons known to be especially vulnerable for ischemic cell death, for example, in hippocampal CA1 and neocortex. The same brain regions showed significant neuroprotection when 3-NPA was given 24 hours before the ischemic cerebral insult in a previous study using the same paradigm (Brambrink et al., 1998). The findings agree with earlier reports on differential gene expression after classical ischemic preconditioning (Shimazaki et al., 1994; Abe and Nowak, 1996), showing increases particularly in gene products involved in neuronal survival. Additional evidence emerged in recent observations suggesting other possible neuroprotective strategies to modulate the neuronal bcl-2 and bax balance (Zhu et al., 1999), and also suggesting that bcl-2 overexpressing mice showed an increased resistance to cerebral ischemia (Martinou et al., 1994).
In some brain regions, however, Bcl-2 protein expression was not significantly different from baseline values after a single dose 3-NPA treatment, that is, in the hippocampal CA3 and dentate gyrus. The author's finding that bcl-2 expression in CA3 was already higher at baseline (naive control animals) than in other brain regions is in agreement with earlier reports (Krajewski et al., 1995) and may explain the relatively strong resistance of this brain region to ischemia. The observed low baseline levels of Bcl-2 protein in the dentate gyrus, despite the high resistance against ischemia in this region, is also confirmed by earlier reports (Krajewski et al., 1995) and may serve as evidence that the relative proportions of bcl-2:bax levels do not account exclusively for the differential ischemia sensitivity of neuronal populations. There are other potential molecular mechanisms responsible for postischemic apoptotic cell death or survival, and the bcl-2 family consists of several members (for example, bcl-xl) that could play a predominant role in dentate gyrus recovery from ischemia. Moreover, postischemic recovery in the dentate gyrus may be organized completely different from that in other brain regions. This is supported by the observation that granule neurons are reproduced continuously throughout adulthood (Gould et al., 1998).
The authors observed a significant time delay in the occurrence of changes in Bcl-2 and Bax protein expression (24 and 96 hours, respectively). This corresponds to earlier observations showing that sustained ischemic tolerance in the brain requires at least 24 hours to develop after 3-NPA (Brambrink et al., 1998) and classical ischemic preconditioning (Kitagawa et al., 1990). However, other investigators found a 3-day interval necessary to achieve optimal neuroprotection after the same stimuli (Wiegand et al., 1999; Sugino et al., 1999; and Heurteaux et al., 1995; respectively). The authors observed an early increase in Bcl-2 protein expression after 3-NPA injection (24 hours, Fig. 3), compared with a late decrease in Bax protein expression after the same treatment (96 hours, Fig. 3). Increases in the bcl-2:bax ratio have been proposed to prevent apoptotic cell death in the brain (Oltvai et al., 1993). The current data suggest that 3-NPA promotes an early increase in this ratio resulting from elevated Bcl-2 protein expression (approximately 24 hours), whereas reductions in Bax protein levels are responsible for an elevated bcl-2:bax ratio at later time points (approximately 96 hours). This may, in part, explain the range of protection (1 to 3 days) reported for 3-NPA-induced ischemic tolerance. However, reports on a delayed and short window of ischemic tolerance after 3-NPA (72 hours, Wiegand et al., 1999) need further investigation to be confirmed.
Effects of single dose 3-NPA in animals recovering from global cerebral ischemia
Three hours after global cerebral ischemia, bcl-2 mRNA was higher and bax mRNA was lower in brains pretreated with 3-NPA, compared with saline treated animals. Similar to findings reported for other neuroprotective strategies (Zhu et al., 1999; Martinou et al., 1994), these data show that differential gene expression in favor of bcl-2 after 3-NPA is maintained in the rodent brain during postischemic recovery. However, it should be taken into consideration that the authors used whole brain preparations for the analysis of mRNA expression; thus, the observed differences may underestimate the degree of regulation reached in individual neurons of various brain regions, as suggested by the data from the immunohistochemical analysis.
The authors' observation of high Bax protein expression in single neurons with evidence of ischemic damage during early postischemic recovery confirms results of other reports in which high levels of Bax protein were associated with imminent cell death (Krajeweski et al., 1995; Chen et al., 1996). The late (96 hours) increase in Bcl-2 protein in viable neurons without simultaneous elevations of Bax protein seem to be associated with the successful postischemic recovery, indicating that changes in the bcl-2:bax balance (Krajeweski et al., 1995) may also play a pivotal role for the postischemic fate of neurons in the authors' experimental model.
Proposed model for and clinical implications of chemical preconditioning using 3-NPA
The results of the current study suggest a signal transduction pathway leading to an increased ischemic tolerance in neurons, which includes temporarily elevated ROS levels and de-novo protein synthesis. The ROS burst, which peaks 3 hours after 3-NPA (Wiegand et al., 1999), is generated upstream of the succinate dehydrogenase and may be a direct result of partial respiratory chain inhibition (Garcia-Ruiz et al., 1995), or alternatively, from an indirect excitotoxic pathway (Kim et al., 2000). Increased levels of ROS may initiate transcription of Bcl-2, which may also operate as a free radical scavenger (Hockenberry et al., 1993). During subsequent periods of cerebral ischemia–reperfusion the Bcl-2 protein surplus may neutralize postischemic free radicals, protecting neurons against their deleterious effects (premitochondrial activity of Bcl-2;Ellerby et al., 1997). In addition, Bcl-2 appears to block the opening of mitochondrial transition pores, either directly (Shimizu et al., 1999) or through heterodimerization with Bax, or both (Zamzami et al., 1998), thereby preventing cytochrome c release and subsequent activation of caspase 9 (Jürgensmeier et al., 1998). Bcl-2 may also suppress calcium store depletion in the endoplasmic reticulum through ion-conducting channel formation, thereby preventing periischemic disturbances of endoplasmic reticulum function (Paschen and Dutheil, 1999). Antioxidant and antiapoptotic activities of Bcl-2 result in increased resistance against subsequent ischemia and delayed neuronal damage. However, there are some death signals that are Bcl-2 independent, and Bcl-2 does not influence postmitochondrial steps of apoptosis (Ellerby et al., 1997). The reduced abundance of neuronal Bax protein 4 days after 3-NPA may be partially responsible for the prolonged state of ischemic tolerance reported in the literature.
The suggested pathway unquestionably represents only parts of one possible signal transduction cascade. Other members of the bcl-2 family with antiapoptotic activity (for example, Bcl-xl) may also be induced by 3-NPA and add to the resulting ischemic tolerance. In addition, chemical preconditioning may depend on mechanisms independent of those described above, and furthermore, some of these appear to be interconnected. Potential pathways include blockade of Ca2+-entry into mitochondria (Pérez-Pinzón et al., 1997), or differential expression of, for example, the neuronal apoptosis inhibitory protein, neurotrophic or vascular growth factors, or cytokine and receptor proteins that modulate the postischemic immune response (Barone et al., 1998). The authors' preliminary data suggest a potential role for certain immediate early genes and other cellular antioxidants. Expression studies on a genome-wide basis are needed to recognize the whole spectrum of changes in gene regulation after chemical preconditioning.
Notably, the induction of ischemic tolerance by means of a cell toxin (3-NPA) does not present a clinically applicable strategy; however, it may serve as a tool to analyze the efficacy of chemical preconditioning and to understand the underlying mechanisms. Identifying the central intracellular signal cascades during tolerance induction will enable one to find or design other, safer substances for chemical preconditioning in patients. Patients undergoing surgical procedures with a risk of cerebral blood flow impairment (for example, during cardiovascular-and neurosurgery) may benefit from preconditioning. Induction of ischemic tolerance, by means of a specific and safe drug given before surgery, may improve postoperative outcome and the long-term quality of life in these patients.
In conclusion, this is the first report showing that chemical preconditioning using 3-NPA in rats is associated with a differential expression of the bcl-2 and bax genes during ischemic tolerance with and without ischemia–reperfusion. Application of both a highly sensitive real-time RT-PCR method for the quantification of transcriptional changes and classical immunohistochemistry enabled the localization of the respective protein expression in brain areas vulnerable to ischemia, that is, the hippocampus and neocortex. In preconditioned animals, bcl-2 mRNA and protein were elevated, and this was maintained during postischemic recovery. Bax mRNA expression after cerebral ischemia was reduced in the same animals, whereas nonischemic rats had reduced neuronal Bax protein levels four days after 3-NPA treatment.
The current results suggest that changes in the bcl-2:bax ratio are involved in 3-NPA-induced ischemic tolerance in rats. The observed postischemic changes in bcl-2 and bax expression confirm the importance of these genes for neuronal survival in the applied model of cerebral ischemia. Differential bcl-2 and bax transcription and translation show that both are inducible genes, and bcl-2 targeting strategies could be of potential benefit for patients for which brain ischemia could be anticipated.
Footnotes
Acknowledgements
The authors thank Michael Malzahn and Andrea Schollmayer for their excellent assistance.