
Abstract
Transient and permanent focal cerebral ischemia results in a series of typical pathophysiologic events. These consequences evolve in time and space and are not limited to the lesion itself, but they can be observed in perilesional (penumbra) and widespread ipsi- and sometimes contralateral remote areas (diaschisis). The extent of these areas is variable depending on factors such as the type of ischemia, the model, and the functional modality investigated. This review describes some typical alterations attributable to focal cerebral ischemia using the following classification scheme to separate different lesioned and perilesional areas: (1) The lesion core is the brain area with irreversible ischemic damage. (2) The penumbra is a brain region that suffers from ischemia, but in which the ischemic damage is potentially, or at least partially, reversible. (3) Remote brain areas are brain areas that are not directly affected by ischemia. With respect to the etiology, several broad categories of remote changes may be differentiated: (3a) remote changes caused by brain edema; (3b) remote changes caused by waves of spreading depression; (3c) remote changes in projection areas; and (3d) remote changes because of reactive plasticity and systemic effects. The various perilesional areas are not necessarily homogeneous; but a broad differentiation of separate topographic perilesional areas according to their functional state and sequelae allows segregation into several signaling cascades, and may help to understand the functional consequences and adaptive processes after focal brain ischemia.
Keywords
The brain more than any other organ depends on the supply of oxygen and glucose for oxidative phosphorylation. These substrates are used primarily to maintain the ionic gradients of glial cells and neurons and to feed glutamatergic neurotransmission (Sokoloff, 1999; Magistretti and Pellerin, 1999a; Magistretti et al., 1999b). If the blood supply to the brain is interrupted by an embolism of intracerebral vessels, local thrombosis, or other means, a cerebral infarction develops. With complete obstruction of a vessel, clinical symptoms usually occur rather fast, thus giving the appearance typical of a stroke.
The stroke-induced cellular cascades develop fast but gradually (Small et al., 1999; Lipton, 1999; Hamann et al., 1999; Siesjo et al., 1999), starting with loss of oxygen and changes in metabolic activity, followed by a hyperexcitatory phase, increasing intracellular calcium, and then activation of calcium dependent enzymes such as constitutive nitric oxide synthases (NOS), caspases, and various proteases. Concomitant conversion of vascular xanthine dehydrogenase into xanthine oxidase (McCord, 1998) and activation of cyclooxygenases (COXs) produce superoxide radicals (O'Banion, 1999). Nitric oxide (NO) and superoxide form cytotoxic peroxynitrite that reacts further to produce even more toxic radicals (Beckman et al., 1996; Bredt, 1999). Finally these reactions cause inhibition of protein function by tyrosine nitration, impairment of membrane function because of lipid peroxidation, disturbance of mitochondrial energy metabolism, and calcium sequestration as well as DNA damage (Farooqui et al., 1998; Dawson et al., 1998; Love, 1999).
Clinically, the symptoms and signs of a stroke are indicative of the site of the lesion. The ability to attribute certain functions to certain brain areas, and certain deficits to certain lesions is the basis of clinical neurology. Studies relating specific deficits to lesion topology have been the basis for much of the facts known about brain function. However, there are many instances in which focal brain lesions also seem to have an impact on the function of surrounding or remote brain areas. Beck et al. (1995) cite Gall as the first who, as early as 1835, wrote “a part being wounded or irradiated, wounds or irradiates all the rest” (Feeney, 1998).
The brain has to be considered as a network with multiple and intricate connections. Therefore, it is very important to analyze in detail which symptoms are a direct consequence of the lesion, the perilesional area, or the reaction of the surrounding brain to the lesion. Remote functional changes have been assumed to play a role in postlesional recovery as far back as the late nineteenth century. At that time von Monakow coined the term diaschisis, purely from a clinical standpoint (von Monakow, 1914). von Monakow described temporary clinical deficits that were ascribed to remote effects from the lesion onto structurally intact but neuroanatomically connected regions and suggested that restitution would reflect resolving diaschisis. This concept has been expanded not only with respect to topography but also in terms of different neurophysiologic phenomena that had been monitored (Hovda et al., 1987; Dobkin et al., 1989; Ginsberg et al., 1989; De et al., 1995,1997; Rizzo et al., 1996; Juhasz et al., 1997; Kim et al., 1997; Mielke et al., 1997; Nguyen and Botez, 1998; Yamauchi et al., 1999; Mochizuki et al., 2000; Rubin et al., 2000). Baron was the first to use the term “crossed cerebellar diaschisis” for cerebellar hypometabolism (Baron et al., 1980, 1992; Dettmers et al., 1995), and “transhemispheric diaschisis” was used by Andrews (1991) for cortical changes of excitability and metabolism observed contralateral to ischemic strokes. Buchkremer-Ratzmann et al. (1996) described long lasting alterations of cortical inhibition in the photothrombosis model of focal ischemia that were called “transcortical electrophysiological diaschisis.” Taken together the term diaschisis today is used for any remote effect initiated by a focal lesion or ischemic event to the brain (Nguyen and Botez, 1998; Seitz et al., 1999; Abe et al., 2000; Rubin et al., 2000). As this definition is rather vague, diaschisis cannot be regarded as a unique, well-demarcated area, but it must be described as a highly variable phenomenon that necessitates a more detailed description. In the current review the authors will differentiate separate topographic perilesional areas according to their functional state and sequelae.
SIGNALING CASCADES AND PERILESIONAL AREAS
Several strategies may be employed to understand the different processes activated in the succession of focal brain lesions. Unraveling the molecular cascades initiated by the ischemia can, for example, be achieved by pharmacologic intervention studies and experiments with transgenic animals. Molecular alterations after brain ischemia have been comprehensively reviewed by Sharp et al. (2000). Another approach is to focus on the differentiation of processes in various perilesional areas as the authors will describe them in this review.
Several strategies may be employed to separate perilesional areas. In some instances, the use of different lesion models (for a comparison of different experimental models of brain ischemia see Hossmann, 1998) is helpful. Experimentally, typical focal brain ischemia, mimicking pathophysiology in humans, can be obtained using the filament model or other models with occlusion of the middle cerebral artery in rodents—in this model the stroke is surrounded by a typical penumbra. Disadvantages of the model include a considerable variability of lesion size, and often indirect effects in remote brain areas because of massive brain edema. For certain inquiries it is advantageous to use the photothrombosis model (Watson et al., 1985; Domann et al., 1993). The dye Rose Bengal is injected intravenously as simultaneously a small area of the cortex is illuminated through the intact skull. Consequently, ischemic lesions, with an average diameter of approximately 2 mm develop, extending through the complete depth of the cortex leaving the underlying corpus callosum intact. The resulting penumbra is very small and extends only approximately 500 μm from the lesion (Schroeter et al., 1994; Jander et al., 1995; Witte et al., 1997; Bidmon et al., 1998a). This model shows good reproducibility of lesion size and location and a clear boundary between brain tissue undergoing ischemic damage and surrounding, nonischemic tissue. Secondary alterations caused by brain edema are not present in the contralateral hemisphere with small photothrombotic cortical stroke because these are small and do not produce pressure effects on the contralateral brain. Pressure induced edema effects can, however, be seen in the ipsilateral hippocampus (Bidmon et al., 1998a). In the photothrombosis model, waves of spreading depression (SD) occur only on the side of the lesion, resulting in a very characteristic distribution pattern in the rodent brain, affecting almost all of the ipsilateral, but not the contralateral, hemisphere (Schroeter et al., 1995). The same is probably true for small or reversible middle cerebral artery occlusion (MCAO), whereas it is conceivable that with permanent MCAO giving rise to pronounced pressure-induced alterations contralateral to the lesion SD may also occur in the contralateral hemisphere. To our knowledge, this has not been systematically analyzed yet.
Another example of how different lesion models can be used to separate the causal sequences of alterations caused by focal brain lesions is the observation made by (Napieralski and coworkers 1996, 1998). They reported that ischemic brain lesions (with a penumbra) have a different effect on remote brain plasticity than suction lesions (without a penumbra; see below).
Some remote changes after focal brain ischemia can also be separated by use of behavioral approaches. Changes caused by use-dependent reactive plasticity may be identified by causing either increased activity (for example, by enriched environment), or by preventing movement of the respective limb (for example, by casting) (Kozlowski et al., 1996; Humm et al., 1998).
Pharmacologic approaches also help to separate different perilesional areas. Effects caused by SD can be isolated by application of MK801, a competitive N-methyl-
In the following sections the authors will describe some typical alterations after focal brain ischemia using the topographic relation and the probable signaling cascades in different lesioned and perilesional areas as a classification frame.
LESION CORE—IRREVERSIBLE ISCHEMIC DAMAGE
The lesion core can be defined as the brain area that suffers irreversible structural damage after brain ischemia. In studies on animals and humans it has been shown that this occurs if blood flow drops below approximately 10 mL/100 g·min or 20% of control (Hossmann, 1994,1998). In this brain area anoxic depolarization develops. This is associated with a breakdown of the transmembraneous ionic gradients. There is a massive increase of extracellular potassium concentration and a reduction of extracellular sodium and calcium concentrations; the cells swell, the size of the extracellular space decreases, protein synthesis is inhibited, mRNA transcription is partly suppressed, and ATP pools are depleted. Glutamate transport is inverted and glutamate is released from glial cells into the extracellular space (Lin et al., 1998). The intracellular calcium loading is thought to play a major role in the subsequent processes of activation of proteolytic enzymes, degradation of cytoskeletal proteins, mitochondrial swelling, lipid peroxidation, and membrane damage.
With respect to the ensuing damage, there is a close link between the degree and the duration of hypoperfusion. First, selective neuronal cell death occurs after ischemia of only 2 to 5 minutes. After approximately 5 to 10 minutes one can observe the so-called no reflow phenomenon in some brain areas (Carden et al., 2000). The thresholds for irreversible tissue damage were systematically investigated by Jones and colleagues in 1981 (Jones et al., 1981) who produced MCAO in awake primates. Cerebral infarction occurred when cerebral blood flow dropped below 17 to 18 mL/100 g·min after permanent cerebral artery occlusion and below 10 to 12 mL/100 g·min after 2 to 3 hours of MCAO. These experiments show that it is difficult to define a lesion core in the first two hours after onset of ischemia.
After occlusion of a brain vessel a characteristic sequence of electrophysiologic events can be observed (van Harreveld, 1947; Creutzfeld, 1957; Krupp, 1972; Caspers et al., 1980). Acute complete obstruction of a vessel will produce symptoms within less than 2 to 3 seconds. There is an early period of normal EEG activity with a duration of a few seconds to minutes depending on the degree of ischemia. This initial interval is followed by increased brain activity initially in the alpha, then beta, and finally delta range. Without reperfusion this period of increased EEG activity lasts approximately 30 to 60 seconds. Thereafter EEG activity is completely depressed. This corresponds to the anoxic depolarization—when the direct current (DC) potential is registered one records a large negative shift of the brain potential in the order of a 10 to 20 mV.
A discussion currently exists as to what extent a lysis with rTPA may rescue the lesion core, or penumbral tissue (Young et al., 1997; Brinker et al., 1999; Hacke et al., 1999; Heiss et al., 1999; Neumann-Haefelin et al., 1999b; Pantano et al., 1999; Kaufmann et al., 1999; Fisher et al., 2000).
PENUMBRA—REVERSIBLE ISCHEMIC DAMAGE
Hossmann defined the penumbra as the brain region that suffers from ischemia, but in which energy metabolism is preserved (Hossmann, 1994). There has been some discussion as to which perilesional areas energy metabolism is preserved and to what extent (Ginsberg et al., 1994). Furthermore, in many instances it will not be possible to definitely ascertain the energy state. Therefore, the authors express penumbra in more general terms as a brain region that suffers from ischemia but in which the ischemic damage is potentially or at least partially reversible.
The description of different viability thresholds has been very helpful to analyze the penumbra (Hossmann, 1998). Protein synthesis is already inhibited by reductions of blood flow to approximately 55 mL/100 g·min and is completely suppressed below 35 mL/100 g·min. mRNA synthesis begins to decline below 35 mL/100 g·min. Notably, mRNA transcription and translation into protein may be still observed for the immediate early gene products. Below a flow rate of 35 mL/100 g·min, glucose consumption first increases, and below 25 mL/100 g·min it sharply declines. The upper level of glucose activation corresponds to incipient acidosis and the early accumulation of lactate. The lower flow rates correspond to pronounced tissue acidosis, and both phosphocreatinine and ATP begin to decline.
Functionally, first a slowing of the EEG appears followed by a reduction of EEG and evoked potentials (Mauguiere, 1998). Complete suppression of EEG activity occurs between 23 and 15 mL/100 g·min. Together with the EEG, evoked potentials also disappear. Neurologic studies indicate that reversible hemiparesis appears in monkeys at approximately 23 mL/100 g·min. Thus, functional deficits are already present with blood flow rates that do not necessarily result in irreversible deficits.
Within the penumbra, repetitive periinfarct depolarizations appear within the first 6 to 8 hours after stroke onset (Hossmann, 1996; Martins-Ferreira et al., 2000). These are similar to the anoxic depolarization in the core but are reversible and appear repetitively. They impose a large metabolic stress on this tissue, thus progressively recruiting much of the penumbra into the lesion. These periinfarct depolarizations are associated with a transient depression of the EEG that lasts, however, up to 10 minutes, that is, considerably longer than with SDs in healthy tissue.
The penumbra is not a homogeneous area. Back et al. (2000) described that in areas with only a minor reduction of blood flow (approximately 20% to 40% of control), areas with preserved tissue ATP, but displaying tissue alkalosis, may appear a few hours after onset of ischemia as a consequence of the periinfarct depolarizations. Such an alkalosis is not seen in the normal brain after waves of SD, possibly because of reactive changes of astrocytes in this brain area. The penumbra is also not homogeneous with respect to its fate; whereas most of it is often recruited into the lesion, peripheral parts may be rescued by spontaneous or induced recanalization or improvement of collateral blood flow.
There have been several attempts to monitor the extent of the penumbral brain area with modern imaging techniques. For a more comprehensive review of this the reader is referred to the review by (Heiss et al. 2000; Neumann-Haefelin et al., 2000). The perfusion deficit can be monitored clinically with positron emission computerized tomography, xenon computerized tomography, and magnetic resonance tomography using perfusion imaging. Neumann Haefelin et al. (1999b) showed that the area in which the blood perfusion is delayed by approximately 4 seconds shows a correlation to the size of the final deficit. De Crespigny et al. (1999) demonstrated that the anoxic depolarization is the main correlate of the increased diffusion-weighted imaging (DWI) signal that corresponds to a decrease of the apparent diffusion coefficient (ADC). In addition, some gradual ADC changes occur earlier that may not be caused by such a massive loss in ion homeostasis (Harris et al., 2000).
Experimental studies have shown that in the penumbra a signal similar to that associated with the anoxic depolarizations is caused by periinfarct depolarizations (Gyngell et al., 1994; Busch et al., 1996). However, these signals only appear transiently; each episode causes a small increase in the size of the lesion core. In accordance with the authors' statement that within the first few hours there is no final lesion core it was shown that with reperfusion of the brain after 10 or 30 minutes the DWI signal is reversible (Fig. 1). Kidwell et al. (2000) reported marked regression of the size of DWI and ADC lesions by approximately 50% 2.5 to 9.5 hours after thrombolysis in humans (Fig. 1). In the authors' experience, the reduction is often less spectacular. This is possibly because the initial recovery may be only in part transient and independent of the continuously ongoing histologic damage (Miyasaka et al., 2000; Harris et al., 2000). During the ischemic episode, cells exhibit a rise in intracellular Ca2+ and become inexcitable. After reperfusion they initially appear morphologically normal, exhibit normal intracellular Ca2+ (Silver et al., 1990), and are again able to generate action potentials for 24 to 72 hours (Gorter et al., 1997). Ultimately, however, intracellular Ca2+ again rises in vulnerable neurons and cell death ensues, exhibiting a number of the hallmarks of apoptosis (Braun et al., 1996; Snider et al., 1999; Colbourne et al., 1999). A similar time course is also observed with DWI MRI recordings; after transient ischemia there is an initial recovery of the DWI lesion that, however, reappears a few days later (van Dorsten et al., 1999; Li et al., 2000).
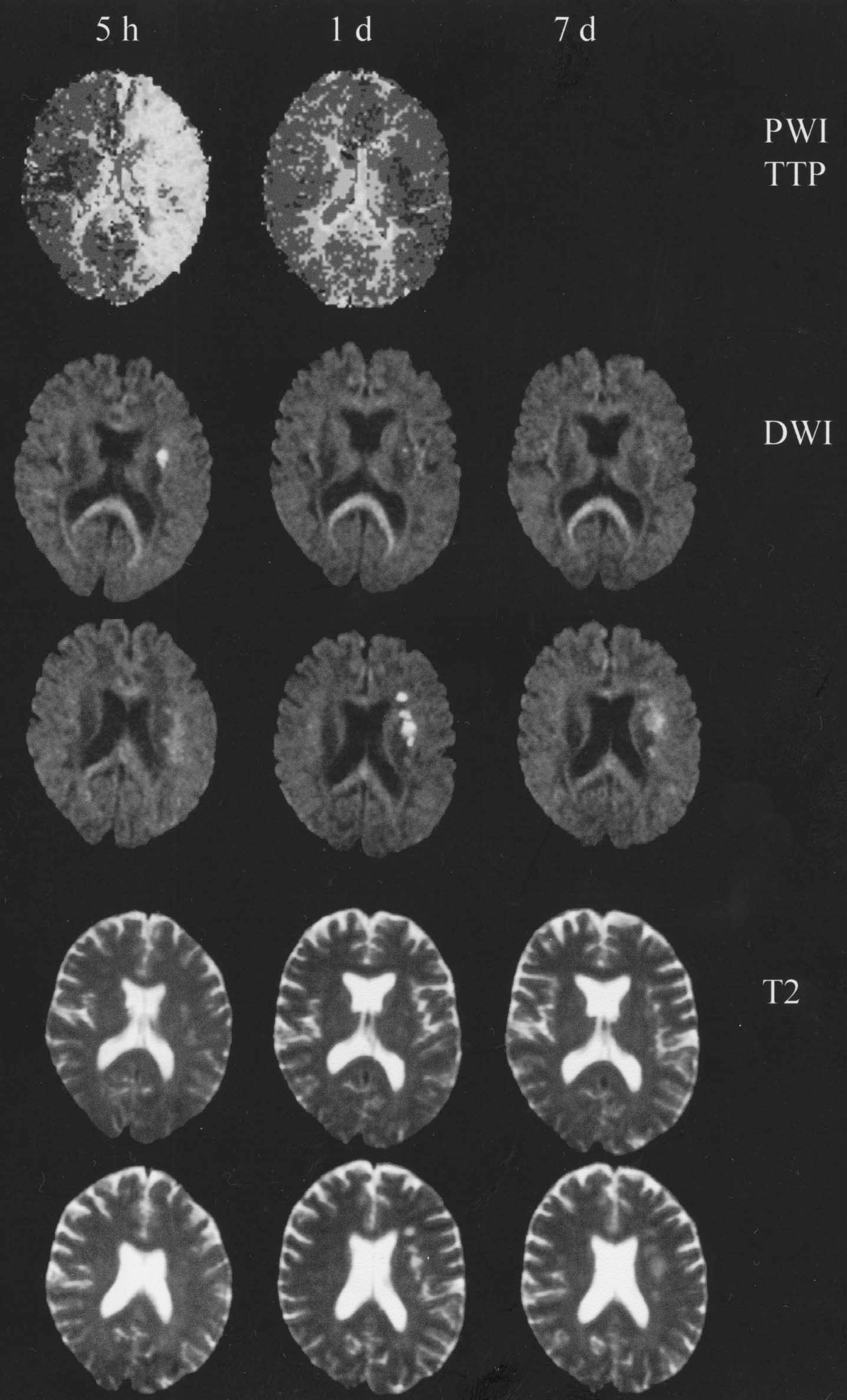
Imaging of ischemic brain injuries with magnetic resonance imaging (MRI). Upper row, perfusion-weighted images (PWI), time to peak (TTP), arbitrarily adjusted contrast. Second and third rows show diffusion-weighted images (DWI). Fourth and fifth rows show T2-weighted images. All images are of the same patient. Images in the first column were recorded 5 hours after symptom onset with aphasia and a mild left sided paresis. Note large mismatch between area with altered perfusion and small DWI lesion. The symptoms recovered in the following hours but reappeared in the evening with another episode. The next day they had nearly completely recovered again. A second MRI now revealed a normal brain perfusion, a resolution of the DWI lesion seen in 5 hours, and new DWI lesions in neighboring sections. These were also visible in the T2 images at this time. Both DWI and T2 lesions partially recovered in the following week. In this patient, the recurrent emboli originated from a dissection of the internal carotid artery, which reopened approximately 12 hours after the first scan. This case demonstrates that the MRI images can change in a very dynamic manner corresponding to the symptomatology. Data obtained in cooperation with U. Moedder and H.J. Wittsack, Department for Diagnostic Radiology, and U. Junghans and M. Siebler, Department of Neurology, University of Duesseldorf.
Much of the attention has been devoted to the salvation of the penumbral brain tissue, and it is indeed fascinating to see how a timely performed lysis may promptly cause a functional recovery in patients. One should, however, keep in mind that the penumbra contains different parts with different fates: one part is recruited into the lesion (for example, by SD), another portion will survive (for example, because of reperfusion or increased collateral blood flow), though possibly with partial damage. Incomplete ischemia causes a differential loss of selectively vulnerable neurons (Schmidt-Kastner et al., 1991). Transient ischemia primarily affects the middle and lower cortical layers, whereas in the penumbra primarily the superficial cerebral layers undergo secondary cell death (Luhmann, 1996). These neurons may die hours to days after the ischemia. The delayed cell death observed after ischemia is at least partially because of apoptosis. There are indications that it is related to a reduction of GluR2 mRNA and protein associated with an increased calcium permeability of the AMPA receptors (Pellegrini-Giampietro et al., 1997). AMPA receptors containing an edited form of the glutamate receptor subunit GluR2 (or GluR-B) are relatively impermeable to divalent cations. Receptors lacking this subunit are much more permeable to Ca2+ (and Zn2+).
Unfortunately, little is known about the function of the brain tissue in the penumbra region that survives. It has been reported that 5% to 25% of patients develop seizures after brain ischemia. In a fraction of the patients, periodic lateralized epileptiform discharges appear in the perilesional brain area in the first days after stroke (Kotila and Waltima, 1992; Lee et al., 1990; Beaumanoir et al., 1996; Ono et al., 1997). These periodic lateralized epileptiform discharges seem to be most prominent with borderzone infarctions, rarely do they disappear spontaneously within a few days or weeks after onset. Luhmann et al. (1996) reported that the penumbral area displays a reduction of functional inhibition, with decreased gamma-aminobutyric acid (GABA) receptor binding, and increased NMDA responses even after 30 days. In a study from the authors' group, in addition to the reduced inhibition, the authors found reduced excitatory responses (Neumann-Haefelin and Witte, 2000) (Fig. 4). Congar et al. (2000) reported that after a reversible ischemia, several months later CA3 neurons in the hippocampus displayed a more depolarized resting membrane potential and had a lower threshold to generate synchronized bursts. Furthermore, Mori (1998) and Aoyagi et al. (1998) found a decrease of long-term potentiation in such brain areas (Crepel et al., 1998).
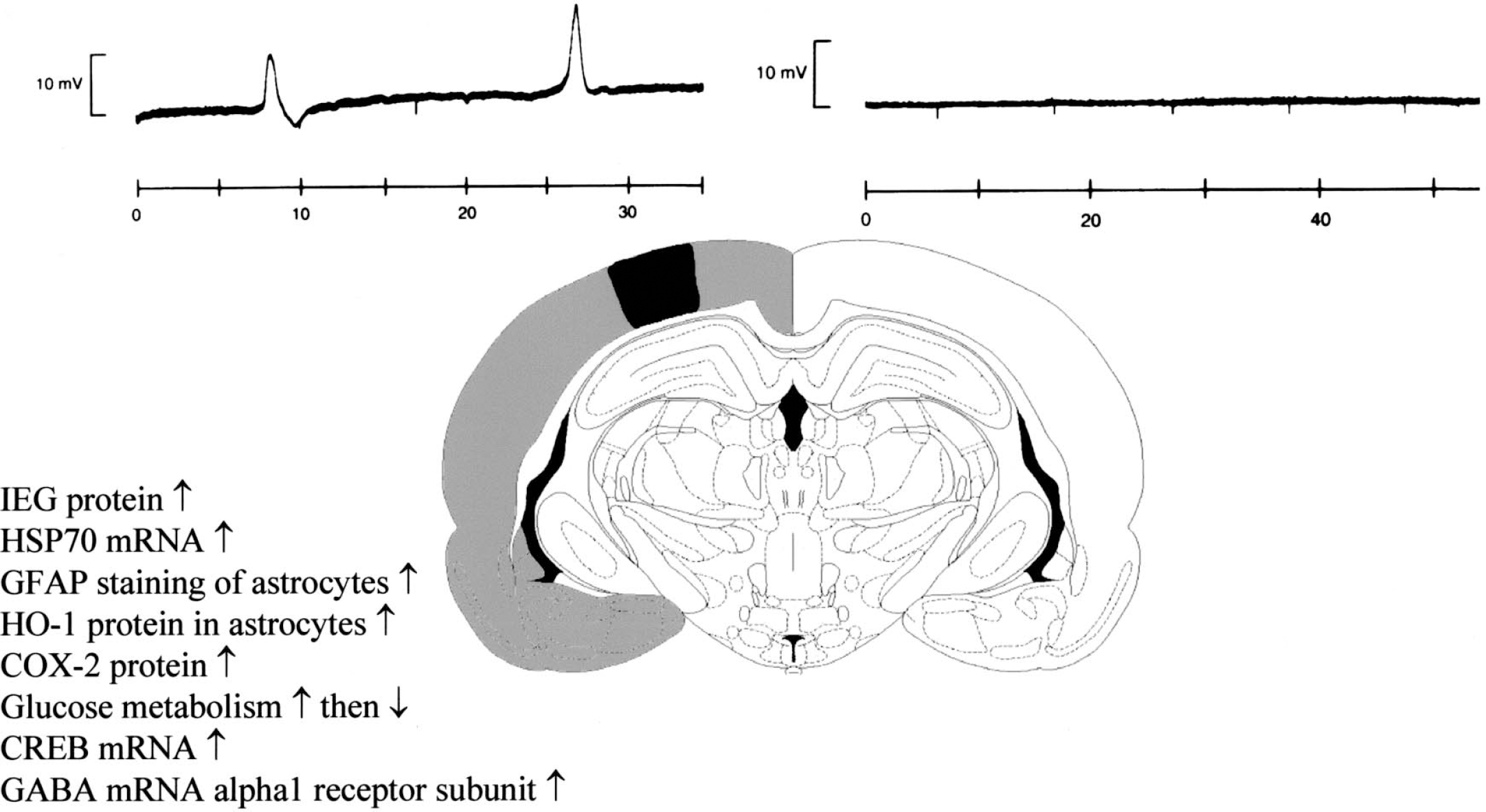
Remote changes caused by spreading depression. Topographic pattern of changes caused by waves of spreading depression (SD) in the cortex of the rat. Spreading depression originates from the lesion (black area) in the first 6 hours after lesion induction and travels repetitively across the whole ipsilateral hemisphere. The list summarizes some of the changes initiated by the SDs. (inset) A typical recording of SDs from the perilesional cortex and their absence in the contralateral hemisphere (horizontal axes represent time in minutes).
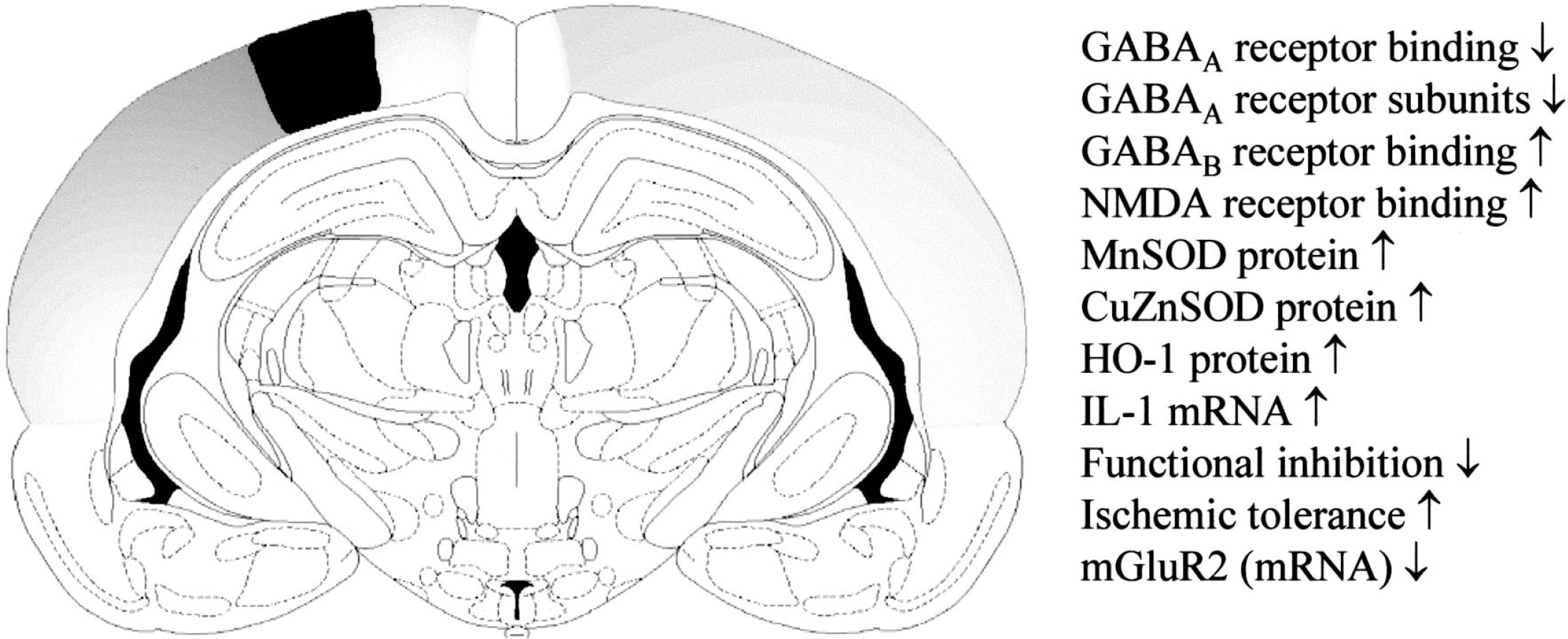
Remote changes in projection areas. Topographic pattern of changes in projection areas in the cortex of the rat. After induction of a small photothrombotic lesion (black area) several processes are initiated in both hemispheres. Some of the changes occurring in these areas are listed. These bilateral changes follow the projections from and to the lesion.
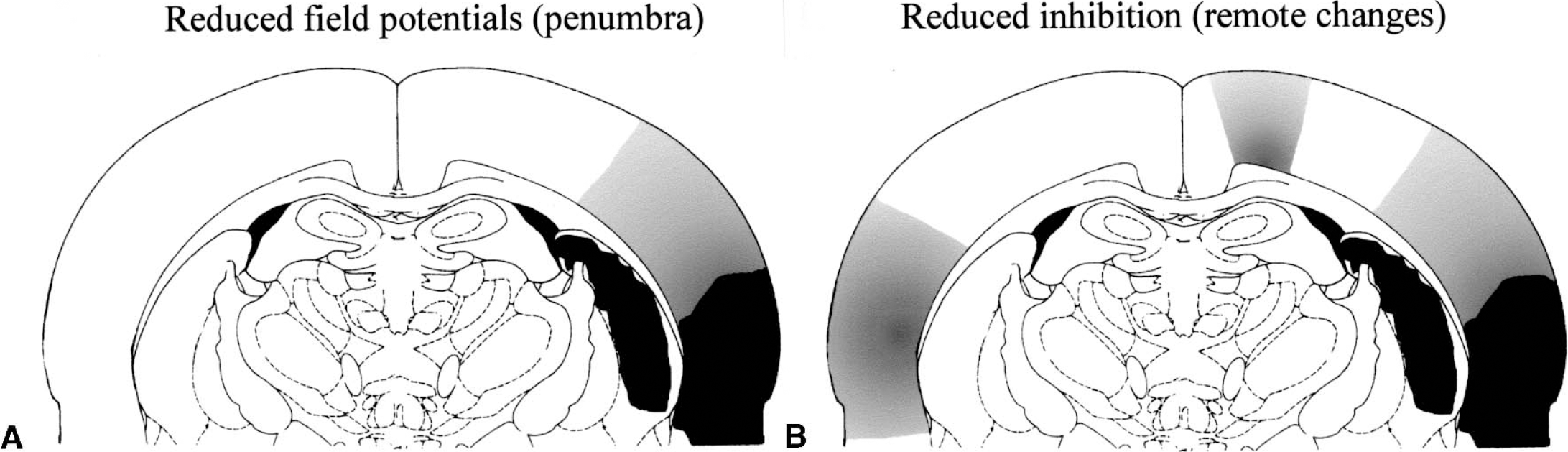
Topographic pattern of changes in the penumbra and the projection areas of a stroke caused by reversible occlusion of the middle cerebral artery.
The functional alterations in the penumbral brain tissue may be of considerable importance for the final functional outcome. (Nudo et al. 1996; Nudo, 1999) observed that perilesional brain areas may progressively loose function if no training to preserve the function occurs. However, (Kozlowski et al. 1996; Humm et al., 1998, 1999) reported that intensive training started too early may actually enhance the size of the lesion, which shows that the perilesional tissue is sensitive to exaggerated activation. These experiments were done with electrolytic brain lesions. A similar observation was also observed after ischemic brain lesions (Johansson, 2000). Risedal et al. (1999) reported that housing the animals in an enriched environment produced functional improvement without additional lesion growth. Additional forced use of the impaired limb led to further functional improvement, which was however, associated with a growth of the lesion size.
REMOTE BRAIN AREAS—NONISCHEMIC CHANGES IN PERILESIONAL AND REMOTE BRAIN AREAS
Brain lesions may cause functional and structural alterations in remote brain areas, that is, in brain areas that were not directly affected by ischemia. With respect to the etiology, several broad categories may be differentiated: first, remote changes caused by brain edema; second, remote changes caused by waves of SD; third, remote changes in projection areas; finally, remote changes caused by reactive plasticity and systemic effects.
Remote changes caused by brain edema
Brain ischemia is associated with edema. Initially, there is cellular swelling; after approximately 9 hours, a breakdown of the blood—brain barrier is observed after MCAO (Huang et al., 1999; Hatashita et al., 1990b). Within a few hours after onset of ischemia, a progressive net uptake of water develops with the increase of total tissue water. This swelling has a remarkably prolonged time course. In the photothrombosis model, in which blood—brain barrier breakdown occurs even faster than with MCAO (Forsting et al., 1994), it reaches its maximum after 24 to 72 hours; in experimental MCAO models and after MCAO in humans, it reaches the maximum extent at approximately 72 hours (Rosenberg, 1999).
With circumscribed focal photothrombotic lesions of the cortex, nearby perilesional cortex and sublesional corpus callosum are compressed resulting in an restricted expression of hippocampal heme oxygenase 1, following exactly the transient, temporal pattern of lesion induced cortical edema (Bidmon et al., 1998a; Fig. 5D). With MCAO, brain edema is one of the primary causes of death after stroke in animals and in humans (Forsting et al., 1995; Doerfler et al., 1996). The tissue swelling produces massive secondary damage by compression of the contralateral brain and other remote brain areas, with an associated secondary ischemia caused by compression of low resistance vessels. If a craniotomy is performed on rats exposed to occlusion of the middle cerebral artery, the lethality declines from approximately 30% to 0%, regardless of the time point (up to 36 hours after stroke) the procedure is performed. Also in patients, death and possibly secondary damage can be prevented by large trepanations of the skull.
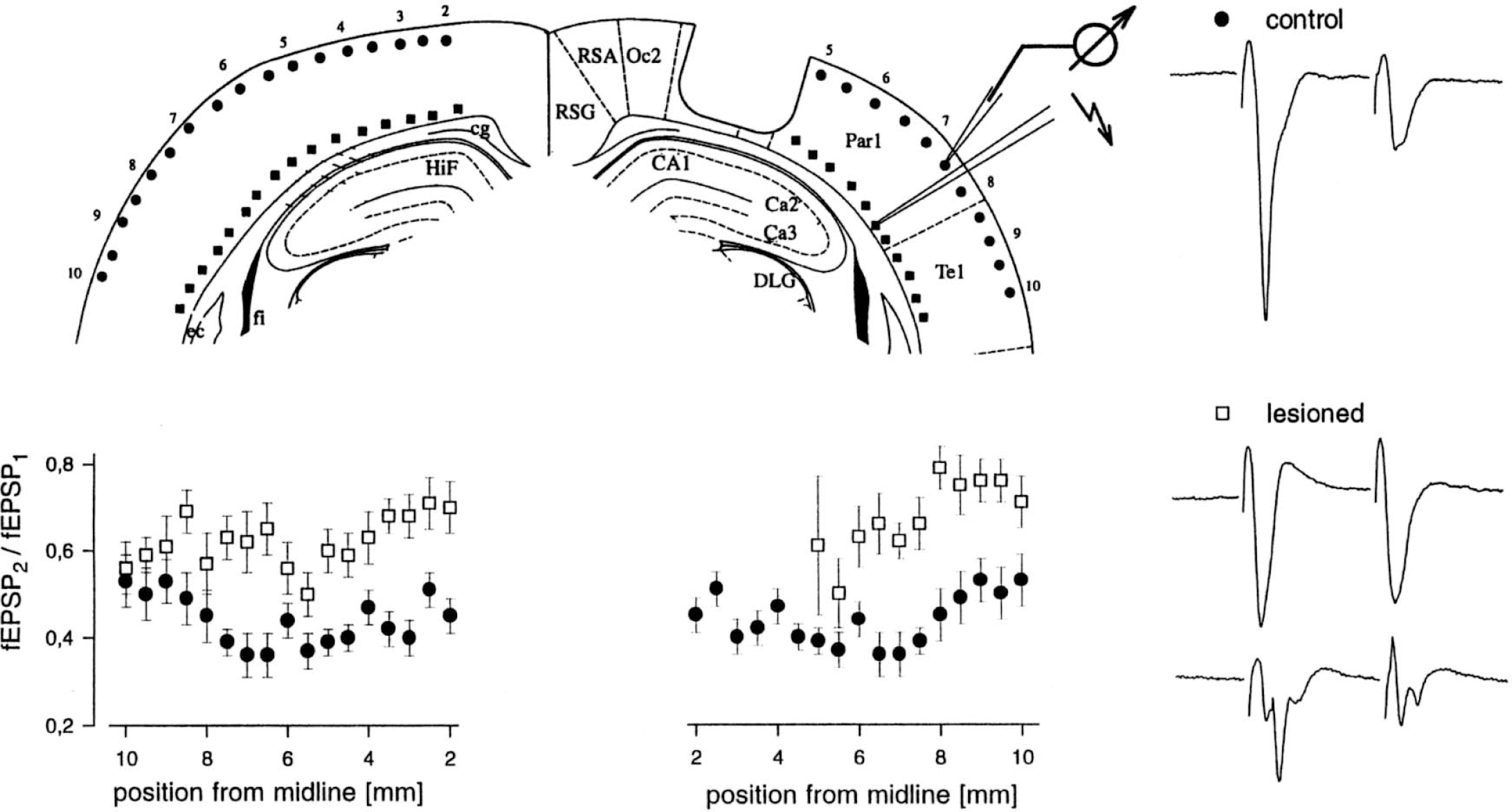
Bilateral alteration of cortical inhibition after cortical photothrombosis. (top left) Schematic drawing of brain slice (bregma −3.8 mm). The lesion is indicated by an indentation. Positions of the stimulation electrode are marked by squares, those of the field potential electrode by circles. Numbers indicate the distance from midline in millimeters. Field potential and stimulation electrodes were moved in parallel from the lesion border down to the rhinal fissure. (bottom left) Spatial profile of ratio of field potential amplitudes fEPSP2/fEPSP1. Mean and SEM values from recordings in control (filled circles) and lesioned (open squares) animals are shown. Abscissa corresponds to positions indicated in the schematic drawing of the brain slice. Lesioning led to a significant increase in the ratio ipsilaterally (right panel) and contralaterally (left panel), indicating an increase in excitability. (right) Typical extracellular recordings of response to paired-pulse stimulation. Top tracing is in unlesioned controls with a ratio of approximately 0.4; middle tracing is in lesioned animals with a ratio near 1; bottom tracing is in lesioned animals with multiple discharges. Abbreviations indicate the cortical areas according to Paxinos and Watson (1986). ec, corpus callosum; fi, fimbria hippocampi; HiF, hippocampal fissure; cg, cingulum; RSG, retrosplenial granular cortex; RSA, retrosplenial agranular cortex; Oc2, occipital cortex—area 2; DLG, dorsal lateral geniculate nucleus; Par1, parietal cortex—area 1; and Te1, temporal cortex—area 1. From Buchkremer-Ratzmann et al. (1996). Reprinted with permission from Lippincott Williams & Wilkins.
The mechanisms of stroke-induced brain edema are only partially understood. A simple explanation would be that it is related to net water uptake because of breakdown of the blood—brain barrier. However, this breakdown of the blood—brain barrier is faster than the edema (Hatashita et al., 1990a). Furthermore, the apparent diffusion coefficient is usually still decreased three days after stroke onset, indicating that the intracellular edema prevails over the extracellular water accumulation. After three days, the edema starts to resolve. With photothrombotic cortical stroke, perilesional cortical thickness (indicating edema) returns almost to baseline during the first 2 weeks, and decreases below normal between 1 and 2 months after injury because of currently unknown degenerating effects accompanied by a partial enlargement of the ipsilateral lateral ventricle (Bidmon et al., 1998a). The edema with MCAO shows a similar time course.
No systematic data on the consequences of edema on the remote brain areas affected by compression are available. Preliminary data in patients indicate that the compressive damage is associated with increased slowing of the EEG in these areas (M. Siebler, personal communication).
Remote changes caused by spreading depression
The authors have discussed in the previous sections that brain ischemia elicits repetitive ischemic depolarizations in the penumbra for up to 6 hours (Nedergaard and Hansen, 1988,1993; Schroeter et al., 1995; Busch et al., 1995; Hossmann, 1996; Nedergaard, 1996). At least in animals, these depolarizations do not stop at the border to normal brain tissue but travel further across the normal brain as waves of SD. They travel across the whole ipsilateral cortex and do not stop at the borders of vascular territories. In patients, all attempts to demonstrate waves of SD after stroke have been unsuccessful as yet; this is possibly because of technical difficulties. If tissue specimens of human brain are recorded in vitro, no problems exist eliciting waves of SD, similar to those produced in rodent brain tissue, by local application of KCl.
In the photothrombosis model with cortical ischemic lesions, waves of SD only occur on the side of the lesion, not in the contralateral cortex (Schroeter et al., 1995) (Fig. 2). This may be different with complete occlusion of the middle cerebral artery. It is conceivable that this model will also cause SD in the contralateral cortex that may sustain massive damage because of edema induced compression. Pharmacologically, SD can be prevented when the animals are treated with the NMDA antagonist MK801 after lesion induction. This allows differentiation of effects that are caused by SD from those that are not.
Also, in the healthy brain not compromised by ischemia, waves of SD cause a massive metabolic stress. They strongly increase brain metabolism for a few hours, followed by a hypometabolism lasting even longer. Consequently, they activate many molecular cascades in widespread brain areas in which they occur—immediate early genes and heat shock protein 70 are up-regulated, there is a short but massive increase in the brain derived neurotrophic factor, and there are transient increases of IL-1, IL-6, COX-2, HO-1, CREB, FOS, JUN, KROX, nerve growth factor (NGF), fibroblastic growth factor (FGF), tissue plasminogen activator (t-PA), neuronal NOS activity (citrullin), and protein kinase C (Fig. 2) (Comelli et al., 1992; Herdegen et al., 1993; Nimura et al., 1996; Witte and Stoll, 1997; Kawahara et al., 1997a,b, 1999; Kariko et al., 1998; Matsushima et al., 1998; Hermann et al., 1999; Shen and Gundlach, 1999; Koistinaho et al., 1999; Mancuso et al., 1999; Koponen et al., 1999; Dietrich et al., 2000; Kato et al., 2000). Astrocytes show signs of activation with increased expression of glial fibrillary acidic protein (Schroeter et al., 1995). The SD induced cyclooxygenase 2 (COX2) up-regulation in remote brain areas (Koistinaho et al., 1999) (Fig. 5A) is also observed in human stroke patients (Sairanen et al., 1998), suggesting that waves of SD do indeed occur in humans.
In addition, there are more indirect effects of SD. Thus mRNA for the alpha subunit of the GABAA receptor is massively up-regulated selectively on the side of the lesion. This unilateral up-regulation in structurally intact brain tissue suggests that it is caused by waves of SD (Neumann-Haefelin et al., 1999a). At the same time the concomitant decrease of GABA receptor protein and binding suggests that there is a block of translation (Fig. 3). A similar mechanism may explain why in the first week after lesion induction NMDA receptor binding increases only in the hemisphere contralateral to the lesion; at later time points it is increased bilaterally (Que et al., 1999a).
In remote ipsilateral cortex devoid of neuronal damage a transient response of microglia with increased complement receptor 3, but not by major histocompatibility complex class II and CD4 expression, can also be observed. Furthermore, the total number of reactive microglia increases. This remote response could be partially blocked by MK801, implicating a causal role of SD (Schroeter et al., 1999).
The functional consequences of the signal cascades induced by SD have only partially been analyzed. In healthy brain, waves of SD do not cause cell death. However, they do induce tolerance to a subsequent ischemia for up to a week (Kobayashi et al., 1995; Kawahara et al., 1997a). During the SD the EEG is interrupted, but subsequently returns to normal activity. The effects caused by SD do not usually last longer than one week.
Remote changes in projection areas
As an example of remote changes not related to edema or SD there are changes of GABAergic inhibition after focal brain lesions (Figs. 3, 4, and 6). For monitoring changes in brain excitability induced by photothrombotic lesions, a so-called in vitro paired pulse paradigm was used. When paired pulses were applied with intervals of 20 milliseconds, the response to the second stimulus was smaller than that of the first stimulus under control conditions indicating that the first stimulus, in addition to the excitatory response, had activated an inhibition (Domann et al., 1993). In animals with a photothrombotic lesion, this paired pulse inhibition was impaired in the whole cortex laterally extending to the rhinal fissure (Buchkremer-Ratzmann et al., 1996,1998; Buchkremer-Ratzmann and Witte, 1997a). Further experiments using intracellular recordings revealed that this was indeed accompanied by a reduction of GABAA dependent inhibition (Neumann-Haefelin et al., 1995). In recordings from animals in vivo an increased spontaneous activity of cells surrounding the photothrombotic lesion was noted (Schiene et al., 1996). This increase in spontaneous discharge frequency appeared with a characteristic delay after lesion induction. It was not yet present 4 hours after lesion induction, but it was manifest after 24 hours and increased up to 5 to 7 days. In the following three months it recovered to a large extent, though incompletely. A further series of experiments indicated that the impairment of GABAergic inhibition was not only present on the side of the lesion, but also in the contralateral brain. Blocking waves of SD that appear after lesion induction by application of MK 801 did not affect the disturbance of inhibition (Buchkremer-Ratzmann and Witte, 1997b). Investigations with quantitative GABAA receptor autoradiography indicated that the decrease of GABAergic inhibition was associated with a downregulation of GABAA receptor binding (Zilles et al., 1995; Schiene et al., 1996; Qu et al., 1998a; Que et al., 1999b). A downregulation of GABA receptor subunits was noted with immunohistochemistry, although this seemed to be less intensive than the reduction of GABAA receptor binding (Neumann-Haefelin et al., 1998). The downregulation of GABAergic inhibition in brain areas not directly affected by ischemia could also be reproduced in animals after MCAO (Qu et al., 1998b; Reinecke et al., 1999). GABAergic inhibition was decreased not only in the penumbra but also in remote areas from the lesion. This impairment of GABAergic inhibition followed the topographic relationship of brain areas connected to the lesion area. (Liepert et al. 2000; Liepert and Weiller, 1999) also reported remote changes in paired pulse inhibition in patients with stroke using transcranial magnetic stimulation.
A relation of these remote alterations of GABAergic inhibition to the neuroanatomic connections of the lesion area was also suggested by another series of experiments. If only superficial lesions were produced, the reduction of a GABAergic inhibition was reduced in an area extending 1 to 2 mm in the surround of the lesion. However, if lower cortical layers that give rise to large amounts of intracortical collaterals were lesioned, and widespread changes of impaired inhibition were found (Buchkremer-Ratzmann and Witte, 1997a). This could also explain why Eysel and coworkers (Luhmann et al., 1996; Eysel, 1997), with small lesions in superficial cortical layers, only noted circumscribed changes in the (nonlesioned) cortical tissue (however, they also used a different lesion model). Silver impregnation studies in the photothrombosis model confirmed that there was widespread degeneration of nerve fibers extending to most of the ipsi- and contralateral cortical areas that showed a change of GABAergic inhibition (unpublished observations, 1998).
Other bilateral functional changes after focal brain lesions are an up-regulation of GABAB receptor binding and of NMDA receptor binding (Que et al., 1999b; Crespi et al., 1997). Also, a bilateral up-regulation of the inducible superoxide dismutase (MnSOD) and the constitutive superoxide dismutase (CuZnSOD) can be observed (Bidmon et al., 1997; Bidmon et al., 1998b). In addition, a rapid contralateral activation of NOS-I and a longer lasting up-regulation of βAPP and neurofilament 68 have been found in rodents (Pierce et al., 1996), whereas in humans a transient contralateral phosphorylation of neurofilament H occurs (Hedreen et al., 1994). The latter could be because of an increase of brain derived neurotrophic factor (BDNF) that has recently been shown to induce neurofilament H phosphorylation (Tokuoka et al., 2000). Furthermore, the inducible heme oxygenase I is selectively up-regulated in oligodendrocytes contralateral to the lesion. On the lesioned side, it is strongly up-regulated in astrocytes, microglia, and neurons, probably caused by SDs (Bidmon et al., 2000). Interestingly, in correspondence with the location of argyrophilic fibers and other markers within the contralateral hemisphere, changes in DWI have been reported after cortical photothrombosis (De Ryck et al., 2000).
Other remote effects can be observed in subcortical structures (Fig. 6B). Thus, secondary degeneration and neuronal loss is observed in several subcortical structures, for example, in the thalamus with MCAO. In secondarily degenerating fiber tracts and nuclei with retrograde neuronal loss, microglia are activated with a delay of days and show increased expression of complement receptor 3 and major histocompatibility complex class II and CD4 molecules, but low phagocytic activity. Also, an increase in the number of reactive astrocytes along such fiber tracts down into the spinal cord after unilateral focal MCAO have been reported (Jones and Schallert, 1992b; Wu and Ling, 1998a,b; Schroeter et al., 1999).
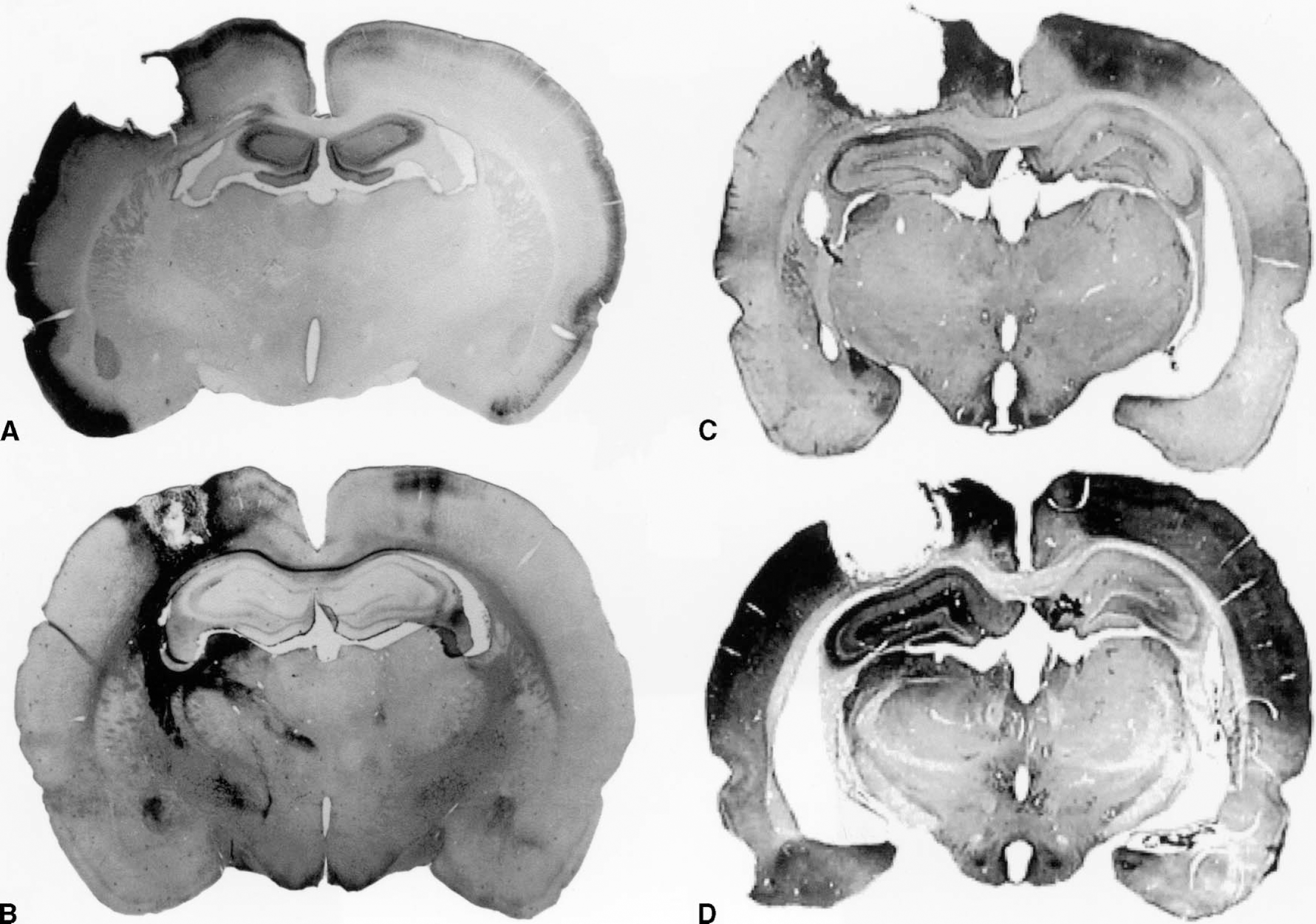
Different patterns of remote changes caused by focal brain lesions. The focal ischemic lesion was induced by photothrombosis.
The functional changes in the projection areas are not necessarily homogenous. Functional recordings indicate that in the primary projection areas homotopic to the lesion a deafferentation induced disfacilitation may prevail over the disinhibition seen in other areas (Divanach et al., unpublished observations, 2000; Rema and Ebner, personal communication, 2000).
The cascades of events leading to the pronounced remote effects are not proven. That they follow the extension of collaterals from the lesion site suggests that they are caused by deafferentation and degeneration of collaterals. Furthermore, retrograde degenerations may be induced by synaptic signaling mechanisms that have been identified as an initial step of distant neuronal apoptosis (Mattson, 2000). Generally, the remote changes that follow the projections from and to the lesion area show a prolonged time course from weeks to months. During the first 2 weeks after ischemia these remote effects (for example, within the contralateral hemisphere) are most pronounced in areas homotopic to the core, but also spread into regions that surround the homotopic area, indicating that at least part of the penumbra and edema affected perilesional areas contribute to these effects. In addition to degeneration, stretch induced by edematous swelling and stretch-related injury may play a role in causing such remote bilateral alterations along the path of traceable fibers (Ahmed et al., 2000).
Remote changes caused by reactive plasticity and systemic effects
Brain lesions will cause adaptive processes, which in turn can initiate functional and structural alterations in the brain. This can either be induced by systemic effects or by more localized behavior-related mechanisms. Examples of systemic effects are increased stress of the animals with brain lesions, or spillover of substances from the lesion into the cerebrospinal fluid and blood (Fassbender et al., 1997). Thus, increased production of several cytokines, including interleukin (IL)-1β, IL-6, IL-8, IL-10, tumor necrosis factor (TNF-α), and granulocyte-macrophage colony-stimulating factor, has been demonstrated intrathecally in patients with acute ischemic stroke (Kostulas et al., 1999; Tarkowski et al., 1999,2000). Increased synthesis of cytokines was also detected systemically, that is, in the blood (Fassbender et al., 1994). These interleukins and tumor necrosis factor-α are also produced by peripheral blood cells after infarction showing that the plasma levels of cytokines do not solely derive from the injured central nervous system (Ferrarese et al., 1999). These cytokines may transmit systemic effects throughout the whole central nervous system (Shibata et al., 1996; De Simoni et al., 1998). Within the injured cerebral tissue these proinflammatory cytokines are regulated by a wide variety of other factors and represent regulators for a large and still increasing number of other factors including radical producing enzymes such as NOS I, NOS II, and NOS III (Strijbos, 1998; Vincent et al., 1998).
Reactive or compensating behavior can be observed in animals and patients—if one upper extremity is affected, they will depend much more on the other one. Agreeing with this, (Schallert et al. 2000; Jones and Schallert, 1992a, 1994; Bland et al., 1999;) described a sprouting of neuronal apical dendrites in the hemisphere contralateral to the lesion that was not present in animals prevented from using the healthy limb. In another series of experiments the authors observed a suppression of proteasome C2 only in the hemisphere contralateral, but not ipsilateral, to the lesion (Keyvani et al., 2000). It has been described earlier that brain plasticity processes are associated with a suppression of proteasome C2 (Wu et al., 2000). (Stroemer et al. 1993,1995) described increases of GAP43 and synaptophysin as indicators for structural plasticity in both hemispheres after unilateral ischemia.
FUNCTIONAL CONSEQUENCES OF PERILESIONAL CHANGES FOR RECOVERY FROM BRAIN ISCHEMIA
von Monakow speculated that recovery from a brain lesion may be caused at least partially by an attenuated depression of brain function in remote brain areas, which he called diaschisis. Glassmann (1971) speculated that “the tissue immediately surrounding the destroyed region suffered temporary loss of function and regained its original excitability during the course of recovery.” It is now known from reperfusion experiments, both under experimental and clinical conditions, that recovery of partially or reversibly ischemic tissue may indeed result in considerable and fast recovery of neurologic function within minutes to hours.
Remote functional alterations may also contribute to the clinical deficit. It is well known, although only anecdotically analyzed, that epileptiform alterations in the surround and even in the contralateral hemisphere may transiently contribute to the neurologic deficits of a patient. Carmichael and Chesselet (2000) reported that this perilesional dysfunctional activity may also have a positive effect. They observed a much stronger structural plasticity in the surround of suction lesions with slow brain activity than without, and they attributed this to the stimulating effect of the perilesional dysfunction on brain plasticity.
Remote changes caused by SD are known to cause an ischemic tolerance to subsequent ischemic events (Kobayashi et al., 1995; Kawahara et al., 1997a). Recently, the authors found that an ischemic tolerance can even be observed in the contralateral hemisphere after a photothrombotic brain lesion (M. Lutzenburg et al., unpublished observations). Among other factors, this is possibly related to the bilateral up-regulation of MnSOD described above.
Remote increases in brain excitability would be expected to increase brain plasticity (Witte et al., 1997; Witte and Stoll, 1997; Witte, 1998; Witte and Freund, 1999) (Fig. 7). In accordance with this, the authors did indeed observe an increase of brain areas activated by stimulation of a vibrissa in perilesional areas (Schiene et al., 1999). A similar increment of sensory representation could recently also be verified for contralateral brain areas (unpublished observations). Experimental markers for brain plasticity are also increased in the surround of the lesion; thus, the authors found that long-term potentiation is more easily induced in the (nonischemic) surroundings of the lesion than in normal brain (Hagemann et al., 1998). Schallert and coworkers demonstrated that contralateral to the lesion, activity can also induce dendritic sprouting, which is not observed in nonlesioned brain (Jones and Schallert, 1992a, 1994; Jones et al., 1999). Thus there are indications for a lesion-induced brain plasticity (Witte, 1998). It should emphasized that this seems to be different in the penumbra in which long-term potentiation is impaired (Mori et al., 1998; Aoyagi et al., 1998).
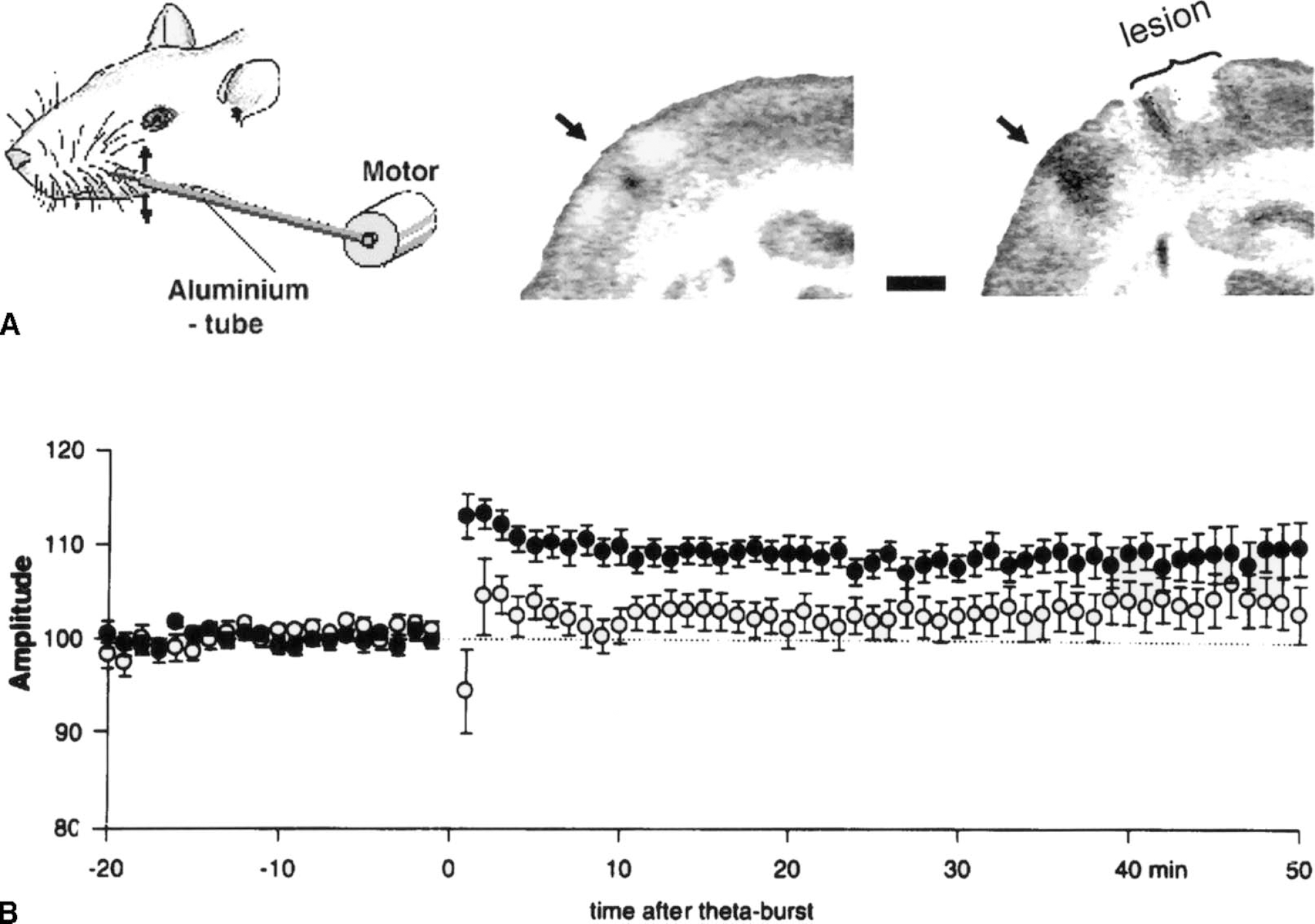
Lesion-induced brain plasticity.
In conclusion, a focal brain ischemia has consequences for the whole brain. The remote effects may critically determine the process of recovery and compensation. This perilesional area is not homogeneous, but it comprises functionally and etiologically different areas. A differentiation of functionally different perilesional areas may support the analysis of the mechanisms underlying recovery.