
Abstract
In a model of glial-specific chemical anoxia, we have examined how astrocytes influence both synaptic transmission and the viability of hippocampal pyramidal neurons. This relationship was assessed using electrophysiological, pharmacological, and biochemical techniques in rat slices and cell cultures, and oxidative metabolism was selectively impaired in glial cells by exposure to the mitochondrial gliotoxin, fluoroacetate. We found that synaptic transmission was blocked shortly after inducing glial metabolic stress and peri-infarct-like spreading depression (SD) waves developed within 1 to 2 h of treatment. Neuronal electrogenesis was not affected until SD waves developed, thereafter decaying irreversibly. The blockage of synaptic transmission was totally reversed by A1 adenosine receptor antagonists, unlike the development of SD waves, which appeared earlier under these conditions. Such blockage led to a marked reduction in the electrical viability of pyramidal neurons 1 h after gliotoxin treatment. Cell culture experiments confirmed that astrocytes indeed release adenosine. We interpret this early glial response as a novel safety mechanism that allocates metabolic resources to vital processes when the glia itself sense an energy shortage, thereby delaying or preventing entry into massive lethal ischemic-like depolarization. The implication of these results on the functional recovery of the penumbra regions after ischemic insults is discussed.
Introduction
Neuronal activity and viability rely on the continuous supply of metabolic substrates by the vascular system. It is now well known that an imbalance between the rate of energy production and utilization causes the collapse of ionic gradients and leads to massive cell depolarization. This is a common scenario in stroke, anoxia, hypoglycemia, and concussion, which initiate the signaling cascades leading to loss of function and cell death (Somjen et al, 1990). Also well known is the tight metabolic coupling between neurons and glial cells (reviewed in Araque et al, 2001), which, in the context of energy shortage, is indicated by the disruption of tissue homeostasis in parallel with the gradual decline of the electrical activity and irreversible injury to neurons on selective blockage of the glial Krebs cycle (Largo et al, 1996). In this process, a rapid loss of excitatory synaptic transmission is followed by the spontaneous development of peri-infarct-like waves of spreading depression (SD) (Hossman, 1996; Canals et al, 2005b). Although SD has little impact in normal tissue (Herreras and Somjen, 1993), it kills neurons in brain regions deprived of glial activity according to a spatiotemporal pattern, which closely reproduces the sequence of events in the penumbra of an ischemic focus (Fabricius et al, 2006). These results underscore the fundamental role of astrocytes in maintaining normal physiologic activity. However, less is known about their active participation in the protective mechanisms activated during energy imbalance (for a review see Ames, 2000).
One such protective mechanism involves the accumulation of adenosine in the extracellular space (Dunwiddie and Masino, 2001). Several studies have shown that during periods of energy shortage, such as ischemia, hypoxia, or hypoglycemia, the concentration of interstitial adenosine increases several folds and it reduces neuronal activity (Pull and Mcllwain, 1972; Gribkoff et al, 1990; Frenguelli et al, 2007). Adenosine may be an ideal molecule to fight against the functional collapse of neurons as it can block synaptic transmission by decreasing neurotransmitter release and hyperpolarizing the postsynaptic membrane (Greene and Haas, 1991), while increasing the local energy supply by its potent vasodilatory activity (Phillis, 2004). Therefore, not surprisingly, blockage of the adenosine A1 receptor during an ischemic episode restores synaptic transmission, precipitates the occurrence of anoxic depolarization, and increases the infarct size (Fowler, 1989; Rudolphi et al, 1992). Adenosine-mediated neuroprotection also occurs in experimental models in which cellular respiration has been pharmacologically depressed (chemical anoxia; Ilie et al, 2006).
In this study, we have examined the relationship between neurons and astrocytes and the effects of adenosine released under conditions of metabolic stress in rat slices and cell cultures. This issue was investigated using electrophysiological and pharmacological techniques in a model of glial-specific chemical anoxia in which fluoroacetate (FAc), a mitochondrial gliotoxin, selectively impaired the oxidative metabolism of glial cells. Fluoroacetate and its active metabolite fluorocitrate inhibit the mitochondrial enzyme aconitase, which catalyzes the conversion of citrate into isocitrate (Clarke, 1991). The specificity of these agents is because of their selective uptake by the acetate transporter present only in glial cells (Clarke et al, 1970; Waniewski and Martin, 1998). Indeed, they have been used in numerous experimental models to study the role of astrocytes or neuro-glial coupling on synaptic transmission and neuron survival (e.g., Largo et al, 1996; Hulsmann et al, 2000; Willoughby et al, 2005; Zielke et al, 2007).
Our results indicate that synaptic transmission is blocked shortly after selective glial metabolic stress because of the rapid extracellular accumulation of adenosine as an active response of astrocytes. Thus, while adenosine A1 receptor antagonists preserve synaptic transmission, they also promote the development of SD waves and accelerate the loss of neuronal viability. Importantly, cell culture experiments show that adenosine is directly produced by astrocytes. We conclude that in periods of energy shortage, glia participates in the overall adjustment of neuronal activity via the adenosinergic blockage of synaptic transmission as a general mechanism to prevent or delay massive depolarization and death. We believe our data unveil a glial-controlled and adenosine-mediated safety mechanism, which could serve as a new therapeutic target in the context of brain ischemia.
Materials and methods
All experimental procedures conformed to ECC regulations for animal care and every effort was made to minimize the suffering and the number of animals used.
Slice Preparation and Treatments
Hippocampal slices (450 μm thick) were obtained from female Sprague—Dawley rats (120 to 150 g) and placed in a holding chamber (35°C) at the interface between humidified air (95% O2/5% CO2) and artificial cerebrospinal fluid (ACSF). Slices were incubated for at least 90 mins and then transferred to a recording chamber (ʻOsloʼ type) continuously superfused by ACSF at 2 mL/min.
To block Krebs cycle in glial cells, we used 10 and 20 mmol/L FAc (Sigma-Riedel de Haen, Seelze, Germany) in ACSF. The effect of increased osmolarity was compensated by adding mannitol to the ACSF for 30 mins before FAc (final osmolarity 315 and 335, respectively).
The general adenosine receptor antagonist, 8-(p-sulfophenyl)-theophylline (8-SPT; Sigma, St Louis, MO, USA), and the A1-selective antagonist, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX; Sigma), were added to the ACSF before or after exposure to FAc.
Cell Culture
Rat glial cultures were prepared as described previously (Canals et al, 2001). Briefly, the hippocampus from 1-day-old neonatal rats was dissected, and the meninges were carefully removed and digested with 0.25% trypsin for 15 mins at 37° C. Trypsinization was stopped by adding an equal volume of culture medium (Dulbecco's modified Eagle's medium with 15% fetal bovine serum) to which 0.02% DNase I was added. The solution was pelleted, resuspended in culture medium, and a single-cell suspension was prepared by repeated pipetting. Cells were seeded at a density of 35,000 cells/cm2 in P-24 multiwell dishes and cultured at 37°C in humidified 5% CO2. The medium was replaced every 6 to 8 days and the cultures reached confluency after 7 to 10 days in vitro. After 10 to 15 days in culture, the number of astrocytes (GFAP+ cells) was about 80% to 90% of the total population of cells.
Hippocampal glial cultures were treated with 20 mmol/L FAc or solvent for 1, 3, 6, and 24 h at 11 DIV. At the end of the incubation, the culture medium was collected in two aliquots, one was used for high-performance liquid chromatography (HPLC) determination of the adenosine concentration and the other aliquot was used to evaluate the lactate dehydrogenase (LDH) activity. The cells were homogenized and used for protein determination.
Adenosine Determination
The samples were pretreated before the injection in the HPLC. The proteins were eliminated by acid treatment (perchloric acid) followed by centrifugation at 13,000 r.p.m. for 10 mins and the supernatants neutralized with KOH. The supernatants were submitted to chromatography through Accell QMA cartridges (from Waters, Milford, MA, USA). These single-use columns retain all the negatively charged nucleotides, permitting the elution of noncharged molecules (nucleosides). Eluates were processed by HPLC, as described in the following paragraph.
Aliquots (25 μL) were analyzed by HPLC on a system made up of a Waters 1515 isocratic HPLC pump, a 2487 dual-wavelength absorbance detector, and a 717 autosampler, which were all controlled by the Breeze software (Waters). The column used was a μbondaPak C18 (22 cm length, 0.4 cm diameter) and the mobile phase was 0.1 mol/L KH2PO4, 5% methanol (pH 6.0). The flow rate was 1.5 mL/min and the column was maintained at room temperature (22°C ± 1°C). Detection was monitored at a wavelength of 254 nm and the system was equilibrated overnight before the injection of the corresponding external standards. External standards were prepared with adenosine purchased from Sigma, prepared in ultrapure water. The quantification of adenosine in the samples was performed by comparing the areas of the peaks obtained in the samples with the corresponding areas of the external standards (Breeze software).
To confirm that the peak that elutes with the same retention time as the standard was actually adenosine, a 24 h sample was enriched with 1 mmol/L commercial adenosine and submitted to chromatography. The analysis of the chromatographic profile showed a single peak with increased size, suggesting the substance contained in the sample was adenosine (Figure 3A, middle panel). To further verify the identity of the measured compound, a sample containing the putative adenosine peak was incubated in the presence of 5' adenylic acid deaminase from Aspergillus sp, which transforms adenosine into inosine. The enzyme (0.12 U) was incubated with the 24 h sample in a final volume of 1 mL of 50 mmol/L HEPES (pH 6.5). Incubation was carried out at 37°C for 1.5 h. The HPLC analysis clearly showed that the compound was adenosine as there was a reduction in the putative adenosine peak with a concomitant increase in the inosine peak (Figure 3A, lower panel).
Electrophysiology
Monopolar stimulating electrodes (40 μm tungsten wires) were placed at the alvear region and the lower third of the stratum radiatum to elicit anti- and orthodromic (synaptic) responses, respectively. Two recording pipettes filled with 150 mmol/L NaCl (3 to 6 MΩ) were placed along the main CA1 pyramidal cell axis to record from the same population. One was located at the stratum pyramidale to record the population spike (PS) and the other at the stratum radiatum to record the field excitatory postsynaptic potential (fEPSP; Canals et al, 2005a). A third pipette was located at the stratum pyramidale of the CA3 region to record the antidromic PS from this region after Schaffer stimulation in the stratum radiatum. The collision test was used to assess whether the ortho- and antidromic stimulating electrodes were essentially activating the same population in the CA1 (Lopez-Aguado et al, 2002). Suitable stimulating/recording arrangements were chosen so that the antidromic PS totally collided with a previous orthodromic PS. After filtering (DC: 5 kHz) and amplifying, signals were recorded on VCR, acquired by a computer (20 to 40 kHz acquisition rate, Digidata 1200; Axon Instruments, Burlingame, CA, USA) and processed using the Axoscope and Clampfit software (Axon Instruments).
In a group of experiments, intracellular recordings were also made using micropipettes (1.5 mm o.d.) backfilled with 4 mol/L potassium acetate (80 to 100 MΩ). The signals obtained were amplified using a bridge circuit amplifier (Axoclamp 2A; Axon Instruments), filtered at 10 kHz, and stored in a VCR for offline analysis. Impalements were made from the somata of electrophysiologically identified pyramidal cells at least 80 μm below the slice surface.
Measurement and Statistics
The antidromic PS in the stratum pyramidale of both the CA1 and CA3 regions was measured as the amplitude of the peak of its negative limb with respect to the preceding baseline. When the artifact partially overlapped the evoked potential, the most positive value between them was used instead. The orthodromic PS in the CA1 was measured as the amplitude from the preceding positive crest and negative peak. The fEPSP was measured as the slope of its negative phase. Data were quantified as the mean±s.e.m. Statistical comparisons were made using the Student's t–test. Multiple comparisons were made with one-way analysis of variance followed by the Newman-Keuls test as a post hoc evaluation. Differences were considered statistically significant when P < 0.05.
Results
The Impairment of Glial Oxidative Metabolism Blocks Synaptic Transmission in the CA1 and CA3 Regions of the Hippocampus
We first characterized the short-term effects of FAc treatment on the electrophysiological properties of pyramidal neurons in the CA1 and CA3 regions of the hippocampus using three recording (R1-3) and two stimulating (S1,2) electrodes in the hippocampus (Figure 1A). The ortho- and antidromic field responses were recorded before and 30 mins after 20 mmol/L FAc treatment (black versus gray tracings, respectively, from a representative experiment; Figure 1B) and the time course of the fEPSP, ortho-and antidromic PS in the CA1, and the antidromic PS in CA3 was established from the pooled data for 20 mmol/L FAc (n = 19 slices, 9 animals; Figure 1C). Synaptic transmission in the hippocampus was strongly depressed after a 30 mins perfusion of FAc. Indeed, the orthodromic PS recorded in the CA1 soma layer and the fEPSP in the stratum radiatum of CA1 decreased to 98.6%±0.5% and 81.4%±6.7% of their control values, respectively (Figure 1C). Orthodromic transmission in the CA3 region also declined, as noted by the selective blockage of the second PS because of recurrent excitation (Figure 1B, arrowhead). This effect commenced 10 to 15 mins after FAc treatment, and once it began, the depression of the orthodromic PS was complete within 10 mins. In sharp contrast, the antidromic field responses in CA1 and CA3 were largely unaffected, as was the fiver volley recorded in the stratum radiatum (arrows in R1-S2 and R3-S1 and asterisk in R2-S1, respectively). Indeed, only a small increase in their amplitude was noted from the onset of the decline in orthodromic transmission (Figures 1B and Figure 1C). Similar blockage of synaptic transmission, and intact action potential initiation and conduction, was observed in intracellular recordings from the CA1 pyramidal cells (Figure 1D). In addition, the intracellular injection of depolarizing current pulses (+ 0.2 nA, 1 sec) elicited very similar responses both before and up to 90 mins after FAc perfusion (in the neuron shown in Figure 1D). A progressive decline of antidromic transmission was only observed after 40 to 90 mins (59±24 mins, mean±s.d.) of FAc treatment, concomitant with the appearance of the first SD waves (see below). These results are in agreement with previous studies using FAc, both in preparations in vitro and in vivo (Stone et al, 1990; Keyser and Pellmar, 1994, Largo et al, 1996; Larrosa et al, 2006), and indicate that the selective impairment of glial oxidative metabolism inhibits synaptic transmission without interfering with the electrogenic membrane function.
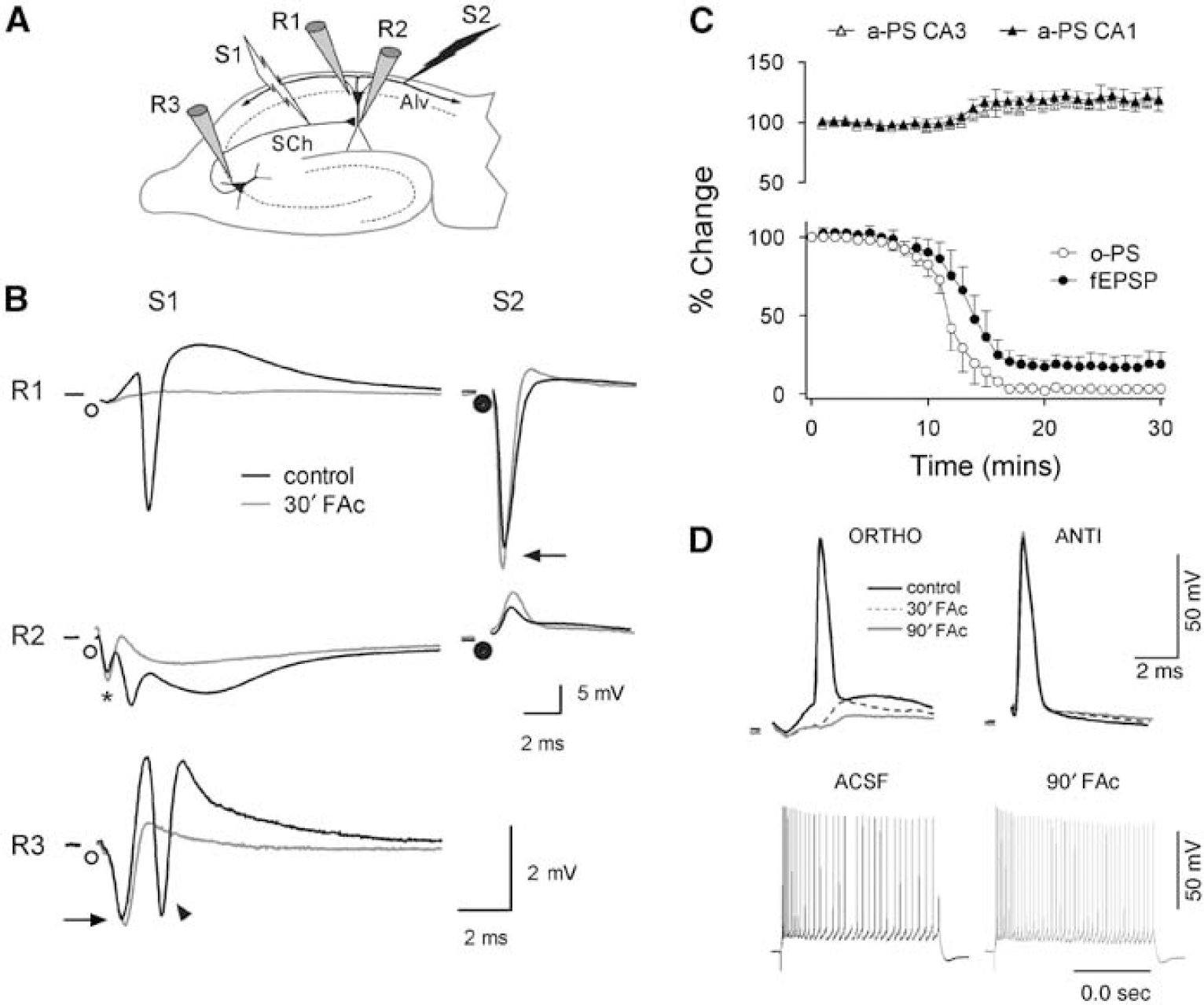
Inhibition of oxidative metabolism in glia blocks synaptic transmission without producing changes in the electrogenic properties of neurons. (
Blockage of Synaptic Transmission is because of the Activation of A1 Adenosine Receptors
Adenosine accumulates in the extracellular space because of the inhibition of mitochondrial respiration during hypoxic and chemical hypoxia (Pull and McIlwain, 1972; Gribkoff et al, 1990; Frenguelli et al, 2007). Hence, adenosine accumulation because of the selective glial intoxication may be responsible for the fast interruption of synaptic transmission in the present model. We tested the effects of general and A1-selective antagonists of adenosine receptors on the blockage of orthodromic potentials in the hippocampus during glial cell intoxication. Synaptic transmission was recovered by adenosine antagonists in both the CA1 and CA3 regions of the hippocampus. This recovery was evident through the temporal evolution of the field potentials, and their quantification on exposure to FAc treatment, both before and after selective inhibition of A1 receptors with DPCPX, is shown in Figures 2A and Figure 2B. A similar effect could be seen in the pooled data for DPCPX (eight slices in five animals) and the general adenosine receptor antagonist 8-SPT (eight slices in five animals; Figure 2C). Likewise, recovery of synaptic transmission was obtained when the slices were exposed to 10 mmol/L FAc in the presence of adenosine antagonists. Indeed, the blockage of synaptic transmission was also prevented when the slices were pretreated with DPCPX or 8-SPT before FAc perfusion (data not shown).
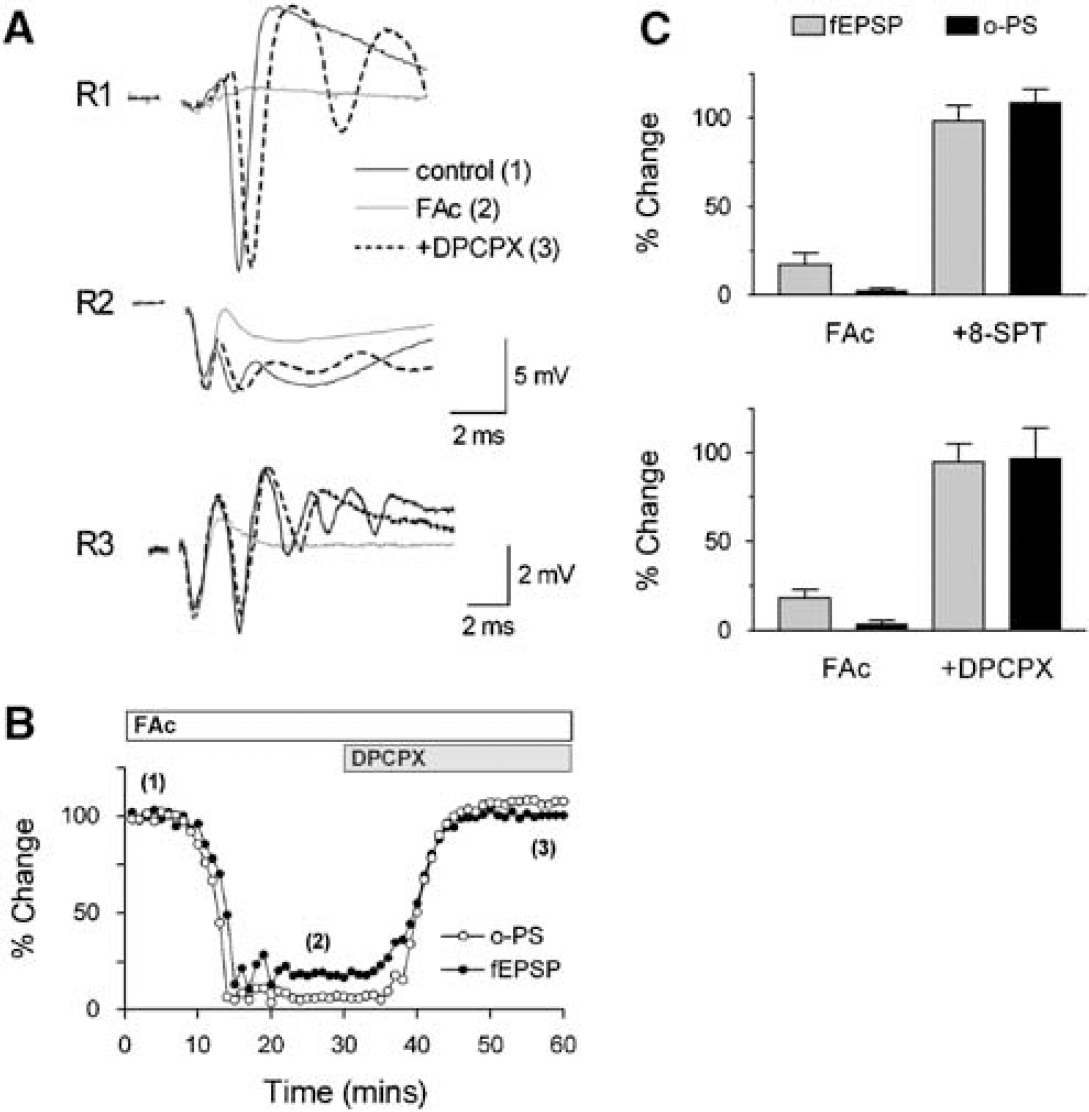
Adenosine A1 receptor antagonism restores synaptic transmission in the hippocampus. Representative tracings (
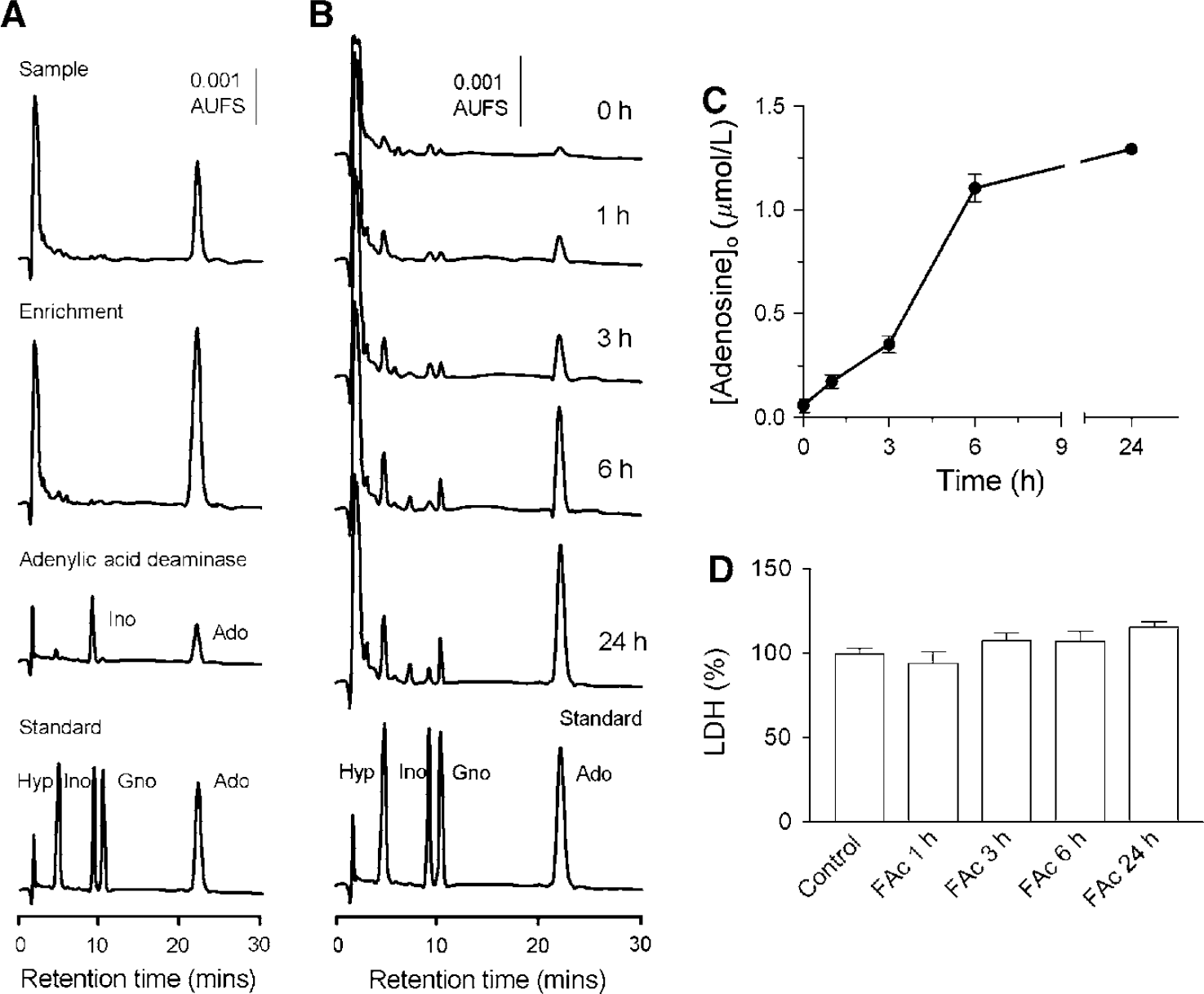
Glial cell-mediated extracellular accumulation of adenosine in response to mitochondrial inhibition. Hippocampal glial cells in culture were exposed to FAc (20 mmol/L) for different periods of time. (
These results show that glial respiration regulates synaptic transmission through adenosine binding to A1 receptors. This mechanism affects different synapses within the hippocampus, as Schaffer collateral-evoked fEPSPs in the stratum radiatum of CA1 and synaptic recurrence in CA3 are regulated in the same way.
Glial Cells and Extracellular Adenosine in Response to Mitochondrial Inhibition
Having shown the mechanistic role of adenosine in synaptic blockade during glial intoxication, we next studied the ability of glial cells in releasing adenosine in response to mitochondrial inhibition. To address this question we cultured glial cells from rat hippocampus and incubated them with 20 mmol/L FAc for different periods of time. We chose the cell culture model to unequivocally show the capability of glial cells from the hippocampus to accumulate adenosine in the extracellular medium in response to a metabolic challenge, independent of the neuron-glia interplay. In addition, the cell culture model allowed us to assay adenosine concentrations in a very robust and reproducible way. The cultures were first characterized immune-cytochemically to show that they consisted mainly of astrocytes (80% to 90% GFAP-positive cells) and almost no neurons were present (less than 1% to 2% β;-tubuline-positive cells). Moreover, cell membrane integrity (LDH release) was assessed in parallel with adenosine determinations. On exposure to FAc, adenosine clearly accumulates in the extracellular space from the first evaluated time point (300% increase) and rises in a time-dependent manner (Figure 3B and Figure 3C). This accumulation was not accompanied by LDH release into the culture medium and thus could not be attributed to the breakdown of the plasma membrane (Figure 3D). Although the time course of adenosine accumulation in a closed system (cell culture) cannot be directly compared with that of an open one (superfused brain slices), these data clearly show that glial cells accumulate adenosine in the extracellular space when their oxidative metabolism is impaired. Our results are in good agreement with previously published data on glial cell cultures showing adenosine accumulation under metabolic stress (Parkinson and Xiong, 2004).
The Glial-Mediated Accumulation of Extracellular Adenosine Delays or Prevents the Occurrence of Lethal SD Waves
Spreading depression waves develop spontaneously when the glial oxidative metabolism is inhibited for long periods leading to neuronal cell death in the hippocampus (Nedergaard and Astrup, 1986; Hossman, 1996; Largo et al, 1996, 1997; Lian and Stringer, 2004). If the blockage of synaptic transmission represents a safety mechanism controlled by glial cells to allocate energy resources to vital processes, then antagonism of A1 receptors should precipitate such processes. We tested this prediction by increasing the time of FAc perfusion (up to 2 h) and evaluating the electrical viability of neurons (n = 6 slices from three rats). Longer exposure to FAc in our model led to the appearance of SD waves (Figure 4) and a decrease of the antidromic PS amplitude (75.6% ± 11.3% of the control 1 h after exposure to FAc). Stepwise reductions in the antidromic PS were observed after each SD episode, and the amplitude of the antidromic PS was inversely correlated with the number of SD events (data not shown). This effect can be appreciated through the evolution of the antidromic PS on exposure to FAc (Figure 4A), where the experiments in which SD waves developed or not can be compared. In the presence of A1 blockers, the restoration of synaptic transmission was accompanied by a hastening of the SD waves (Figure 4B), an increase in its frequency (Figure 4C), and a marked reduction in the electrical viability of pyramidal neurons (Figure 4D). Thus, the antagonic effect of adenosine on SD occurrence during glial intoxication represents a fundamental protective mechanism for neurons.
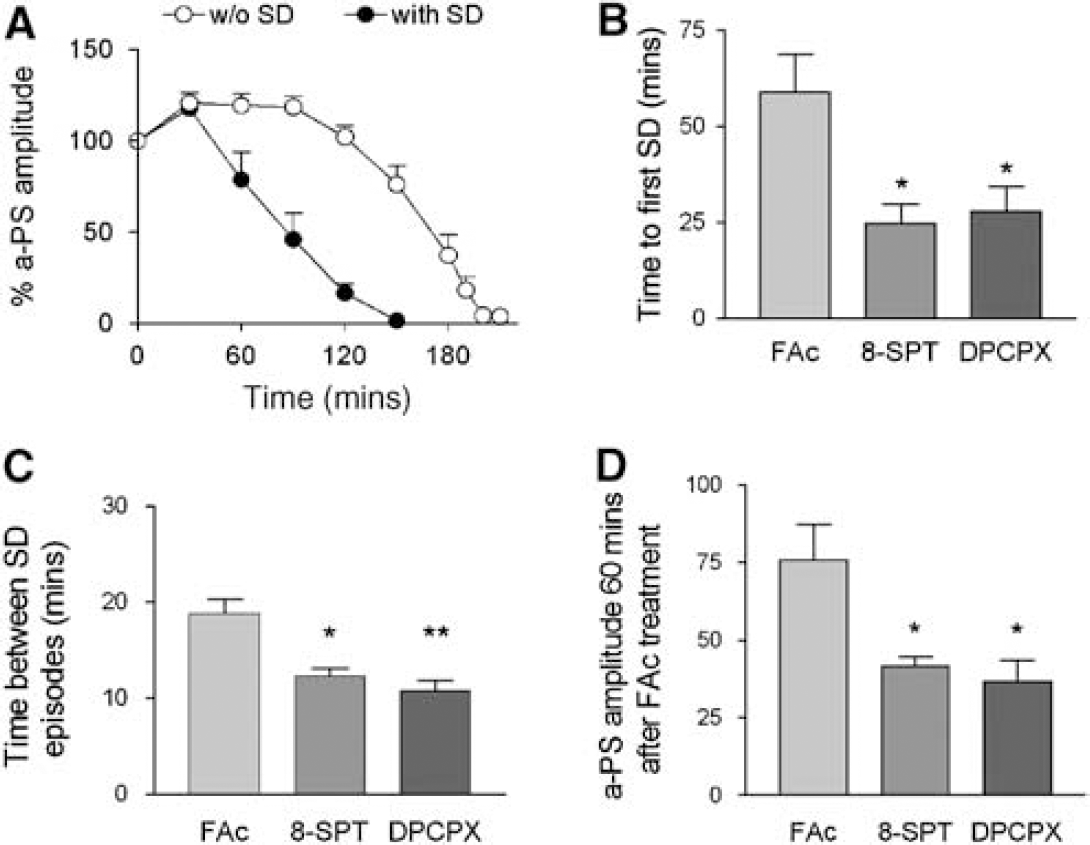
The glial-dependent accumulation of adenosine is a safety mechanism that preserves neuronal viability by opposing SD waves. Cell viability in CA1 is quantified by the amplitude of maximal antidromic PS during the perfusion of FAc (20 mmol/L) (
This phenomenon is also illustrated in Figure 5, where the effects of A1 antagonism were investigated well after the inhibition of glial respiration. In this experiment, FAc treatment produced the described effects on synaptic and antidromic transmission with a stepwise reduction in the a-PS (black arrow) only after the occurrence of a SD episode (black asterisk). The subsequent administration of DPCPX, more than 90 mins after the FAc treatment started, was still very efficient in recovering the synaptic transmission (gray arrow); however, the recovery process was curtailed by the immediate precipitation of a second SD episode (gray asterisk), which was now lethal for the tissue (note the complete abolition of the antidromic response).
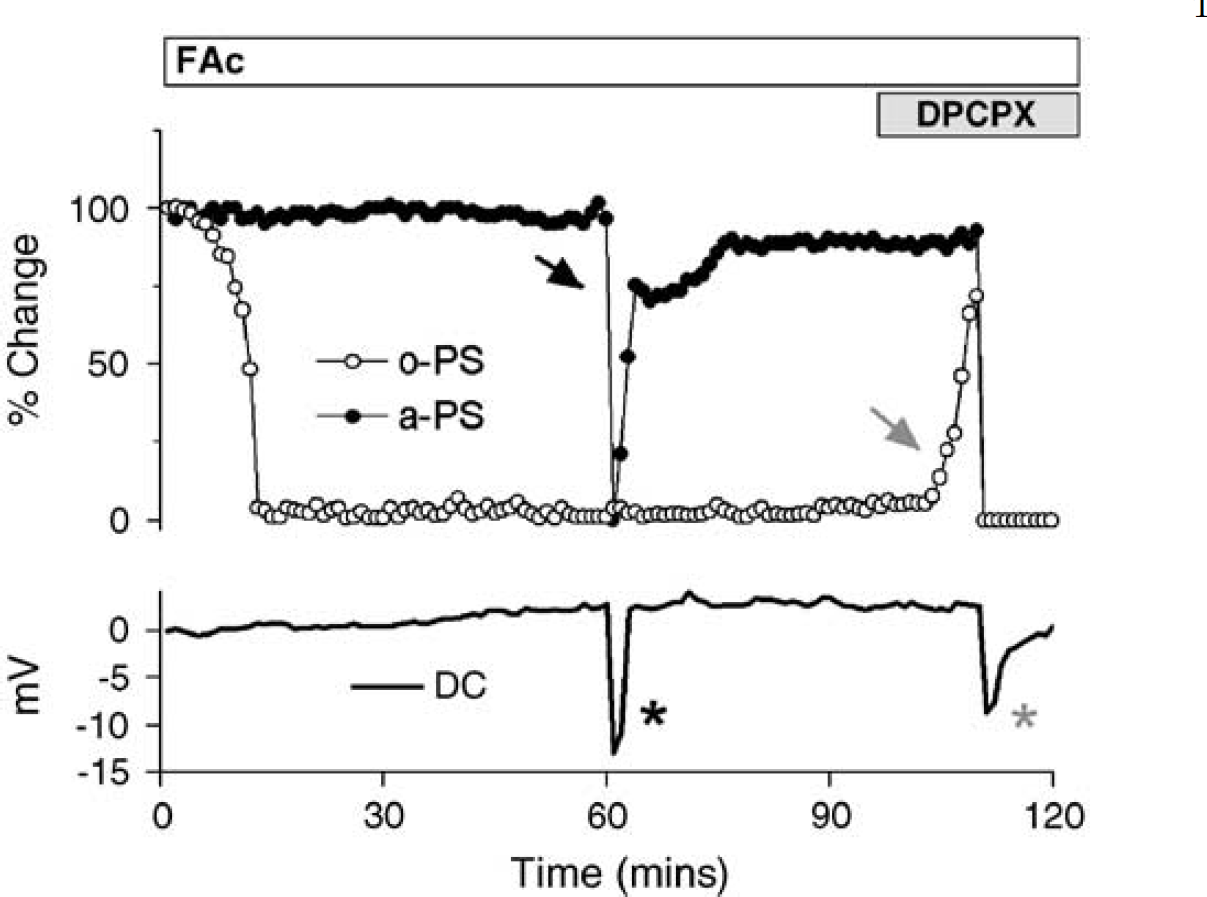
Adenosine released in response to glial intoxication preserves neuronal viability. The time course of the evoked field potentials (anti- and orthodromic PS, black and white circles, respectively) and DC potential (black line) in CA1 during FAc perfusion are shown. At 95 mins of FAc treatment, 0.1 μmol/L DPCPX was introduced in the perfusion medium (gray bar). Note the occurrence of two SD waves, one in a period of only FAc treatment (black asterisk), inducing a transient inactivation of the antidromic transmission (during the time length of the SD episode; black arrow) and followed by the partial recovery of the a-PS amplitude; and a second one after DPCPX inclusion, producing the permanent abolition of electrical activity in the slice. The synaptic transmission was also recovered by the A1 antagonist, shortly before the ignition of the second and lethal SD event.
Discussion
The main finding of this study is that inhibition of oxidative metabolism in glial cells produces an adenosine A1-receptor-mediated blockage of synaptic transmission in the hippocampus, which delays the appearance of SD waves and preserves neuronal viability. We interpret these results as representing a glial-operated safety mechanism that allocates metabolic resources to vital processes when the energetic capacity of the tissue (or the capacity sensed by glial cells) is surpassed. In turn, this delays the onset of SD waves that are lethal to neurons in regions of glial malfunction, increasing the time window for survival.
Specificity of Glial Toxins
Numerous reports indicate that FAc and fluorocitrate are selective inhibitors of glial metabolism. Not only are they selectively taken up by astrocytes (Clarke et al, 1970; Waniewski and Martin, 1998), their initial effects are also unambiguously related to the impairment of functions commonly attributed to glial cells. These include the regulation of pHo, spatial potassium buffering, and the metabolic Glu—glutamine cycle (Berg-Johnsen et al, 1993; Largo et al, 1996; Stringer and Aribi, 2003). Also, their exogenous application causes the gradual selective depolarization of astrocytes (Largo et al, 1997; Hulsmann et al, 2000). In contrast, these toxins have no effect on the antidromic PS (Stone et al, 1990; Keyser and Pellmar, 1994; Largo et al, 1996, 1997; Larrosa et al, 2006) or on the membrane properties of pyramidal neurons (Larrosa et al, 2006; Hulsmann et al, 2000). The neuronal demise observed after long-term exposure to these inhibitors (several hours) is therefore secondary to the disruption of glial homeostatic activity, the unavoidable perturbation of the reciprocal neuro-glia metabolic and signaling pathways, together with the occurrence of SD waves (Largo et al, 1996; Lian and Stringer, 2004).
Mechanisms of Synaptic Blockage
It is generally agreed that the loss of synaptic transmission on FAc treatment is produced by the exhaustion of the neurotransmitter pool of glutamate because of the inhibition of the glutamine/glutamate cycle in glial cells (Quastel, 1978). This view is supported by the experimental observation that the glutamine precursor is depleted after FC treatment (Largo et al, 1996), and that exogenous glutamine maintains synaptic potentials in brain slices (Keyser and Pellmar, 1994). However, this interpretation is weakened by the fact that glial-derived glutamine is not the only source of the neurotransmitter glutamate, as it can also be synthesized from glucose in neurons (Ward et al, 1983). Furthermore, the highly variable rate and degree of synaptic depression observed in different studies (from 30 mins to 5 h and from total suppression to only 85%; Berg-Johnsen et al, 1993; Keyser and Pellmar, 1994; Largo et al, 1996; Martin et al, 2007) raise some doubt whether the lack of transmitter is the main cause of synaptic failure during glial poisoning. One may argue that because of the compartmentalization of metabolites, only the neurotransmitter glutamate pool is affected by glial intoxication and that this represents a small fraction of the total glutamate pool (Hamberger et al, 1979). Even if this was the case, it is also known that glutamine can feed the tricarboxylic acid cycle, bypassing the FAc inhibition (Hassel et al, 1994) and, thus, restoring the energetic status of the glial cells. Indeed, glutamine supplementation prevents the decrease in ATP levels induced in glial cell cultures by FAc (Hassel et al, 1994). Based on the above evidence, it does not appear that neurotransmitter depletion is responsible for the blockage of synaptic transmission during glial poisoning or vice versa. Our results clearly showed that the glutamate pool is available for synaptic transmission during FAc treatment (at least for the length of our experiments, some up to 3 h), as synaptic transmission is restored shortly after it has been fully blocked, and that the mechanism for synaptic blockade is, indeed, the accumulation of extracellular adenosine and subsequent activation of A1 receptors in the pyramidal neuron.
A variety of manipulations that alter energy metabolism, such as oxygen and/or glucose deprivation and chemical anoxia, have been shown to produce adenosine accumulation in the interstitial space as well as adenosine-mediated blockage of synaptic transmission (Latini and Pedata, 2001). Adenosine could be released from both pre- and postsynaptic neurons, as well as from nonneuronal cells. Normally, adenosine accumulation is thought to arise from neural elements by either direct release or through the extracellular metabolism of released nucleotides (Cunha, 2001; Latini and Pedata, 2001). However, our results, together with the data reported by others (Parkinson and Xiong, 2004; Martin et al, 2007), indicate that astrocytes are a source of adenosine. The adenosinergic blockage of synaptic transmission reported here is by far the earliest noticeable effect of FAc challenge (~ 5 to 10 mins) well before any indication of altered ion homeostasis, which occurs in vivoafter at least 1 to 2 h. Thus, it is likely that adenosine is released directly from astrocytes as a direct response to the blockage of the tricarboxylic acid cycle. This interpretation is also supported by the experiments in glial cultures, which show that in absence of neurons, glial cells produce considerable amounts of adenosine when challenged with FAc.
Glial-Mediated Accumulation of Extracellular Adenosine Provides Neuroprotection by Delaying the Occurrence of SD Waves: Implications for Ischemic Brain Injury
For the first time, we have shown that glial-mediated interruption of synaptic transmission is a reversible phenomenon that delays the appearance of SD waves, decreases their frequency, and preserves the electrical viability of pyramidal neurons. It is well known that neuronal damage at the core of a brain infarct cannot be reversed. Massive depolarization is accompanied by the breakdown of membrane integrity and the release of a number of neuroactive agents. However, the surrounding region, the so-called ischemic penumbra, is initially viable and only moderate signs of neuron distress can be detected (Hossman, 1996). A causal relationship between the SD waves arising from the borders of the ischemic nucleus and the neuronal death in the surrounding penumbra has already been established (Iijima et al, 1992; Fabricius et al, 2006). It was previously shown that energy shortage in glial cells is a necessary condition for SD waves to kill the neurons. Accordingly, it was proposed that SD waves and glial dysfunction together are responsible for cell death in the ischemic penumbra (Largo et al, 1996). Here, we show that the specific metabolic stress of glia is by itself a reliable trigger of SD waves, and that their occurrence, as well as the subsequent loss of neuronal function, can be delayed by the activation of A1 receptors. In the context of brain ischemia, we consider that the release of adenosine by glia could function as a protective response against cell death, diverting energy from processes that are not essential for cell survival to vital processes, and delaying the occurrence of lethal SD waves in the ischemic penumbra. Such a mechanism could increase the survival time window of any brain region confronted by an ischemic episode.
It is also well established that glial cells participate in the regulation of cerebral blood flow (Harder et al, 1998) and that adenosine fulfils a key role in the so-called neurovascular coupling (Phillis, 2004). Together, these observations suggest that by concomitantly decreasing synaptic transmission and increasing local cerebral blood flow, adenosine from glial origin could restore the ratio of energy production and consumption during periods of energy imbalance in vivo. It is also plausible that in the ischemic penumbra, adenosine release alleviates hypoxic episodes (Lauritzen et al, 1982; Shin et al, 2006; Strong et al, 2007) and the excessive workload associated to the passage of peri-infarct depolarizations. Hence, the neuroprotective mechanism described here may be envisaged as a safeguard against the otherwise lethal outcome of breaking the normal interplay between neurons and glia when these are unable to maintain the normal flow of energy metabolites. In vivo studies have already shown interferences between endogenous adenosine and the generation of SD waves (Kaku et al, 1994). Further studies will be required to understand the mechanism of such an interaction, which is of particular clinical relevance as a potential new target for the treatment of ischemic and traumatic brain injury.
Footnotes
Acknowledgements
We thank Rosa Solano for help with cell cultures, Eduardo Martin for critical reading of the paper, Alfonso Araque for helpful comments, and Mark Sefton (BiomedRed SL) for editorial assistance.
The authors declare no conflict of interest.