
Abstract
Recent evidence suggests that stress-activated protein kinases expressed in glial cells have very important roles during cerebral ischemia. The neuroprotective agent chlomethiazole, which is known to enhance the conductance at the GABAA receptor complex, is presently in clinical trials for the treatment of severe stroke. Here the authors suggested that chlormethiazole has anti-inflammatory properties because it potently and selectively inhibited p38 mitogen-activated protein (MAP) kinase in primary cortical glial cultures. The inhibition of p38 MAP kinase resulted in the attenuation of the induction of c-fos and c-jun mRNA and AP-1 DNA binding by lipopolysaccharide (LPS). In addition, chlomethiazole inhibited the activation of an AP-1-dependent luciferase reporter plasmid in SK-N-MC human neuroblastoma cells in response to glutamate. Chlomethiazole inhibited the p38 MAP kinase activity as revealed by the decrease in the LPS-induced phosphorylation of the substrates ATF-2 and hsp27, whereas the phosphorylation status of the p38 MAP kinase itself was unaffected. Interestingly, chlomethiazole exhibited an IC50 of ~ 2 μmol/L for inhibition of c-fos mRNA expression, indicating 25 to 75 times higher potency than reported EC50 values for enhancing GAB AA chloride currents. The results indicated a novel mechanism of action of chlomethiazole, and provided support for a distinctive role of p38 MAP kinase in cerebral ischemia.
The p38 and c-Jun N-terminal kinases (JNK) subgroups of mitogen-activated protein (MAP) kinases are important transducers of stress signals in cells and have been implicated in regulation of cytokine release and in cell death mainly of apoptotic nature (Karin, 1998). JNKs and p38 MAPKs were shown to be activated downstream of cytokine receptors, such as tumor necrosis factor (TNF) and interleukin-1 (IL-1) receptors (Saklatvala et al., 1996; Yuasa et al., 1998), and other forms of stress like UV irradiation, H2O2, osmotic stress, and ischemia—hypoxia (Woodgett et al., 1995; Mizukami et al., 1997). In particular, p38 MAP kinase activity, a target of a group of cytokine-suppressant anti-inflammatory drugs (CSAIDs), such as the pyridinyl imidazole compound SB203580 (Lee et al., 1994; Kumar et al, 1999), has been correlated with apoptotic cell death in cerebellar granule cells and other neuronal cells in response to glutamate (Xia et al., 1995; Kawasaki et al., 1997), and its inhibition resulted in protection of cultured cardiac myocytes from ischemic injury (Mackay and Mochly-Rosen, 1999). In a recent review, Barone and Feuerstein (1999) raise the therapeutic potential for p38 MAP kinase inhibitors in stroke because of the inhibition of the production of the inflammatory cytokines IL-1β and TNF-α, and their effect against neuronal apoptosis.
Only recently has the role of inflammatory mediators in ischemic injury in cell death and survival been realized. Direct trauma, ischemia, neurotoxicity, viral or bacterial infection, or immunologic challenge share a common denominator—the brain's inflammatory response, namely activated glial cells which secrete cytokines. For instance, bacteria-derived lipopolysaccharide (LPS) triggers the inflammatory response by activating the transcription factor NF
Lipopolysaccharide administration also induces, through MAP kinases, the expression of the immediate early genes fos and jun resulting in the activation of the transcription factor AP-1 (Han et al, 1997; Hazzalin et al, 1997). Both fos and jun family genes have been studied in models of experimental ischemia, mostly in rodents, in which inhibition of c-fos expression by antisense oligonucleotides and targeted disruption of the JNK-3 gene have resulted in a substantial degree of neuroprotection from excitotoxic insults (Chiasson et al., 1997; Lu et al., 1997; Yang et al., 1997).
Chlomethiazole (CMZ) is a hypnotic sedative and anxiolytic agent (Cross et al., 1989; Moody and Skolnick, 1989) that has neuroprotective properties in several models of focal and global ischemia in rodents (reviewed by Green, 1998). Most importantly, CMZ has been efficacious when administered four hours after the ischemic insult. Recently, a report of a clinical trial of CMZ suggested a clinical benefit of treatment with the compound of patients with severe stroke (Wahlgren et al., 1999).
The authors reported here that CMZ attenuates the activation of c-fos/c-jun/AP-1 through inhibition of p38 MAP kinase activity. In view of the important roles of p38 MAP kinase in ischemic injury, the authors suggested that the anti-inflammatory properties of CMZ might be of relevance for the neuroprotection seen by CMZ in vivo.
MATERIALS AND METHODS
Chemicals
Chlomethiazole was a gift from Dr. Ylva Terelius, AstraZeneca, Södertälje, Sweden. Lipopolysaccharide (from E. Coli 0127:B8) and TPA were purchased from Sigma, St. Louis, MO, U.S.A. SB203580 (4-(4-Fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole) was purchased from Calbiochem, CA, U.S.A.
Cell culture
Rat primary cortical glial cultures were established from cortices of newborn rats (6 to 12 hours) as previously described (Tindberg et al., 1996). The cells were grown in minimum essential medium supplemented with 20% fetal calf serum, amino acids, vitamins, D-glucose (5 mmol/L), penicillin (100 IU/mL), and streptomycin (100 μg/mL). SK-N-MC human neuroblastoma cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, U.S.A.) and grown in minimum essential medium supplemented with 10% fetal calf serum, 2.2 g/L of sodium pyruvate, 0.1 mmol/L nonessential amino acids, penicillin (100 IU/mL), and streptomycin (100 μg/mL). All media for cell culture were from Life technologies Rockville, MD, U.S.A. Primary cortical glial cells were fixed with 2% paraformaldehyde and incubated with two different antibodies reactive towards glial fibrillary acidic protein (GFAP; Dakopatts A/S, Denmark) or CD11b (Biosource international); subsequently, a secondary TRITC-labeled antibody (Boehringer Mannheim, Germany) was used. Cells were also stained with propidium iodide and the primary cortical glial cultures were found to contain ~85% GFAP+ cells (astrocytes) and ~3% CD11b+ cells (microglial cells). All experiments involving the establishment of primary cortical glial cultures were approved by the ethical committee for animal studies at Karolinska Institutet.
Quantitative RT-PCR and Northern blot
Total RNA was isolated from primary cortical glial cells cultured on 150-mm culture dishes by the guanidium isothiocyanate method (Chomczynski and Sacchi, 1987). The RNA was subjected to electrophoresis on a 1.2% agarose—formaldehyde denaturing gel (40 μg per lane), transferred onto nitrocellulose filter, and hybridized with radiolabeled cDNA probes for c-fos, c-jun, and β-actin, as described by Struhl (1993b). The probes were obtained by polymerase chain reaction (PCR) amplification of cDNA derived from the same cells and were labeled with (α-32P(dCTP (3000 Ci/mmol/L, Amersham, U.K.) using the Radprime DNA labeling system (Life). The primers used to obtain the probes were the same as for the quantitative PCR analysis (see below).
For the quantitative reverse transcriptase polymerase chain reaction (RT-PCR) analysis, 1 μg of total RNA was reverse transcribed using an oligo-dT primer (18-mer). The RT reaction was performed at 42°C using the 1st™ strand cDNA synthesis kit (CLONTECH). Quantitative PCR analysis of the relative amount of the c-fos or c-jun mRNA in differently treated primary cortical glial cells was performed using the PCR mimic 228 kit (CLONTECH, Palo Alto, CA, U.S.A.). The primers used in the PCR analysis corresponded to the exon 1 DNA sequences of the rat c-fos gene, 5'- CTT GAA GAC GAG AAG TCT GCG −3' for the upstream primer, and 5'- GGT CAT TGA GAA GAG GCA GG −3' for the downstream primer (also described by Yu et al., 1995). The c-fos cDNA fragment, 226 bp long, was amplified in the presence of decreasing concentrations of a 546 bp DNA standard (mimic DNA, constructed by PCR amplification of a BamHI- EcoRI restriction fragment of the v-erbB gene, as described in the PCR mimic™ construction kit). The primers for c-jun corresponded to the following DNA sequences in exon 1 of the rat c-jun gene: 5' - AAC AGA TCC CGG TGC AGC AC − 3' for the upstream, and 5' - CCA CCT GTT CCC TGA GCA TG −3' for the downstream primer. The c-jun fragment of 279 bp, was coamplified with a mimic DNA fragment of 546 bp as described also for c-fos. The PCR amplification was conducted in a Perkin Elmer amplifier (Perkin Elmer, Norwallk, CT, U.S.A.) under the following thermal cycle conditions: 45 seconds denaturing at 94°C, 45 seconds annealing at 57°C, and 1 minute extension at 72°C for 37 cycles, followed by 7 minutes final extension at 72°C. The PCR products were analyzed on a 1.6% agarose/TBE gel, and the negatives of Polaroid photographs (Polaroid, Cambridge, MA, U.S.A.) of the Ethidium Bromide-stained gels were scanned using a laser densitometer (Molecular Dynamics, U.S.A.). The levels of c-fos and c-jun mRNAs were calculated as the concentration of the mimic DNA required to reach an equal density of the target and mimic bands by examining the relative intensities between target and mimic in reactions carried out at 3 to 4 different mimic DNA concentrations. The IL-1β primers used corresponded to the following sequences of the rat IL-1β cDNA: 5'- GAC CTG TTC TTT GAG GCT GAC −3' and 5'- TTC ATC TCG AAG CCT GCA GTG −3' for the upstream and the downstream primer respectively, and the quantitation procedure followed the same principles as described above. The TNF-α primers used corresponded to the following sequences in the rat TNF-α gene: 5'-ACG CTC TTC TGT CTA CTG-3' (exon-1) and 5'-GGA TGA ACA CGC CAG TCG-3' (exon 4) for the forward and the reverse primer respectively. The results of all PCR quantitations were normalized by the β-actin content of each sample which was also quantified by RT-PCR mimic system (for primer sequence and experimental setup see Fang et al., 1998).
Cell extracts and Western blot
Rat primary cortical glial cultures were stimulated with LPS (100 ng/mL). After stimulation, cells were immediately put on ice, washed, and harvested in lysis buffer containing 20 mmol/L Tris-HCl pH 7.4, 137 mmol/LNaCl, 10% glycerol, 1% Triton X100, 1 mmol/L Na3VO4, 5 mmol/L NaF, 10 mmol/L Na2MoO4, 2 mmol/L EDTA and 1 mmol/L phenylmethyl sulphonylfluoride (PMSF). Lysed cells were left at +4°C for 30 minutes with slight shaking, followed by centrifugation of the material for 15 minutes at 10.000x g, at +4°C. The protein content of the Triton X-100 soluble fractions was determined according to the Lowry method. Proteins were separated in a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a Bio-Rad Protean II apparatus at 100 V (Biorad, CA, U.S.A.). The proteins were subsequently transferred to nitrocellulose. After the transfer, the filters were washed in phosphate buffered saline, pH 7.4, dried, and blocked in 5% dry milk in Tris buffered saline, pH 7.4 containing 0.2% Tween 20 (TBS-tween) overnight at +4°C. Filters were then either incubated for 2 hours at room temperature or overnight at +4°C with an antiphospho-JNK or antiphospho-p38 antibody, or an antibody to phosphorylated extracellular signal regulated kinase (ERK) (Calbiochem) diluted 1/1000 in TBS-tween with 5% bovine serum albumin. The filters were subsequently washed three times and incubated for 120 minutes with a horseradish peroxidase secondary antibody (Dakopatts A/S) diluted 1/2000 in TBS-tween with 5% milk. After thorough washing in TBS-tween (5 times for 30 minutes), proteins were visualized with enhanced chemiluminescence (Amersham, U.K.). Molecular weight standards used for molecular weight estimations were from Bio-Rad (Hercules, CA, U.S.A.).
In vitro phosphorylation assays and immunocomplex kinase assay
GST-c-Jun fusion protein (amino acids 1 to 79) and recombinant hsp27 were obtained from Calbiochem, and Stressgen Biotechnologies, Victoria BC, Canada, respectively, and phosphorylated as described by Sanghera et al. (1996). Thirty to forty μg protein from cell extracts in 40 μL of lysis buffer (described above), were incubated with 1 μg of GST-c-Jun fusion protein or 0.5 μg recombinant hsp27, 10 mmol/L MgCl2, 1 mmol/L MnCl2, 50 μmol/L ATP, and 6 μCi [32P]-γATP (3000 Ci/mmol·L; Amersham Pharmacia Biotech, Sweden) in a volume of 50 μL. After samples were incubated for 30 minutes at +30°C, the reaction was stopped with the addition of SDS loading buffer. The proteins were separated on a 12% SDS-PAGE and transferred to supported nitrocellulose. Membranes were exposed to autoradiography film from 3 to 24 hours. Alternatively, the gel was dried and directly exposed to autoradiography film. For the immunocomplex kinase assay 500 μg protein from total cell extracts were immunoprecipitated for 2 hours at +4°C with the appropriate titer of an agarose-conjugated anti-p38 antibody (Santa Cruz, Santa Cruz, CA, U.S.A.) that was able to precipitate the active kinases. The immunocomplexes were washed twice with lysis buffer, and twice with 10 mmol/L Hepes, 10 mmol/L (CH3COO)2Mg containing 0.1 mmol/L Na3VO4, and then suspended in 35μL lysis buffer. The kinase reaction mix was then added as described above, and the immunoprecipitated active kinases were allowed to phosphorylate 1.6 μg of GST-ATF2 fusion protein (Santa Cruz) for 30 minutes at 30°C. The reaction was stopped by the addition of SDS-loading buffer, and the proteins were separated on a 10% SDS-PAGE which was then dried and subjected to autoradiography for 2 to 4 days. In some experiments, the immunocomplexes were directly electrophorized on SDS-PAGE, transferred to nitrocellulose, and blotted with an anti-p38 antibody (the same as used for the immunoprecipitations but not conjugated to agarose) to verify the equal immunoprecipitation efficiency for all the differentially treated samples. Alternatively, cells were labeled with 35S-methionine for 1.5 hours, treated with LPS ± inhibitors, and total cell extracts were prepared. After immunoprecipitation with an agarose-conjugated anti-p38 antibody, the 35S-labeled proteins were separated on SDS-PAGE, transferred to nitrocellulose, and subjected to autoradiography.
Electrophoretic mobility shift assays
Nuclear extracts were isolated essentially as described by Struhl (1993a) with minor modifications. In brief, the cells derived from two 150 mm petri dishes were washed twice and harvested in ice-cold phosphate-buffered saline, and then pelleted by centrifugation at 1,850× g for 10 minutes. The supernatant was discarded, and the cell pellet was washed once in 5 volumes of hypotonic buffer (10 mmol/L Hepes, pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.2 mmol/L PMSF, 0.5 mmol/L DTT). The cells were resuspended in 3 volumes of hypotonic buffer and allowed to swell on ice for 10 minutes before homogenization in a glass Dounce homogenizer. The nuclei were pelleted by centrifugation at 3,400x g for 15 minutes and resuspended in 250 μL of low salt buffer (20 mmol/L Hepes, pH 7.9, 25% glycerol, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.2 mmol/L EDTA, 0.2 mmol/L PMSF, 0.5 mmol/L DTT). Then, KCl 2.5 mol/L was added dropwise and under continuous mixing to a final salt concentration of 0.4 mol/L; the nuclei were incubated for 30 minutes at +4°C under gentle agitation. The lysed nucleic debris were pelleted by centrifugation at 25,000× g for 30 minutes at +4°C. The nuclear protein extracts remaining in the supernatant were aliquoted and immediately frozen at −70°C. Protein concentration was determined by the Bradford method. The reactions were carried out in a total volume of 20 μL with 4 to 8 μg of nuclear protein and a final concentration of 30 mmol/L Hepes, pH 7.9, 20% glycerol, 2.25 mmol/L MgCl2, 0.2 mmol/L EDTA, 0.12 mmol/L PMSF, 0.5 mmol/L DTT, 0.1 μg/μL poly[d(I-C) d(I-C). The final salt concentrations were 200 mmol/L KCl for AP-1 and 130 mmol/L NaCl for NF-κB incubated reactions. The double-stranded AP-1 or NF-κB probes were 32P-end-labeled using T4 polynucleotide kinase and [32P]-γATP. For competition controls, a 25- or 50-fold molar excess of the AP-1 and the NF-κB double-stranded oligonucleotides was used. The DNA-protein complexes were separated on a 4% nondenaturing polyacrylamide gel in Tris/glycine buffer (50 mmol/L Tris, 380 mmol/L glycine, 2 mmol/L EDTA) at +4°C. The protein components of the retarded AP-1 complex were identified by using specific antibodies against the c-Fos and c-Jun proteins, as well as a Jun antibody recognizing all members of Jun family (mainly c-Jun, and with lower affinity JunB and JunD) in the incubation reaction (the antibodies were purchased from Calbiochem, LaJolla, CA, U.S.A., and Santa Cruz, respectively). The NF-κB retarded complex was investigated using 1 μg per lane of p65 and p50 antibody (Santa Cruz). The probes used had the following nucleotide sequences: 5'- CGCTTGATGACTCAGCCGGAA − 3' for the consensus AP-1 probe, 5'-AGTTGAGGGGACTTTCCCAGGC-3' for the consensus NF
Transient transfections
SK-N-MC human neuroblastoma cells and primary cortical glial cultures were grown to approximately 50% to 60% confluence and were then transfected by means of DMRIE-C agent (Life). Cells were transfected in Optimem medium (Life) with a luciferase reporter plasmid containing seven AP-1 response elements and an expression plasmid encoding mitogen activated protein kinase kinase kinase 1 (MEKK1) (all from Strata-gene, CA, U.S.A.). SK-N-MC cells were transfected with 0.5 to 1 μg of AP-1 firefly luciferase plasmid and, as a positive control, 25 ng of MEKK1 expression plasmid, to cells on a plate with an area of 3.8 cm2. Primary cortical glial cultures were transfected with 0.5 to 1 μg of AP-1 firefly luciferase plasmid and 0.025 ng to 25 ng of MEKK1 expression plasmid, to cells on a plate with an area of 3.8 cm2. A plasmid encoding Renilla luciferase (pRL; Promega, WI, U.S.A.) was used as an estimate of transfection efficiency. The transfection medium containing DNA was replaced by normal growth medium after 4 hours, and SK-N-MC cells were stimulated with glutamate (100 μmol/L) after a further 24 to 36 hours. Chlomethiazole was added 1 hour before stimulation. After harvest, cell extracts were incubated with Firefly/Renilla luciferase substrate and activity measured on a luminometer (Turner Designs, CA, U.S.A.).
Data analysis
For statistical evaluations of the results, Student's unpaired t-test was used as only two parameters (two different treatments in this case) were compared. Each independent experiment constitutes an independent preparation of RNA or cell extract from different cell culture preparations. The value of each experiment is the mean of the replicate samples (usually double or triple) of the same treatment.
For the quantitative RT-PCR mimic analysis, the calculations were at the linear range when target to mimic DNA ratios were between 0.66 to 1.55 (the relative content of target cDNA of each sample was calculated from the linear regression of the target-mimic DNA ratio versus concentration of mimic for 3 to 4 different mimic concentrations). Values obtained for individual samples were always divided with the β-actin content of each sample.
In all experiments, the P values were calculated on the percentage induction or percentage inhibition when the value of the control (no treatment) or induced sample (LPS or glutamate) was taken as 100%.
RESULTS
Effects of chlomethiazole on c-fos and c-jun mRNA expression
The time dependence of c-fos and c-jun mRNA induction upon LPS treatment in primary cortical glial cells was characterized by using a quantitative RT-PCR assay and Northern blot analysis. The expression of c-fos mRNA was induced as early as 15 minutes by LPS treatment and the maximal induction, usually up to 4- to 5-fold, was observed in most experiments at 30 minutes (Fig. 1A). The time dependence of c-fos mRNA in response to LPS was characterized by a rapid and transient increase (15 to 60 minutes) followed by a postinductional suppression phase at 6 hours and a tendency to be up-regulated again at 15 hours, the latest time point examined. The induction by LPS at 15 and 30 minutes was inhibited by CMZ by 60% to 70% when administered 1 hour before LPS at a dose of 40 μmol/L (Fig. 1A). Induction of c-jun mRNA by LPS was up to 4.5-fold and the time point of maximal induction varied somewhat. Maximal induction was usually observed at 30 to 60 minutes and up to 3 hours of LPS treatment. Chlomethiazole, at a dose of 40 μmol/L, potently inhibited the induction of c-jun mRNA at all time points (Fig. 1B).
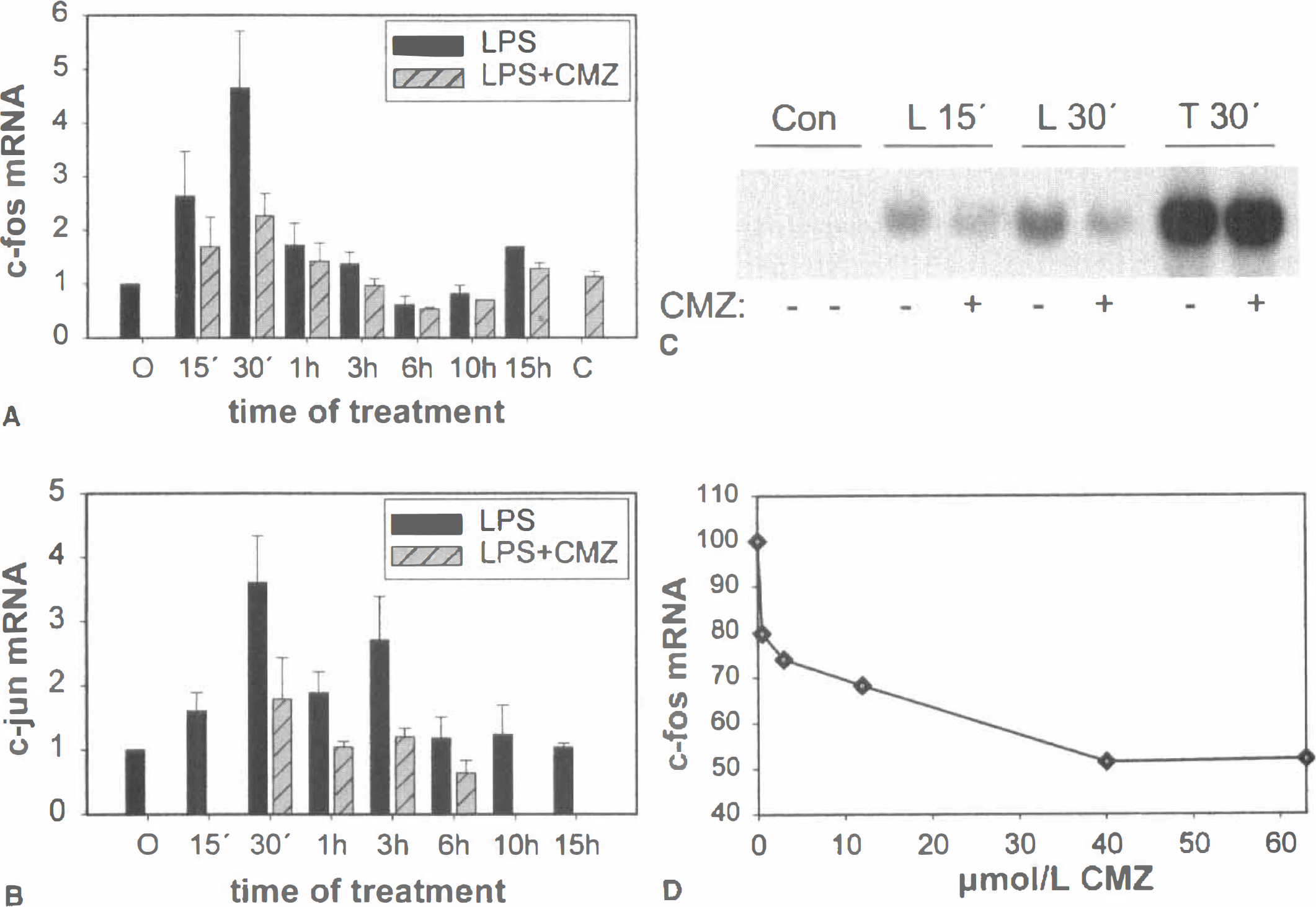
Effect of CMZ on LPS-induced c-fos and c-jun mRNA. Quantitative RT-PCR and Northern blot analysis were performed on isolated RNA from primary cortical glial cultures treated with 100 ng/mL LPS for the indicated time periods. A dose of 40 μmol/L CMZ was administered 1 hour before LPS. Each point of the diagrams
Apart from LPS, in several experiments primary cortical glial cells were also stimulated with TPA, a strong activator of c-fos. TPA treatment for 30 minutes caused a more than 20-fold induction of c-fos mRNA levels, as quantified by RT-PCR mimic, but 40 or 60 μmol/L CMZ could not attenuate this induction (Fig. 1C). The dose-dependence to CMZ was analyzed on c-fos mRNA induction after 30 minutes of LPS treatment, and the IC50 value for CMZ was calculated to equal approximately 2 μmol/L (see Fig. 1D).
To examine the potential involvement of p38 MAP kinases in the induction of c-fos and c-jun mRNA levels by LPS, SB203580, a potent inhibitor of the activity of p38α, p38β, and p38β2 isoforms of p38 MAP kinase was added to cortical glial cultures 1 hour before LPS. In three independent experiments (Fig. 2), 10 μmol/L SB203580 was sufficient to inhibit the LPS-mediated induction of both c-fos and c-jun mRNA by 70% to 80%.
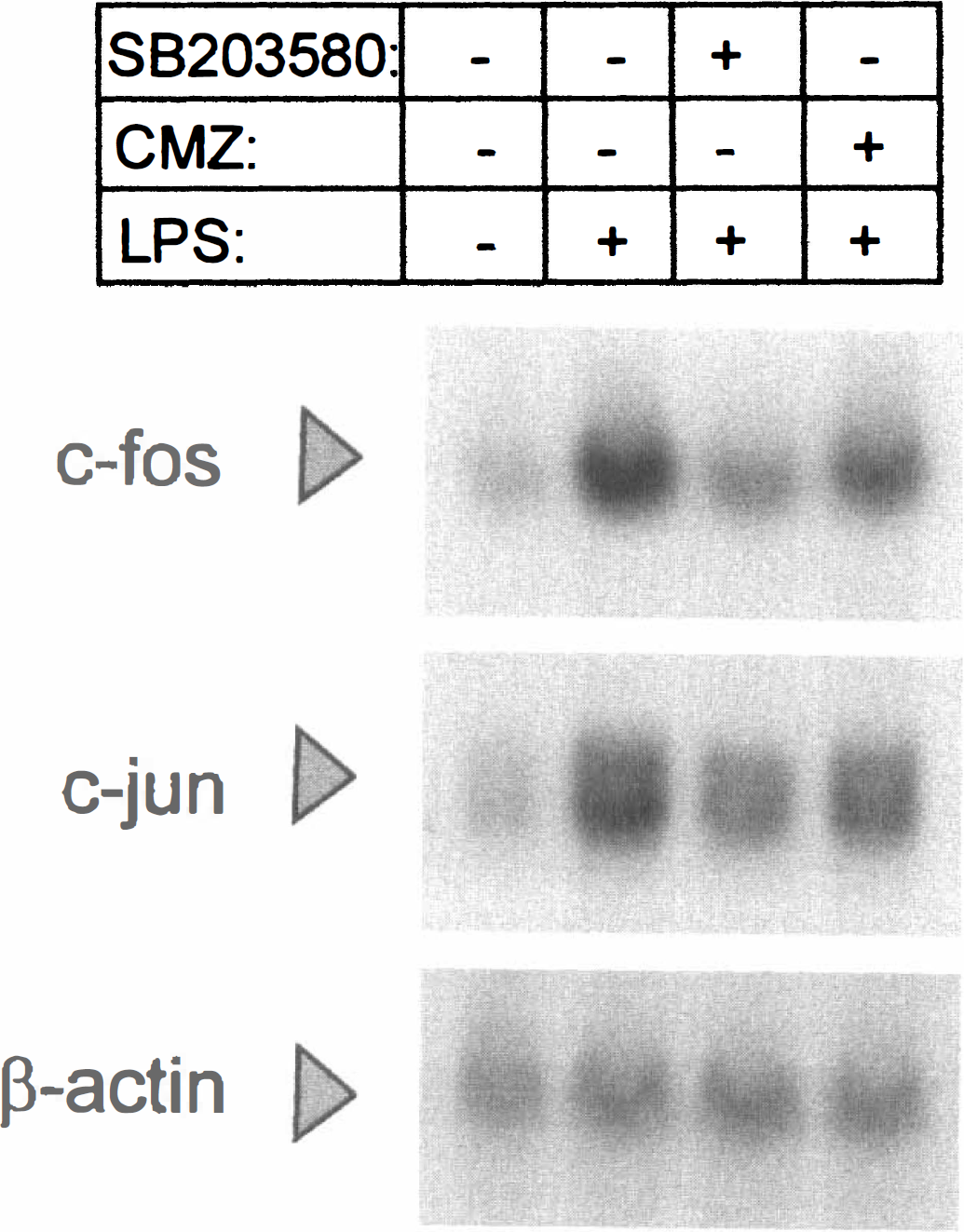
Effect of SB203580 on c-fos and c-jun mRNA. Total RNA was isolated from rat primary cortical glial cells treated with LPS for 30 minutes with or without inhibitors. SB203580 or CMZ was added to the cells 1 hour before LPS at a dose of 10 and 40 μmol/L respectively. After formaldehyde denaturing gel electrophoresis, 40 μg RNA were hybridized to c-fos, c-jun or β-actin 32P-labeled DNA probes. A representative out of three independent experiments is shown.
Chlomethiazole inhibits AP-1 DNA binding and transactivation
The AP-1 DNA binding activity upon LPS treatment was assayed by electrophoretic mobility shift assay in nuclear extracts from primary cortical glial cultures. The DNA probe was a consensus AP-1 element, which is known to bind Fos and Jun heterodimers with high affinity, as well as Jun-Jun homodimeric complexes. As shown in Fig. 3A, there was an increase in the DNA binding detected after 9 hours of LPS treatment which was counteracted by CMZ. Treatment with TPA was also shown as a positive control. The AP-1 complex was found to consist of c-Fos and c-Jun heterodimers, as revealed from antibody interference-supershift analysis, in which the LPS-induced AP-1 band was reduced in the presence of two different c-Fos specific antibodies and a c-Jun specific antibody; whereas, the AP-1 bond was completely abolished when an antibody recognizing JunB and JunD in addition to c-Jun was used. Furthermore, an antibody against GFAP, an astrocyte cytoskeleton protein, did not affect the specific AP-1 binding as expected (Fig. 3B). The specificity of the DNA-protein interactions was confirmed in competition experiments, in which a 25 or 50 times excess of cold AP-1 oligonucleotide probe abolished the binding only of the specific retarded band (Fig. 3B).
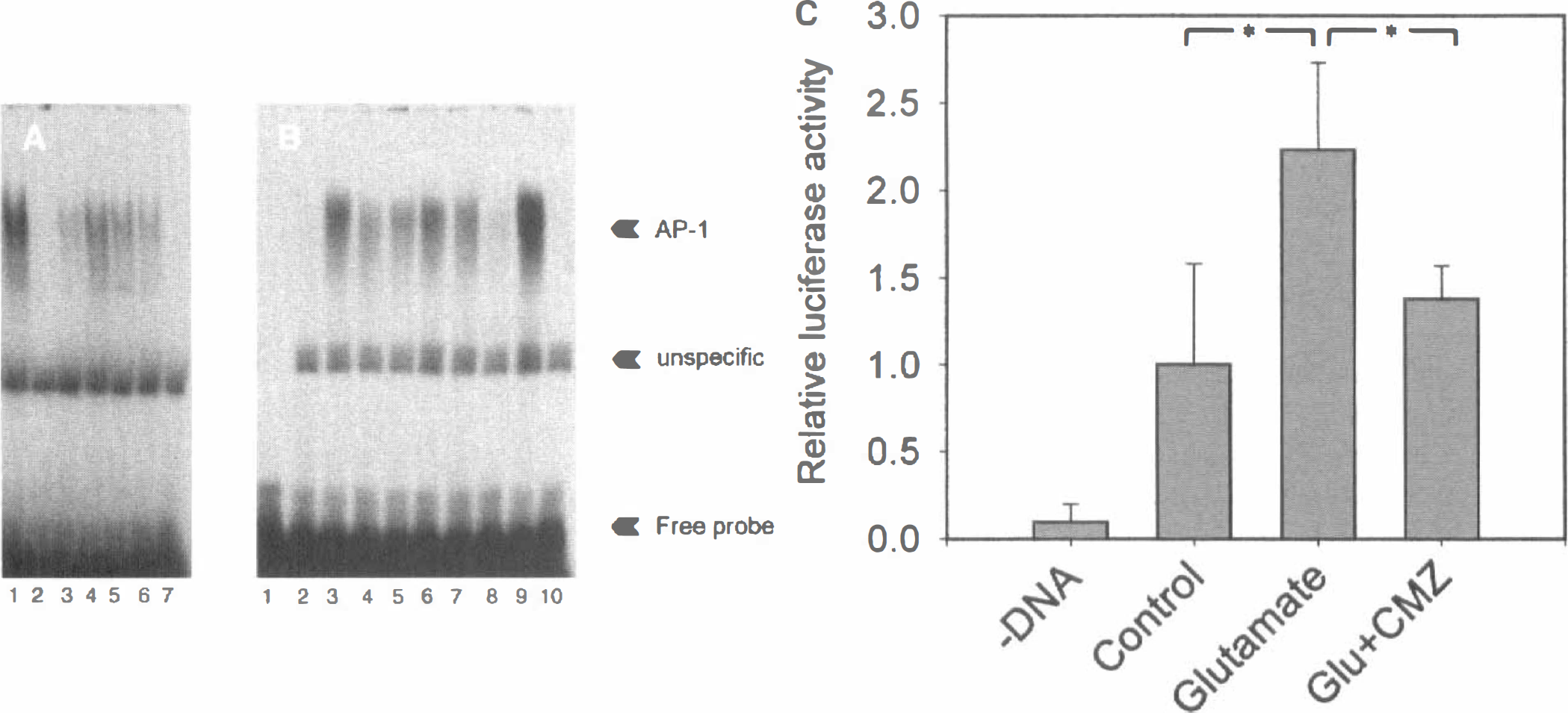
Effect of CMZ on DNA binding of AP-1 and AP-1 transactivating activity. Electrophoretic mobility shift assay was performed on nuclear extracts from primary cortical glial cells using the consensus ds AP-1 element as 32P-labeled probe. Fresh media were added to the cells 1 hour before stimulation. LPS and TPA were used at concentrations of 100 ng/mL and 100 nmol/L respectively, whereas CMZ was added at a dose of 60 μmol/L.
Human SK-N-MC neuroblastoma cells were transfected with an AP-1 luciferase reporter plasmid containing seven consensus AP-1 elements. One hundred μmol/L glutamate gave a slightly greater than twofold increase in luciferase activity that was time dependent and peaked at approximately 3 hours. Chlomethiazole inhibited the glutamate-dependent AP-1 activation to ~70% to 80% at 30 to 60 μmol/L in a highly reproducible manner (Fig. 3C). The inhibition of glutamate induced AP-1 transactivation by CMZ was dose dependent, with an estimated IC50 of ~8 μmol/L.
Chlomethiazole has no effect on NFk B DNA-binding, IL-1β or TNF-α mRNA expression in response to LPS
Compounds such as aspirin and sodium salicylate have been suggested to be neuroprotective through the inhibition of the transcription factor NF
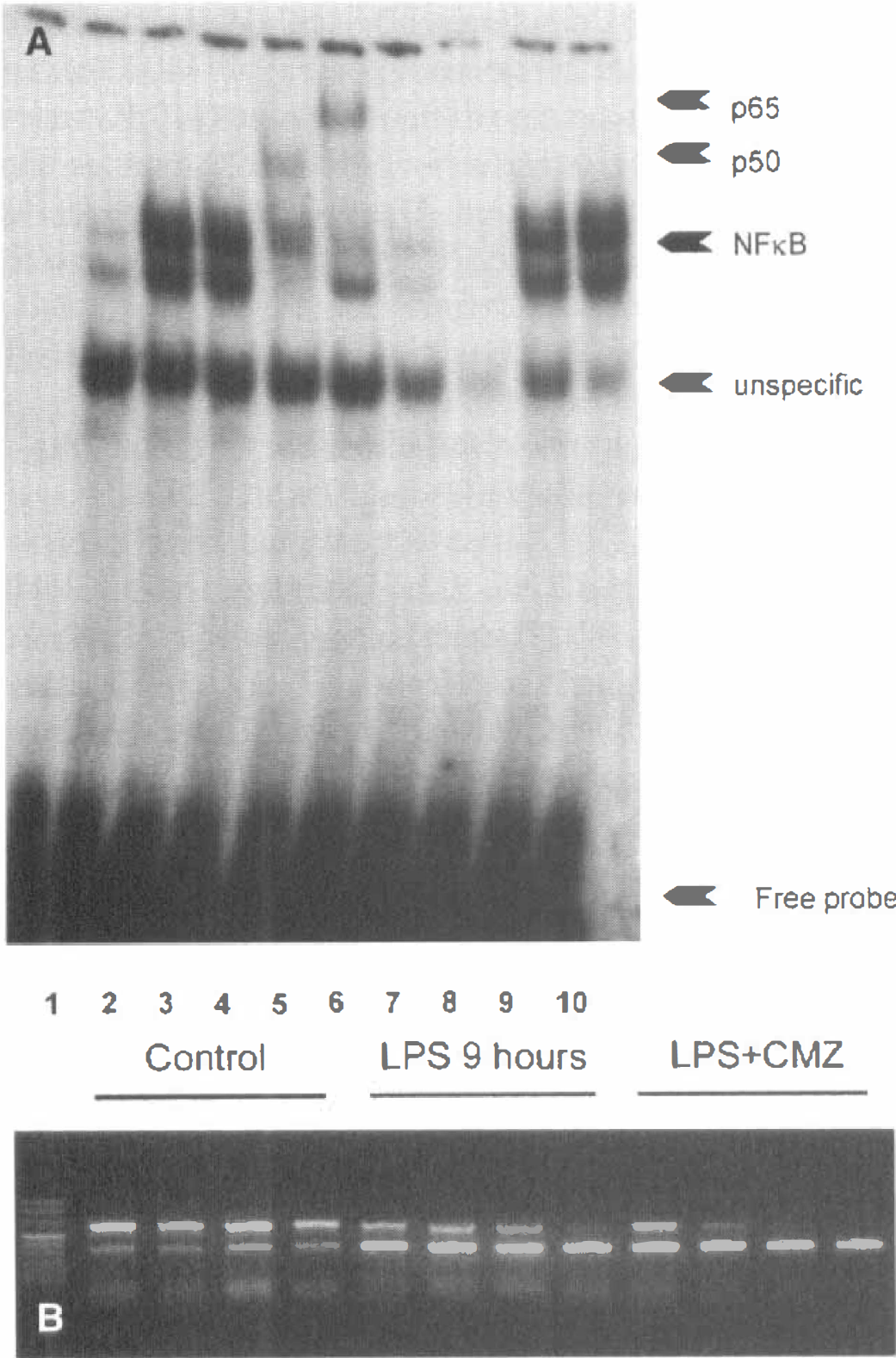
CMZ does not counteract the induction of NF
The effect of CMZ on the mRNA expression of two proinflammatory cytokines, at least partially under the control of NF
Lipopolysaccharide activates JNK and ERK
MAP kinase activation was studied with Western blot analysis of cortical glial cell extracts using antibodies against phospho-JNK and phospho-ERK. These antibodies recognized a 46 kDa band (JNK) and 42 and 44 kDa bands (ERK), respectively; whereas very weak phosphorylation of JNK and ERK by LPS was seen at 10 to 15 minutes, and full activation was detected at 60 to 120 minutes. However, 60 μmol/L CMZ did not show any inhibitory action on the activation of JNK or ERK by LPS when added 30 to 60 minutes before LPS stimulation (not shown). In addition, induction of c-fos mRNA by TPA, which is primarily acting through the ERK pathway, was not inhibited by CMZ (Fig. 1C).
Lipopolysaccharide caused a substantial increase in the N-terminal phosphorylation of a GST-c-Jun, a fusion protein used as a substrate for JNK, in an in vitro kinase assay using primary cortical glial cell extracts. The increase in phosphorylation of GST-c-Jun was not affected by CMZ at 30 or 60 minutes (not shown). Chlomethiazole was ineffective in inhibiting GST-c-Jun phosphorylation when added to the cell culture or in vitro in the phosphorylation assay. In addition, primary cortical glial cultures were transfected with an AP-1 luciferase reporter and a MEKK1 expression plasmid, a MAP kinase kinase kinase upstream of JNK previously shown to be activated by LPS in macrophages (Sanghera et al., 1996). MEKK1 cotransfections resulted in a potent and dose dependent increase in AP-1 luciferase activity. Chlomethiazole (60 μmol/L) did not counteract this induction under conditions in which MEKK1 was limiting (that is, low concentrations of plasmid) nor under any of several different time aspects examined (not shown).
Phosphorylation of p38 MAP kinase is unaffected by chlomethiazole
Phosphorylation of p38 MAP kinase was analyzed with a phospho-p38 antibody in a Western blot assay of primary cortical glial cultures. The antibody recognized a single band with mobility corresponding to ~40 kDa that was found to be reactive within 10 minutes of LPS treatment and remained reactive for at least 120 minutes, the longest time point analyzed. The phosphorylation status of p38 MAP kinase was not altered by the 60 μmol/L CMZ added 45 minutes before LPS (Fig. 5A).
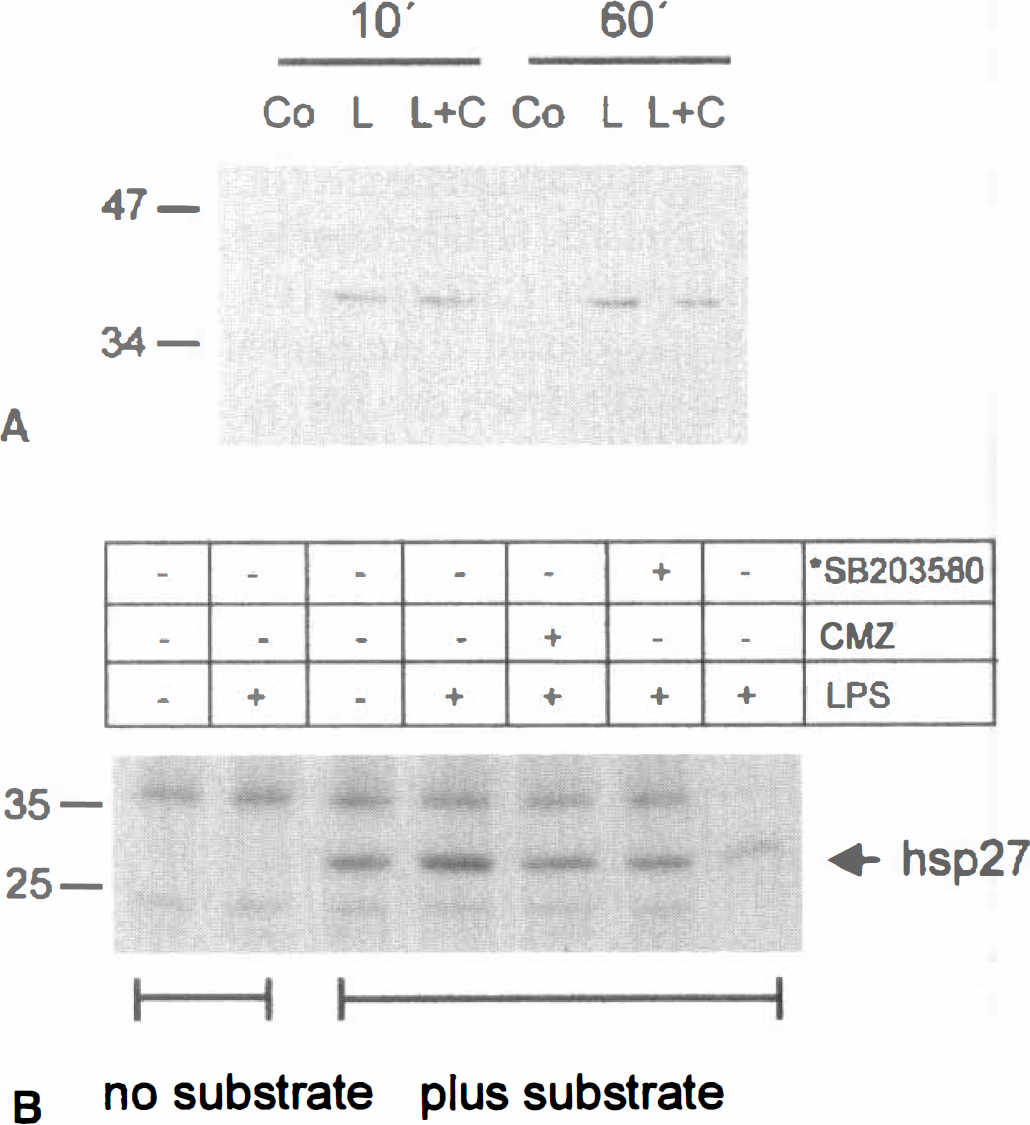
Effects of CMZ on the phosphorylation p38 MAP kinase and hsp27.
Chlomethiazole inhibits the phosphorylation of the p38 MAP kinase substrates hsp27 and ATF-2
Because p38 MAP kinase activity, but not phosphorylation status, was shown to be inhibited by SB203580 and equivalent pyridinyl imidazoles (Kumar et al, 1999), the authors examined the effect of CMZ on p38 MAPK activity towards two downstream effectors—the indirect target hsp27, which lies downstream of MAPKAP-K2, and the direct p38 MAPK substrate ATF-2. To assess the phosphorylation of hsp27, the authors used an in vitro kinase assay in which kinases present in total cell extracts from primary cortical glial cells were allowed to phoshorylate in vitro recombinant hsp27 in the presence of [32P]-γATP. A slightly greater than twofold increase in the phosphorylation of hsp27 was seen after 10 minutes of LPS treatment. Chlomethiazole, at a dose of 40 μmol/L, inhibited this increase to 50% at both 10 and 30 minutes of LPS treatment (Fig. 5B). The LPS-induced phosphorylation of hsp27 was found to be dependent on p38 MAP kinase, as it was inhibited by SB203580 added in vitro in the incubation by almost 90% (Fig. 5B). Lipopolysaccharide further induced the phosphorylation of a GST-ATF-2 fusion protein by 2- to 3-fold maximally at 30 minutes, as assayed in total cell extracts from rat cortical glial cultures. The increased phosphorylation of GST-ATF-2 was also inhibited by CMZ either when the drug was added in cell cultures 1 hour before LPS, or when added in vitro in the kinase assay (not shown). In addition to CMZ, GST-ATF-2 phosphorylation was sensitive to 10 μmol/L SB203580, but only when the compound was added in vitro in the kinase assay (data not shown).
Because ATF-2 is also substrate for JNK, immunoprecipitation experiments were carried out using an agarose-conjugated specific antibody against p38 MAP kinase isoforms α and β. Immunocomplex kinase assays using GST-ATF-2 as a substrate showed an induction at 30 minutes by LPS that was counteracted by 40 μmol/L chlomethiazole (Fig. 6). The phosphorylation of GST-ATF-2 by p38 MAPK immunoprecipitates was sensitive to SB203580 (10 μmol/L) when added in vitro in the kinase reaction, whereas it was sensitive to chlomethiazole (40 μmol/L) when added either in the cell culture media or in the kinase reaction (Fig. 6B). When a GST-c-Jun fusion protein was used as a substrate in the same assay, no phosphorylated band was detected, indicating the absence of JNK in the anti-p38 immunoprecipitates (Fig. 6, lane 6). The quantitative nature of the p38 immunocomplex kinase assay was verified in experiments in which immunocomplexes from differently treated cells were found to contain the same amount of immunoprecipitated kinases when analyzed by Western blot using the same anti-p38 antibody or by autoradiography from 35S-methionine-labeled cells (not shown).
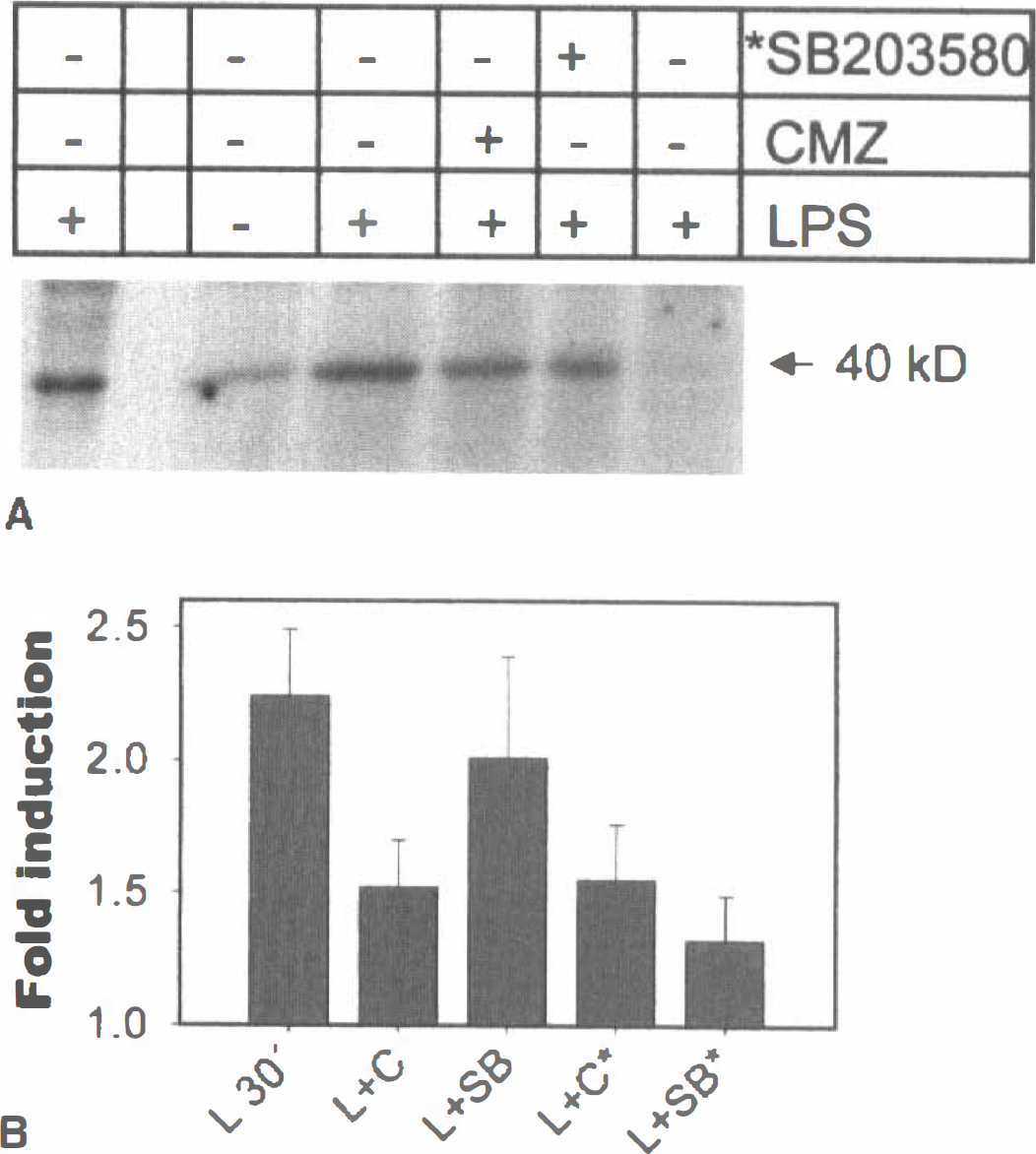
CMZ inhibits the in vitro phosphorylation of GST-ATF-2 by the immunoprecipitated p38 MAPK.
DISCUSSION
We have found that chlomethiazole specifically attenuates the induction of c-fos, c-jun, and AP-1 in response to LPS in rat primary cortical glial cells, mainly consisting of astrocytes. It was also found that chlomethiazole counteracted AP-1 transactivation in response to glutamate in human SK-N-MC neuroblastoma cells. The inhibitory effect was seen in several different assays, including quantitative RT-PCR, Northern blot, electrophoretic mobility shift assay, and transfectional analysis. Using several different experimental approaches we found no evidence for an effect of CMZ in ERK or JNK MAP kinase pathways, or in NF
A mechanism in which CMZ acts to attenuate p38 MAPK activity suggested that the compound has prominent anti-inflammatory properties. It has been shown that CMZ is highly effective as a neuroprotectant in many different animal models of cerebral ischemia (Green, 1998), models in which inflammatory factors such as the cytokine IL-1β might be of importance (Rothwell and Relton, 1993; Yamasaki et al., 1995). It has also been shown that other cytokines such as TNF-α and IL-6 might assume modulatory roles during cerebral ischemia (Maeda et al., 1994; Bruce et al., 1996). In many cells p38 MAP kinase has a role in regulation of biosynthesis of cytokines such as IL-1β and TNF-α, and the p38 MAP kinase inhibitor SB239063 has been found to reduce brain injury and neurologic deficits in models of MCA occlusion (Barone et al, 1999). Importantly, it was recently found that p38 MAP kinase is activated predominantly in astrocytes during cerebral ischemia (Irving et al, 1999), indicating a crucial role for astrocytes in regulation of p38 MAP kinase dependent responses of the CNS during ischemia. Except for glial cells, several reports have also demonstrated the contribution of stress activated protein kinases of the JNK and p38 families in nervous tissue excitotoxicity and apoptosis, and in ischemia of the heart. Both JNK and p38 MAP kinases were found to be activated during nerve growth factor withdrawal or glutamate exposure and were implicated in a proapoptotic pathway (Xia et al., 1995; Mukherjee et al., 1999). Interestingly, the pyridinyl imidazole SB203580 has been found to potently counteract induction of apoptosis by glutamate in cerebellar granule cells (Kawasaki et al., 1997) in which similar concentrations of SB203580 were needed for inhibition of p38 MAP kinase as were for inhibition of apoptosis. Recently, it was also suggested that SB203580 might attenuate cell injury in response to ischemia in cardiac myocytes (Mackay and Mochly-Rosen, 1999). Taken together, much evidence favors a role of p38 MAP kinases in ischemic and apoptotic injury of cells of the central nervous system, and it has been suggested that the ability of CMZ to inhibit p38 MAP kinase activity contributes to the neuroprotective properties of the compound. Based on the above outlined evidence, we believe that the in vitro models employed in the current investigation are relevant for studies of inflammatory responses of cells of the central nervous system during ischemia.
The mechanism employed by CMZ for inhibition of AP-1 in SK-N-MC human neuroblastoma cells is yet to be elucidated. However, it is very interesting that CMZ also affects the same or similar responses in neuronal cells and that it interferes with early events in stroke like glutamate-mediated responses.
The sedative, hypnotic, and anxiolytic properties of CMZ have previously been ascribed to the potentiation of the action of GABA at the GABAA receptor complex (Moody and Skolnick, 1989; Hales and Lambert, 1992). Although the relation of the inhibition of c-fos/c-jun/AP-1 seen to the GABAA mimetic properties of CMZ has not been directly addressed in the current study, we consider it unlikely that GABAA is involved in the action of CMZ seen in these studies for two reasons. First, whereas CMZ inhibited c-fos expression with an IC50 of ~2 μmol/L, CMZ has been reported to enhance picrotoxin-sensitive chloride uptake with an EC50 of 48 μmol/L and to displace binding of t-butylbicyclophosphorothionate, a compound that binds to a site associated with the chloride channel, with an IC50 of 58 to 150 μmol/L in different studies (Cross et al., 1989; Moody and Skolnick, 1989; Green, 1998). Secondly, although GABAA receptors are highly expressed in glial cells in vivo, astrocytes have been reported not to express these receptors at significant levels in vitro (Bureau et al., 1995; Hosli et al., 1997). Notably, CMZ has recently been shown to attenuate the inflammatory response to chronic ethanol treatment of the liver (Fang et al., 1998), a tissue which, to our knowledge, does not express GABAA receptors.
Several different transcription factors known to bind to either c-fos, or c-jun promoters, or both, have been shown to potentially lie downstream of p38 MAP kinases; these include MEF2C (Han et al., 1997), CREB (Tan et al., 1996; Deak et al., 1998; Xing et al., 1998), Elk-1 (Raingeaud et al., 1996) and ATF-2, an excellent in vitro substrate for several JNK and p38 MAP kinase isoforms (Gupta et al., 1995; Jiang et al., 1996; Goedert et al., 1997) although its role in vivo is less clear and may depend on the cell type studied (Hazzalin et al, 1996; Hazzalin et al, 1997; Chen et al, 1998; Cheong et al, 1998). LPS-induced transcription of c-jun has been shown to be, to a large extent, dependent on a MEF2C binding site. Binding to this element is inhibited to 65% by SB203580, and mutation of this site inhibits LPS-induced c-jun transcription (Han et al., 1997). Accordingly, it is a distinct possibility that CMZ inhibits c-jun mRNA induction by inhibiting transactivation through the MEF2C site. Transcription of c-fos in fibroblasts induced by UV radiation or anisomycin, but not by growth factors or phorbol ester, has been shown to be sensitive to SB203580 (Hazzalin et al., 1996). Thus, LPS effects on c-fos in primary cortical glial cells resemble the former group. It has been suggested (Cohen, 1997) that the serum response element and cyclic AMP response element are important for c-fos transcription in response to stressful stimuli as their activation is sensitive to SB203580 (Tan et al., 1996; Ahn et al., 1998). A possible hypothesis is that CMZ effects on c-fos are mediated through the serum response elements, or cyclic AMP response elements, or both. However, further studies are required for direct identification of CMZ sensitive elements of c-fos and c-jun promoters.
In conclusion, we have found that CMZ acted as an inhibitor of p38 MAP kinase catalysis, and that expression of the early genes c-fos and c-jun in response to LPS can be attenuated. Inhibition of p38 MAPK occurred through direct interaction with p38, or an associated protein, as CMZ was effective in in vitro kinase assays but did not affect phosphorylation of p38 MAP kinase. The results described a novel mechanism of action of CMZ that because of dose-response relations was suggested to be of relevance for the neuroprotective properties of CMZ previously seen by others.
Footnotes
Acknowledgments
The authors thank Margareta Porsmyr-Palmertz for excellent technical assistance with some transfection experiments, and Inger Bengtsson for her valuable contribution with the NF-κB gel shifts.