
Abstract
Leukocyte infiltration into the brain contributes to the development of ischemic brain damage and is mediated by endothelial/leukocyte adhesion molecules, cytokines, and chemokines released by ischemic brain cells. In this study, we provide evidence that human astrocytes (FHAs) subjected to in vitro hypoxia produce proinflammatory mediator(s) capable of up-regulating inflammatory genes, including intercellular adhesion molecule-1, interleukin (IL)-1β, tumor necrosis factor-α, IL-8, and monocyte chemotactic protein-1 (MCP-1) in human cerebromicrovascular endothelial cells (HCECs). FHAS were exposed to hypoxia in an anaerobic chamber for 4 hours, followed by reoxygenation for 24 hours. Astrocyte-conditioned media (ACM) collected from normoxic FHAS or FHAS subjected to hypoxia/reoxygenation were applied to HCEC cultures for 4 to 24 hours. Semiquantitative reverse transcription–polymerase chain reaction, immunocytochemistry, and enzyme-linked immunosorbent assay demonstrated up-regulation of intercellular adhesion molecule-1 in HCECs exposed to hypoxic ACM. A pronounced elevation in cytokine IL-1β and tumor necrosis factor-α, and chemokine IL-8 and MCP-1 mRNA, accompanied by increased release of immunoreactive cytokines and chemokines into cell media was observed in HCECs exposed to hypoxic ACM. Hypoxia/reoxygenation induced a transient (4 to 18 hours of reoxygenation) upregulation of IL-1β mRNA in FHAS and a two- to threefold increase in IL-1β levels secreted into ACM. Pretreatment of FHAS with 10 μmol/L dexamethasone inhibited both hypoxia-induced expression/secretion of IL-1β and the ability of hypoxic ACM to induce inflammatory phenotype in HCECs. The ability of hypoxic ACM to up-regulate inflammatory genes in HCECs was inhibited in the presence of IL-1 receptor antagonist (IL-1Ra) and by pretreating ACM with the blocking anti-IL-1β antibody. These findings strongly implicate IL-1β secreted by hypoxic astrocytes in triggering inflammatory activation of HCECs and thereby influencing inflammatory responses at the site of the blood-brain barrier.
Clinical and experimental evidence has implicated brain inflammation in the development of secondary ischemic brain damage in brain ischemia and trauma (del Zoppo, 1994; Feuerstein et al., 1997, 1998; Hallenbeck et al., 1986; Kim, 1996; Kochanek and Hallenbeck, 1992; Ransohoff and Tani, 1998). Brain inflammation after ischemia develops as a consequence of two related processes, i.e., the activation of glial cells and resident perivascular/parenchymal macrophages (Giulian, 1990) and the mobilization (del Zoppo et al., 1991) and infiltration of peripheral inflammatory cells into the brain (Barone et al., 1991; Hallenbeck et al., 1986; Matsuo et al., 1994). Activated glia has been shown to produce numerous proinflammatory mediators (Giulian, 1990; Stoll et al., 1998), including interleukin (IL)-1β (Lee et al., 1993). Cerebral endothelium undergoes a “proinflammatory” activation leading to increased interactions with peripheral leukocytes (del Zoppo, 1994). Mobilization of polymorphonuclear leukocytes in ischemic microvessels occurs as early as 30 minutes after the middle cerebral artery occlusion (MCAO) in rat and is followed by accumulation of circulating monocytes within capillaries after 4 to 6 hours (Garcia et al., 1994). Both neutrophils and monocytes subsequently infiltrate the brain tissue (Barone et al., 1991; Garcia et al., 1994) and can be detected in the brain as late as 7 days after the insult. The process of leukocyte trafficking across the blood–brain barrier is regulated by mediators produced/released by brain parenchymal cells, cerebral endothelial cells (CECs), and leukocytes (Baggiolini, 1998; Barone et al., 1991; Feuerstein et al., 1998; Kishimoto and Rothlein, 1994; Ransohoff and Tani, 1998).
Cytokines, including IL-1β, are central regulators of inflammatory responses in peripheral tissues (Colotta et al., 1998; Dinarello, 1996, 1998). IL-1β is synthesized as a precursor protein, pro-IL-1β, which is then cleaved by IL-1-converting enzyme (ICE) into an active IL-1β (Colotta et al., 1998). IL-1β binds two types of receptors, a signaling type I (IL-1RI) receptor, and type II (IL-1RII) receptor, a decoy that inhibits IL-1 activity (Colotta et al., 1998; Dinarello, 1998). IL-1β has been shown to induce the expression of adhesion molecules including intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1, and E-selectin in human CECs (Stanimirovic et al., 1997b, c ). Up-regulation of both endothelial cell and leukocyte adhesion molecules by ischemia is necessary for leukocyte rolling and adhesion to vascular walls (Kim, 1996; Kishimoto and Rothlein, 1994). Subsequent leukocyte transmigration across the endothelial cell barrier also requires the presence of chemotactic gradients (Baggiolini, 1998; Baggiolini et al., 1997; Hausmann et al., 1998; Rollins, 1997).
Inflammatory activation of CECs in cerebral ischemia probably starts via signals activated by hypoxia/ischemia in CECs themselves and is then sustained by paracrine mediators released by neighboring parenchymal cells. Astrocyte end-feet envelope brain capillaries and close interactions between these two cell types have been implicated in the regulation of phenotypic properties of CECs (Abbott et al., 1992; Cancilla et al., 1993), CEC proliferation and angiogenesis (Cancilla et al., 1993; Stanimirovic et al., 1995), CEC production of anticoagulant factors (Tran et al., 1999), and the blood-brain barrier permeability (Cancilla et al., 1993). Astrocyte/endothelial proximity and interactions may also be important in regulating inflammatory responses at the site of the blood-brain barrier. Astrocytes have been shown to produce cytokines, chemokines, and adhesion molecules when stimulated with cytokines (Aloisi et al., 1995; Lee et al., 1993, 1998). We have recently demonstrated that both human CECs and human astrocytes subjected to a simulated ischemia in vitro secrete bioactive chemokines IL-8 and monocyte chemotactic protein-1 (MCP-1) and other neutrophil chemoattractants (Zhang et al., 1999).
In the present study, we provide evidence that human astrocytes exposed to in vitro hypoxia express and release bioactive IL-1β, which triggers a proinflammatory activation of human brain endothelium in a paracrine manner.
MATERIALS AND METHODS
Cell cultures
The protocols used in these studies have been approved by the Human Research Ethics Committees of the National Research Council of Canada and the Montreal Neurological Institute (McGill University).
Primary human cerebromicrovascular endothelial cell cultures (HCECs) were isolated and maintained by using a modification (Stanimirovic et al., 1996) of the procedures described originally by Gerhart et al. (1988). The cells were grown at 37°C in a 5% CO2 atmosphere in a medium containing 65% Dulbecco's modified Eagle's medium (DME) (4,500 mg/L glucose), 10% fetal bovine serum (FBS; HyClone, Logan, UT, U.S.A.), 5% human serum (Sigma, St. Louis, MO, U.S.A.), 20% murine melanoma cell-conditioned media, 5 μg/mL insulin, 5 μg/mL transferrin, 5 μg/mL selenium, and 10 μg/mL endothelial cell growth supplement (Collaborative Biomedical Products, Bedford, MA, U.S.A.). Both primary and propagated cultures were routinely characterized for the expression of factor VIII–related antigen and the absence of smooth muscle cell α-actin as described previously (Stanimirovic et al., 1996). Passages 2 to 6 of HCECs were used for the experiments in this study.
Primary fetal (10 to 18 weeks of gestation) human astrocyte (FHAS) cultures were generously provided by Dr. J. Antel (Montreal Neurological Institute, Montreal, QC, Canada). These cultures were prepared by using previously described procedures (Yong et al., 1992). FHAS were grown in DME containing 10% FBS (HyClone, Logan, UT, U.S.A.) and antibiotics (GibcoBRL, Burlington, ON, Canada) in an atmosphere of 5% CO2/95% air at 37°C. More than 95% of cells in FHAS cultures were immunopositive for the glial fibrillary acidic protein. Passages 2 to 6 of FHAS were used in this study.
In vitro hypoxia
FHAS were grown on 35-mm dishes to 90% confluence in complete medium. For the experiments, media were removed and cells washed in a Hanks' balanced salt solution (HBSS; GibcoBRL). A serum-free DME was then added to the cells and they were subjected to severe hypoxia (<2% oxygen) for 4 hours in an anaerobic chamber (Anaerobic System Model 1024, Forma Scientific, Fisher Scientific, Nepean, Canada) equipped with a humidified, temperature-controlled incubator directly accessible within the chamber. The entire system was purged with 95% N2/5% CO2 atmosphere. At the end of the hypoxic period, the media were removed, cells washed twice in HBSS, and 1.5 mL of DME containing 1% FBS was added to the cells. The cells were reoxygenated in ambient air at 37°C and both media and cells were harvested after 4, 8, 18, or 24 hours. We have previously shown that these conditions do not affect viability of astrocyte cultures (Stanimirovic et al., 1997a).
Astrocyte-conditioned media
FHAS grown in tissue culture flasks (Nalge NUNC International, Rochester, NY, U.S.A.) were subjected to hypoxia as above, washed twice in HBSS, and reoxygenated for 24 hours in 12 mL of DME containing 1% FBS. The media [astrocyte-conditioned media (ACM)] were then collected and applied immediately to HCECs as described below. Control FHAS were not subjected to hypoxia but underwent the same media changes and ACM collection as hypoxic FHAS.
ACM collected from either normoxic or hypoxic FHAS was applied to HCECs grown in 35-mm dishes (1 × 105 cells/dish) to 90% confluence. HCECs were incubated with ACM for 4, 8, 18, and 24 hours. The media were then collected and HCECs were prepared for immunocytochemistry and biochemical analyzes. The supernatants of media and cell extracts were stored at −80°C until use.
Immunocytochemistry
The expression of ICAM-1 in HCECs was detected by immunocytochemistry and enzyme-linked immunosorbent assay (ELISA) by using a monoclonal antibody to human ICAM-1 (Clone CD54; Upstate Biotechnology, Lake Placid, NY, U.S.A.). The specificity of the antibody has been confirmed previously in studies using purified ICAM-1 (Makgoba et al., 1988). The antibody also recognized a single band on a Western blot of HCEC lysate (data not shown).
For immunocytochemistry, HCECs grown on 10 μg/mL human fibronectin-coated glass coverslips were washed in HBSS and incubated at room temperature with the primary anti-ICAM-1 antibody (2 μg/mL) for 40 minutes. The nonspecific binding was blocked by 4% goat serum. Cells were then washed in HBSS and incubated for 1 hour with a 1:50 dilution of a 4-nm colloidal gold particle-conjugated goat antimouse IgG antibody. Coverslips were then fixed for 30 seconds with 9.25% formaldehyde and 45% acetone in HBSS, washed, and incubated in a silver-enhancing solution (IntenSE™; Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.) for 25 minutes. Coverslips were then washed and counterstained with Giemsa stain for 15 minutes. Nonspecific staining was determined by (1) omitting the secondary antibody from the procedure, and (2) by replacing primary anti-ICAM-1 antibody with the isotype-matched antibody to glial fibrillary acidic protein (Accurate Chemical and Scientific, Westbury, NY, U.S.A.).
ELISA
An ELISA was used to quantify levels of ICAM-1 expression in HCECs and levels of immunoreactive IL-8, MCP-1, and IL-1β in FHAS-conditioned media and in media collected from differently treated HCEC.
ELISA for ICAM-1 was performed essentially as described by Stanimirovic et al. (1997b). In brief, HCEC cultures grown in 96-well microtiter plates (2 × 104 cells/100 μL/well), at 37°C in 5% CO2, were sequentially incubated with a primary monoclonal antihuman ICAM-1 antibody (2 μg/mL; clone CD54; Upstate Biotechnology) for 1 hour at 37°C, followed by 1:500 diluted peroxidase-conjugated goat antimouse IgG in phosphate-buffered saline for 45 minutes at 37°C. Nonspecific binding sites were blocked with 2% bovine serum albumin in phosphate-buffered saline for 30 minutes at 37°C. After each incubation, the plates were washed three times with phosphate-buffered saline. Color was developed by the addition of 100 μL of a 1 mg/mL solution of the horseradish peroxidase substrate, 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonic acid) (Immuno-Pure ABTS; Pierce Chemical, Rockford, IL, U.S.A.). The reaction was stopped after 5 minutes by the addition of an equal volume of 1 % sodium dodecyl sulfate to each well. The optical density of the developed color was read at 405 nm by using a SpectraMAX (Molecular Devices, Menlo Park, CA, U.S.A.) microplate reader.
ELISA assays for IL-8, MCP-1, and IL-1β were performed as described by Zhang et al. (1999). The ACM or endothelial cell media were centrifuged at 14,000 rpm for 5 minutes at 4°C and an aliquot (100 μL) was used in ELISA. The levels of IL-8, MCP-1, IL-1β, and tumor necrosis factor-α (TNF-α) were determined by using commercially obtained ELISA kits (Bio-Source, Camarillo, CA, U.S.A.). ELISA data were corrected for the levels of chemokines/cytokines present in (control or hypoxic) ACM applied to HCECs. The assays were performed according to the manufacturer's instructions in at least three independent experiments.
Reverse transcription polymerase chain reaction
Total RNA was isolated from FHAS and HCECs grown on 35-mm dishes by using 500 μL/dish Tri reagent (Sigma, St. Louis, MO, U.S.A.) following the manufacturer's instruction. The RNA pellets were resuspended in 50 μL of diethyl polycarbonate-treated dH2O and incubated at 55°C for 10 minutes. The quality of the RNA was confirmed for each sample by the use of formaldehyde-agarose gel electrophoresis. RNA (0.5 μg) was then mixed with 0.5 μg of oligo(dT)12–18 primers, heated for 10 minutes at 70°C, and then chilled on ice. First-strand cDNA synthesis (reverse transcription) was performed (20 μL reaction) as described (Zhang et al., 1999) and 20 μL of cDNA was then diluted with 20 μL of dH2O. Specific primers for human IL-1β, IL-1RI, ICE, TNF-α, IL-8, MCP-1, ICAM-1, and the housekeeping gene β-actin were designed according to published sequences from the GenBank (Table 1) and were synthesized by using a PerSeptive Biosystem Synthesizer (Framingham, MA, U.S.A.). The polymerase chain reaction (PCR) amplifications were performed in a final volume of 50 μL containing 1x reaction buffer (Promega), 1.5 mmol/L MgCl, 0.2 μmol/L dNTPs, 0.4 μmol/L each of the primers, 2.5 units of Taq DNA polymerase (Promega, Madison, WI, U.S.A.), and cDNA. All amplifications were performed by using a denaturation step at 94°C for 30 seconds, an annealing step at 55°C for 45 seconds, and a polymerization step at 72°C for 40 seconds.
Sense and antisense sequences of primers used in PCR reactions
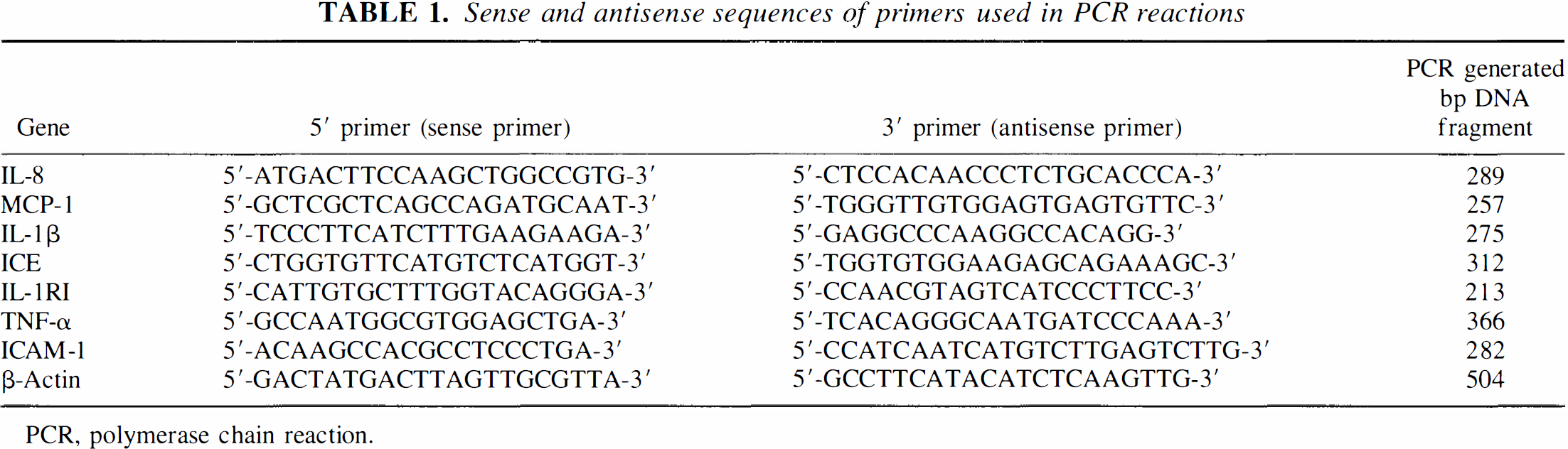
PCR, polymerase chain reaction.
To obtain a linear range of genes listed in Table 1 and β-actin, serial dilutions of cDNA preparation from the cells (HCECs and FHAS) were PCR-amplified for 20 to 45 cycles. The resultant PCR products were run on a 1% agarose gel, stained with ethidium bromide, photographed, and scanned with the use of a laser Computing Densitometer (Model 300A; Molecular Dynamics, CA, U.S.A.). Standard curves of various products and β-actin were then generated, indicating a linear range of each “target” gene and β-actin amplification for each cell type. All experiments are subsequently performed by using conditions optimized for linear amplification.
MCP-1 and β-actin were amplified in one tube by using 2-μL of cDNA diluted as herein described (i.e., 1:1, v/v, with dH2O) in a 50-μL reaction for 25 cycles. IL-8 and β-actin amplification was performed in the same PCR reaction containing 5 μL of diluted cDNA for 35 cycles. In both cases, 10 μL of the PCR product was then subjected to gel electrophoresis as described herein. IL-1β, ICE, TNF-α, and ICAM-1 were amplified by using 10 μL of diluted cDNA, and IL-IRI by using 15 μL of diluted cDNA for 45 cycles. Internal control (β-actin) for these genes was amplified by using 1 μL of diluted cDNA in a 50-μL reaction for 45 cycles. Equal amounts of internal control and IL-1β, ICE, TNF-α, IL-8, or ICAM-1 PCR products were then mixed and subjected to electrophoresis on a 1.5% agarose gel in 1x Tris-borate/EDTA buffer containing 0.5 μg/mL ethidium bromide and then photographed. The relative densities/volumes of the bands on film negatives were measured by using a Computing Densitometer (Model 300A; Molecular Dynamics, Sunnyvale, CA, U.S.A.) and analyzed by using ImageQuaNT, version 4.1, software (Molecular Dynamics). Various gene products were expressed as percentages of the β-actin band volume.
RESULTS
Stimulation of proinflammatory genes in HCECs by hypoxic ACM
To test the hypothesis that hypoxic astrocytes secrete factor(s) capable of triggering proinflammatory activation of HCECs, we used an in vitro paradigm in which FHAS cultures were subjected to hypoxia/reoxygenation, and the effects of media conditioned by these cells (ACM) on the expression of inflammatory genes (i.e., adhesion molecules, cytokines, and chemokines) in HCECs were examined. None of various protocols used affected the viability of either FHAS or HCECs as determined by the exclusion of the vital dye CFDA-AM (Molecular Probes, Eugene, OR, U.S.A.) (data not shown).
ICAM-1
The expression of ICAM-1 mRNA in HCECs exposed to hypoxic ACM was up-regulated at 4, 8, 18, and 24 hours after the addition of hypoxic ACM, compared with ACM from normoxic FHAS (Fig. 1). In parallel, immunocytochemical staining for ICAM-1 demonstrated an up-regulation of ICAM-1 protein in cells exposed to hypoxic ACM for 24 hours (Fig. 2A) shown as both an increased number of immunopositive cells and a higher intensity of cell staining (Fig. 2A). The levels of ICAM-1 protein expression were quantified by ELISA in two independent experiments (Fig. 2B). A twofold increase of ICAM-1 levels was observed in HCECs exposed to hypoxic ACM for 24 hours compared with cells exposed to control ACM for the same period of time (Fig. 2B). ICAM-1 up-regulation is considered a hallmark of proinflammatory endothelial phenotype (Carlos and Harlan, 1994; Kishimoto and Rothlein, 1994). HCECs have previously been shown to up-regulate ICAM-1 when stimulated with cytokines, including IL-1β (Stanimirovic et al., 1997a,b).
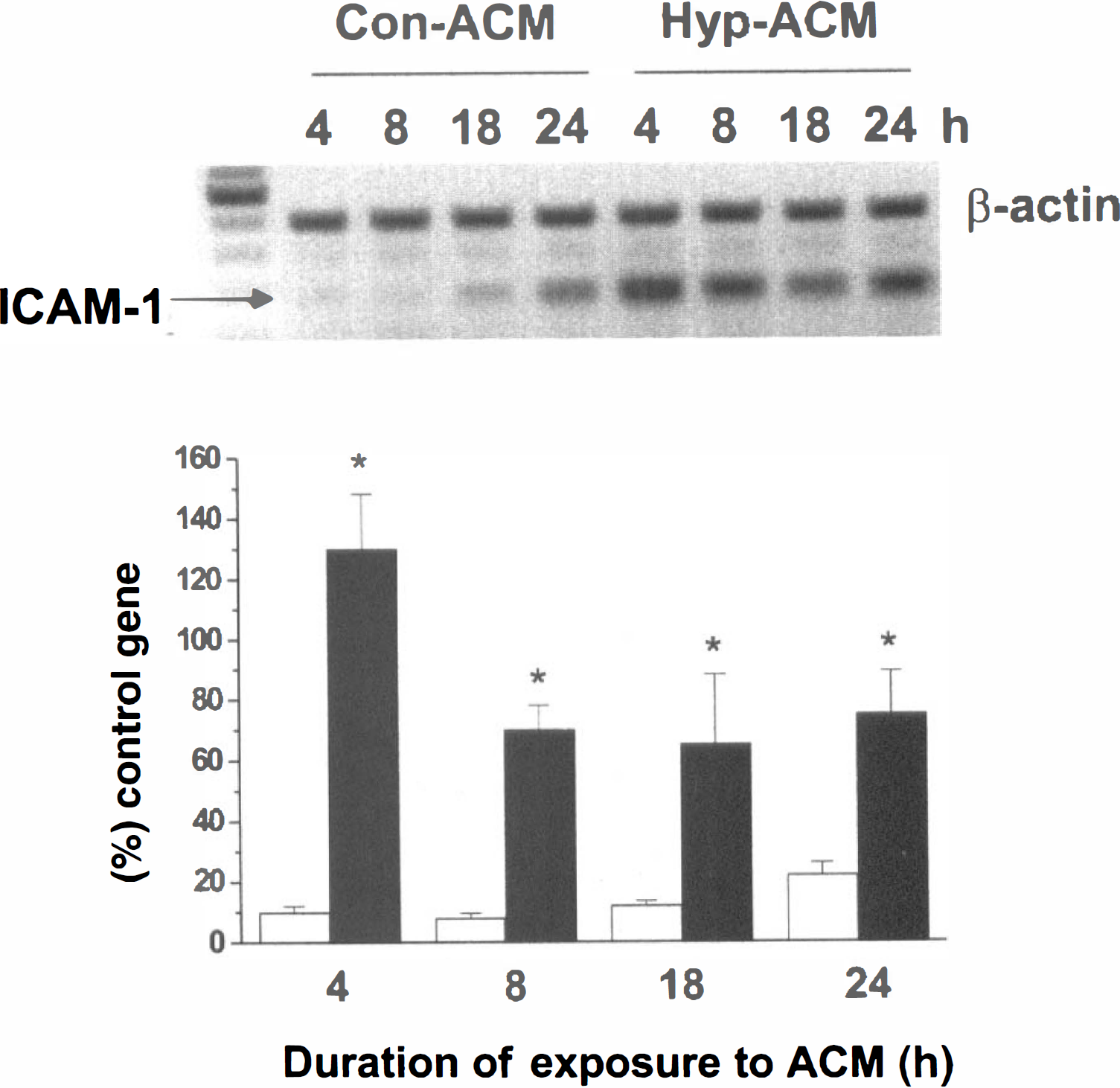
The effects of the media conditioned by either control (Con-ACM) or hypoxic (Hyp-ACM) fetal human astrocytes (FHAS) on the expression of ICAM-1 mRNA in human cerebral endothelial cells (HCEC). HCEC were exposed to Con-ACM or Hyp-ACM prepared as described in Materials and Methods for the indicated periods of time. ICAM-1 mRNA expression was determined by RT-PCR using β-actin as an internal control. Volumes of ICAM-1 bands for Con-ACM (open bars) and Hyp-ACM (closed bars) were expressed as a percentage of β-actin bands. Each bar represents the mean ± SD derived from four separate gels. *indicates a significant difference (P < 0.01; analysis of variance followed by the Fisher's protected least square difference multiple comparisons) from corresponding time-matched Con-ACM levels.
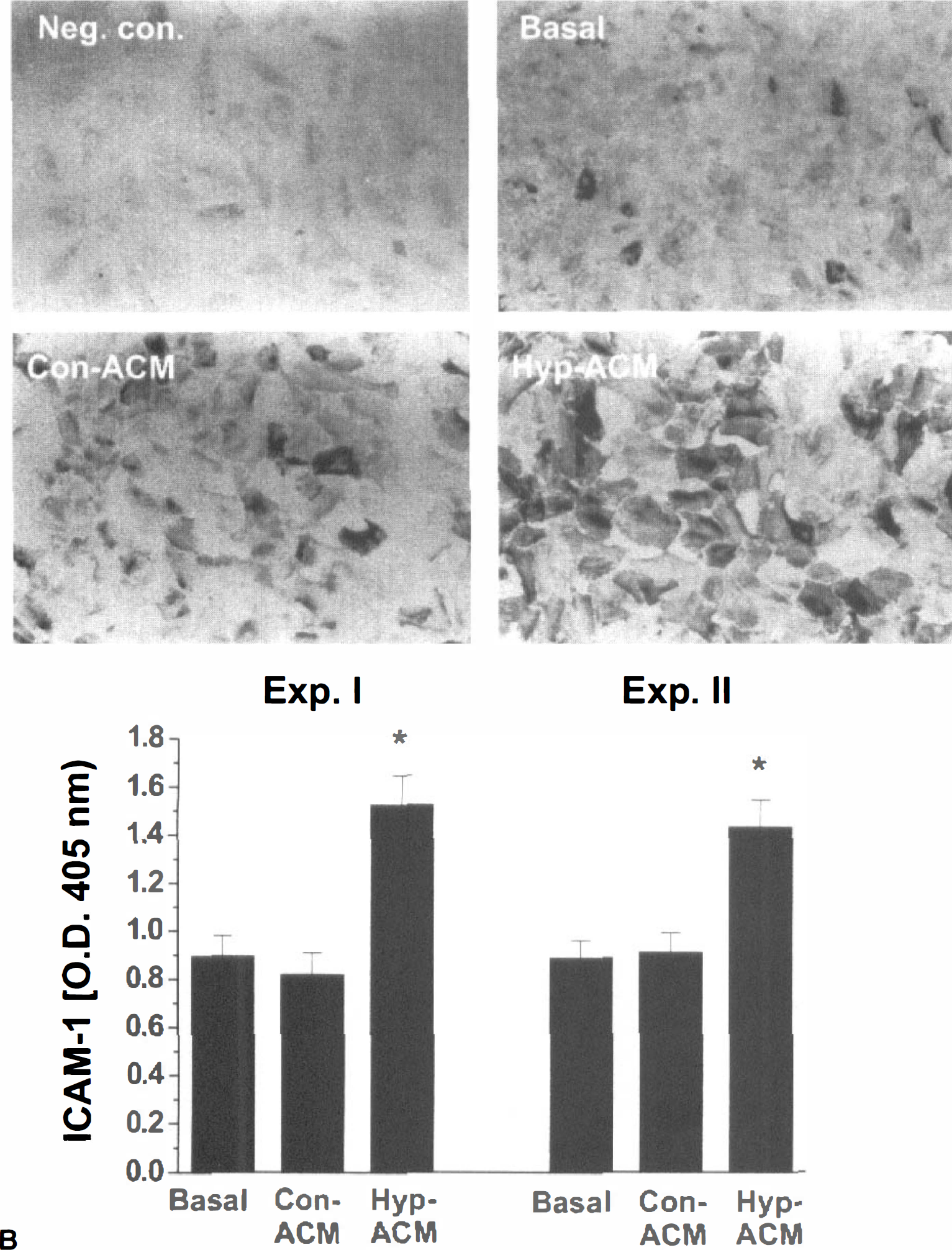
The effects of the media conditioned by either control (Con-ACM) or hypoxic (Hyp-ACM) fetal human astrocytes (FHAS) on the expression of ICAM-1 protein in human cerebral endothelial cells (HCEC) determined by immunocytochemistry
Chemokines
Hypoxic ACM was found to up-regulate both IL-8 (Fig. 3A) and MCP-1 (Fig. 3B) mRNA expression in HCECs compared with control ACM. Both IL-8 and MCP-1 mRNA expression was up-regulated at 4, 8, 18, and 24 hours after addition of hypoxic ACM to HCECs (Fig. 2A & B).
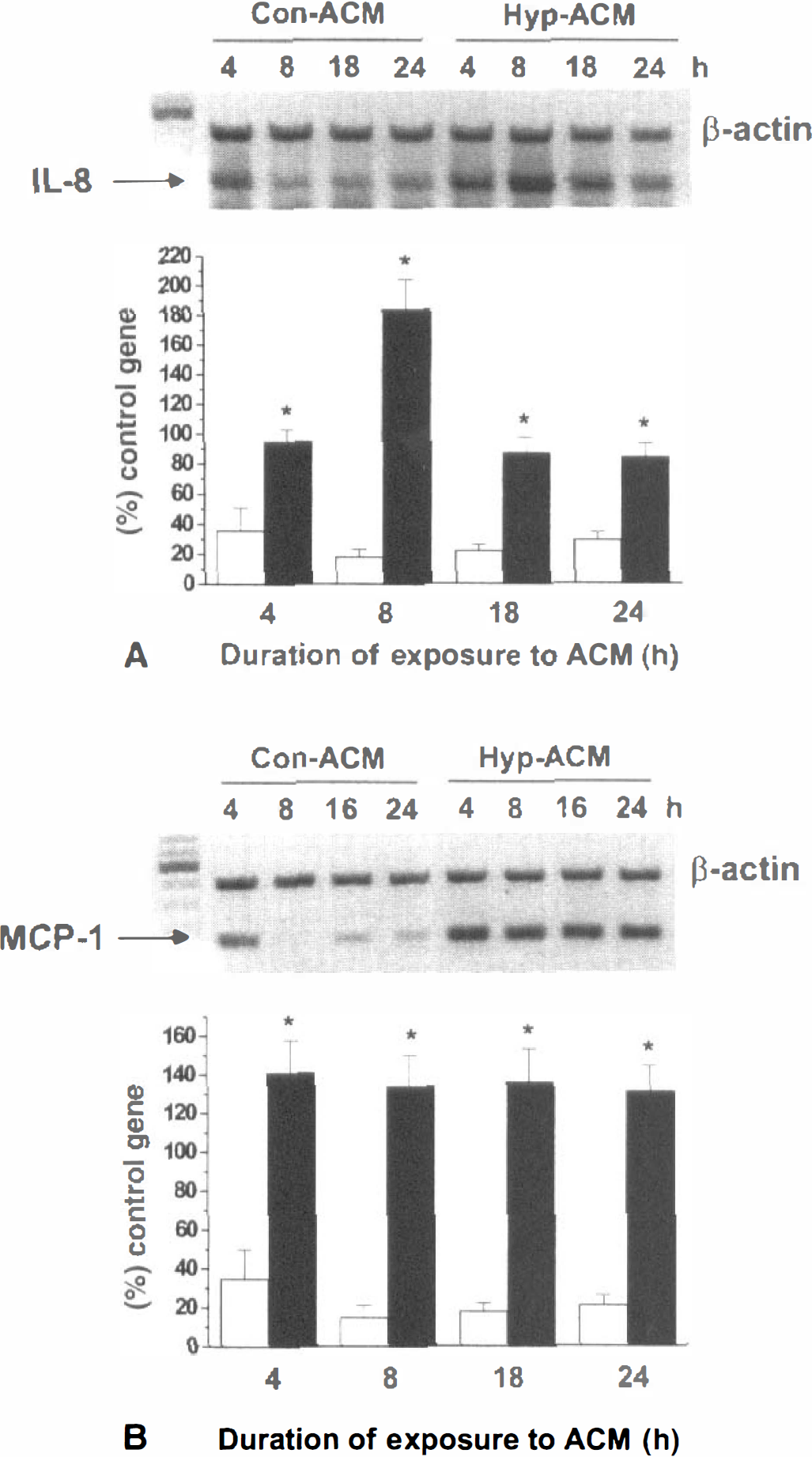
The effects of the media conditioned by either control (Con-ACM) or hypoxic (Hyp-ACM) fetal human astrocytes (FHAS) on the expression of IL-8
The levels of immunoreactive IL-8 secreted in media of HCECs exposed to hypoxic ACM were significantly higher than those measured in HCECs exposed to control ACM (Fig. 4A). The increased release of IL-8 from HCECs exposed to hypoxic ACM was apparent as early as 4 hours, reached maximum at 8 hours, and was continuously elevated to 24 hours after ACM addition. The levels of MCP-1 released by HCECs exposed to hypoxic ACM were five- to sevenfold higher from control at 4 to 8 hours (Fig. 3B) and remained elevated to 24 hours (Fig. 4B).
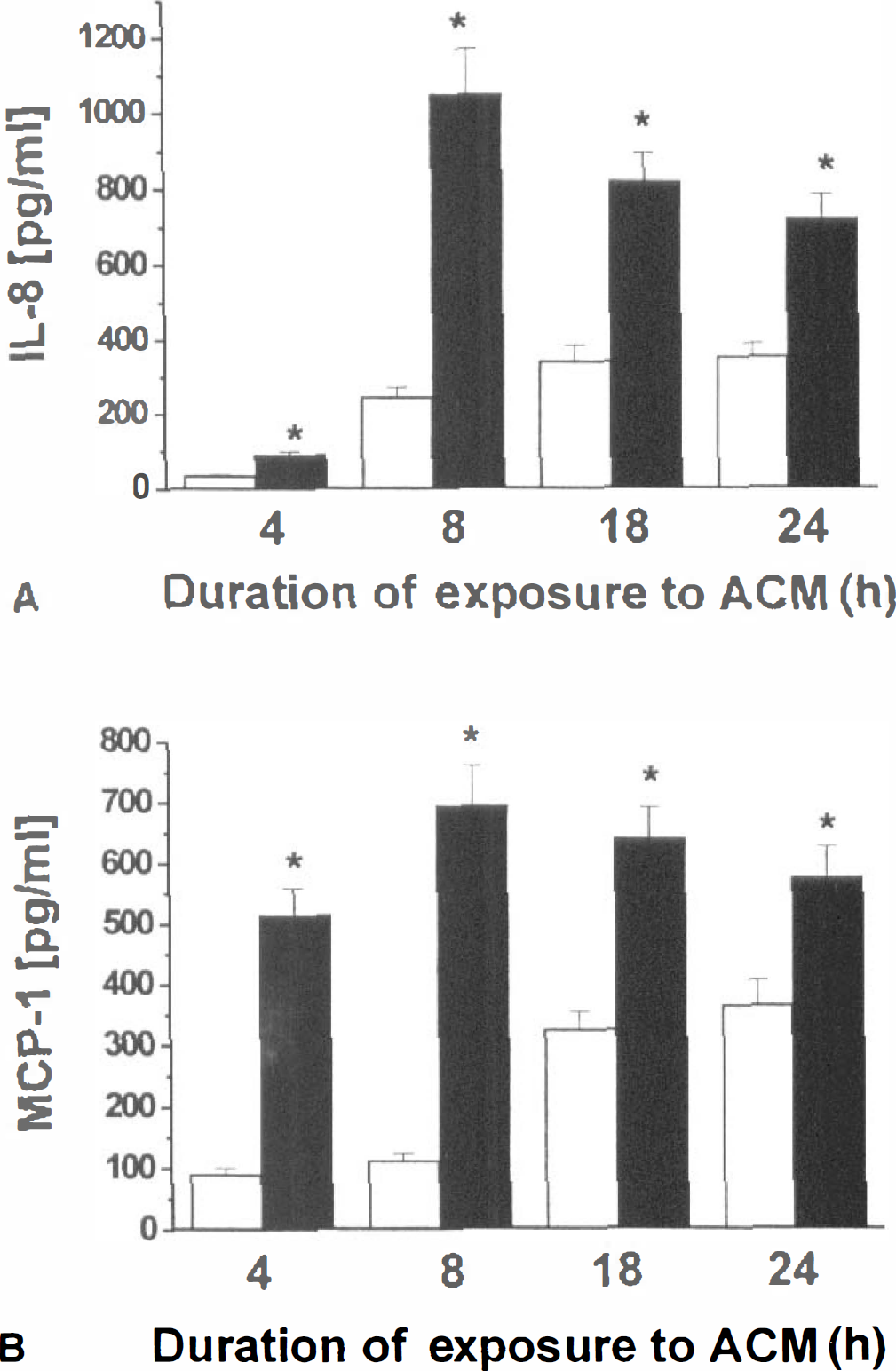
The effects of the media conditioned by either control (Con-ACM; open bars) or hypoxic (Hyp-ACM; closed bars) fetal human astrocytes (FHAS) on the release of immunoreactive IL-8
IL-1β and IL-1RI
The exposure of HCECs to ACM from hypoxic FHAS resulted in the up-regulation of IL-1β mRNA over the entire 4- to 24-hour period examined (Fig. 5A). In contrast, the same hypoxic ACM downregulated mRNA of IL-1RI, a signaling form of IL-1β receptor (Fig. 5B). A similar decrease in the IL-1RI mRNA expression was also seen in HCECs exposed to 100 UNITS/mL IL-1β for the same duration (Fig. 5B). The downregulation of IL-1RI by exogenous IL-1β has previously been shown in other cell types (Yu et al., 1997). These data suggested that mediators released in hypoxic ACM act on HCECs in a manner similar to that of IL-1β.
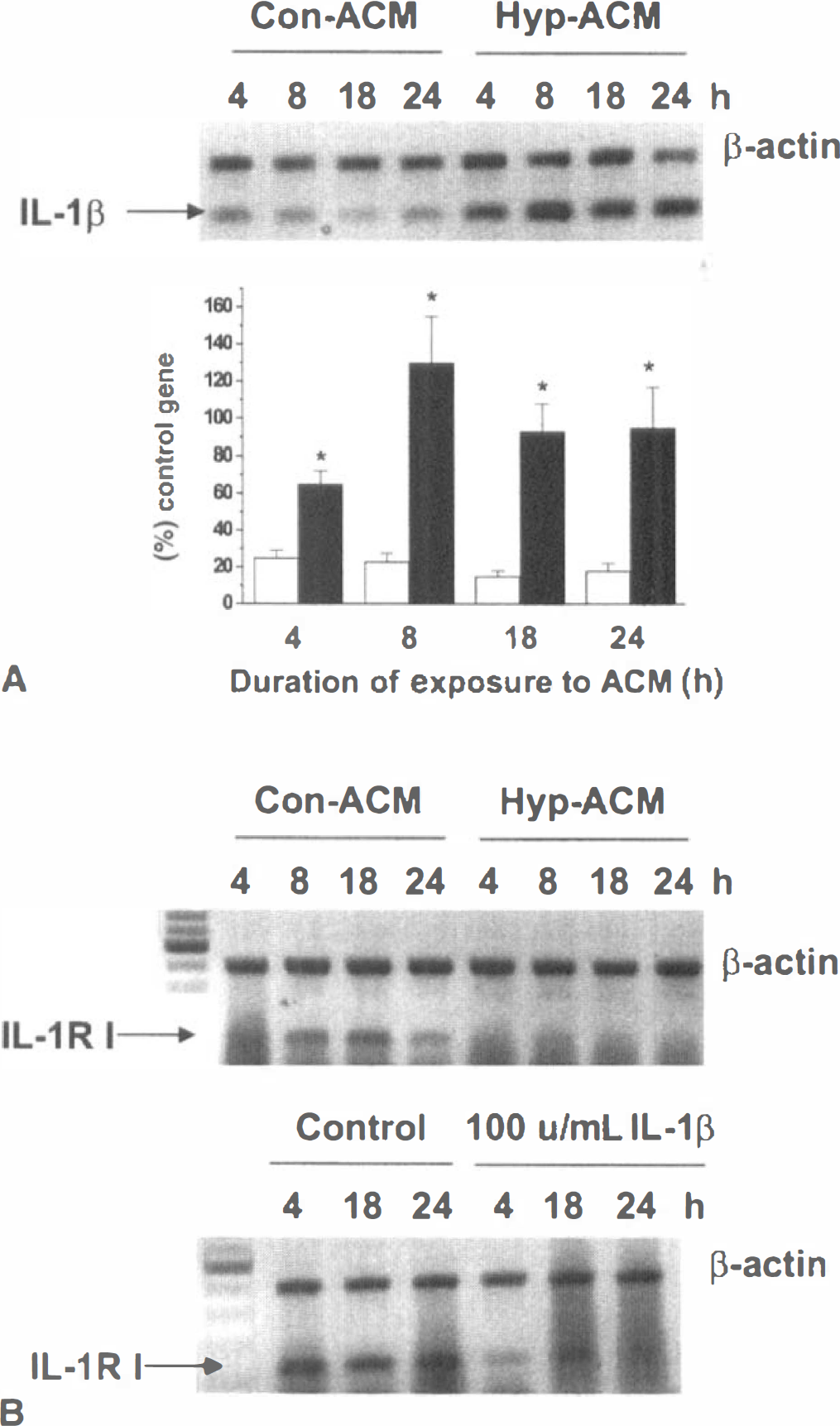
The effects of the media conditioned by either control (Con-ACM) or hypoxic (Hyp-ACM) fetal human astrocytes (FHAS) or IL-1β on the expression of IL-1β and IL-1RI in HCEC. HCEC were exposed to Con-ACM or Hyp-ACM prepared as described in Materials and Methods, or to 100 UNITS/mL of IL-1β for the indicated periods of time. IL-1β
Effects of hypoxia on expression of IL-1β and ICE in human astrocytes
To explore the possibility that IL-1β is an active component of hypoxic ACM that induces inflammatory HCEC phenotype, we examined the effect of hypoxia on the expression of IL-1β and other components of the IL-1β system in FHAS.
Hypoxia/reoxygenation was found to induce ICE mRNA in FHAS (Fig. 6A), which suggests that an active form of IL-1β could be produced/secreted by hypoxic FHAS. Indeed, IL-1β mRNA was found to be transiently up-regulated in FHAS at 4 hours and 18 hours of reoxygenation after a 4-hour hypoxia (Fig. 6B). The levels of immunoreactive IL-1β secreted into FHAS-conditioned media were elevated at 4, 18 and 24 hours of reoxygenation (Figure. 6C).
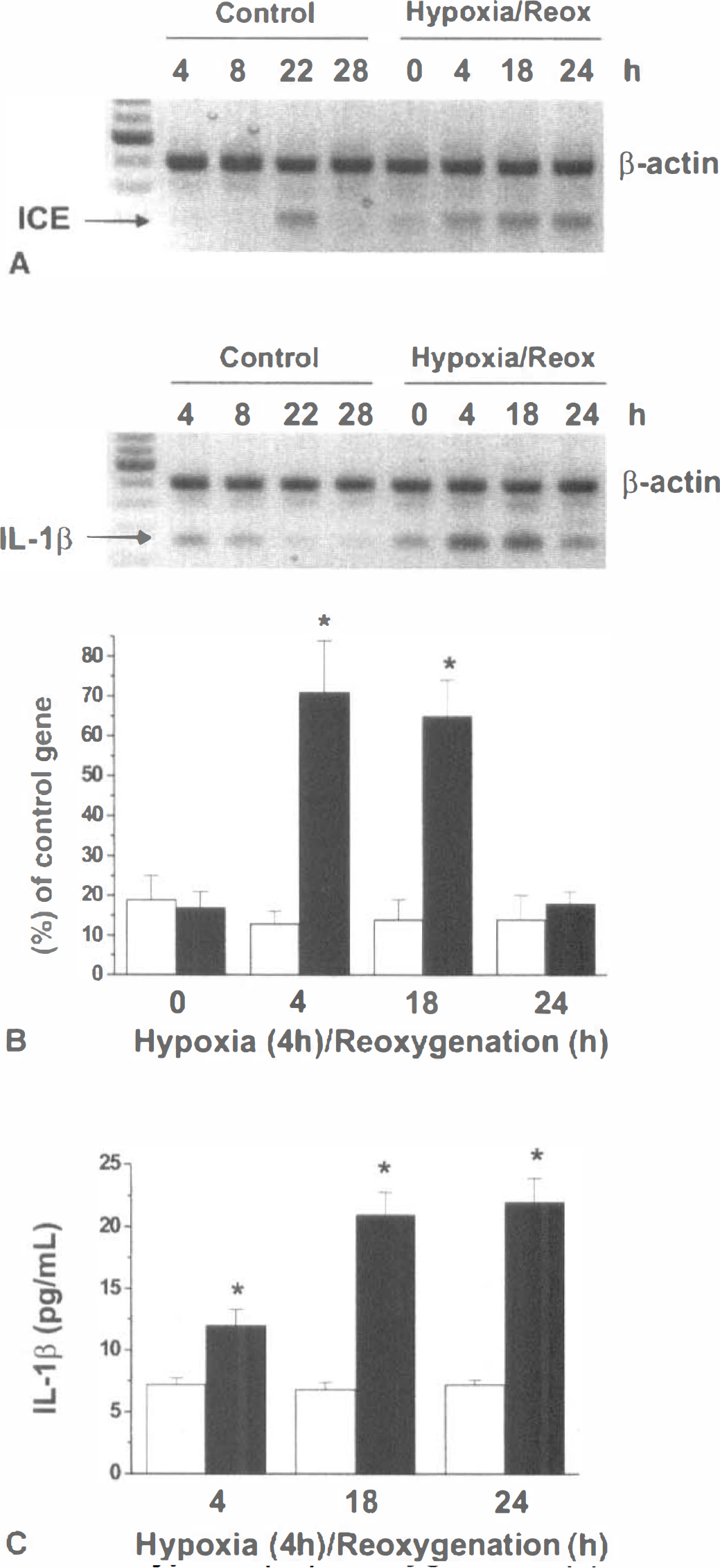
The effects of hypoxia-reoxygenation on the expression of ICE
In addition to IL-1β, TNF-α mRNA was also upregulated in hypoxic FHAS (data not shown). However, we failed to detect immunoreactive TNF-α (detection limit, 50 pg/mL) in ACM from either control or hypoxic FHAS (data not shown).
Effects of dexamethasone on hypoxia-induced IL-1β expression in human astrocytes
Treatment of FHAS with 10 μmol/L dexamethasone (DEX) for 45 minutes before and during hypoxia resulted in a downregulation of IL-1β mRNA expression in control FHAS, and a significant inhibition of the hypoxia-stimulated IL-1β mRNA expression in FHAS (Fig. 7A). The levels of immunoreactive IL-1β secreted in hypoxic ACM of DEX-treated FHAS were reduced to control values at 24 hours of reoxygenation (Fig. 7B). DEX has been shown to selectively inhibit the expression of IL-1β at both transcriptional and posttranscriptional levels in other cell types (Colotta et al., 1998; Kern et al., 1988; Knudsen et al., 1987; Lee et al., 1988).
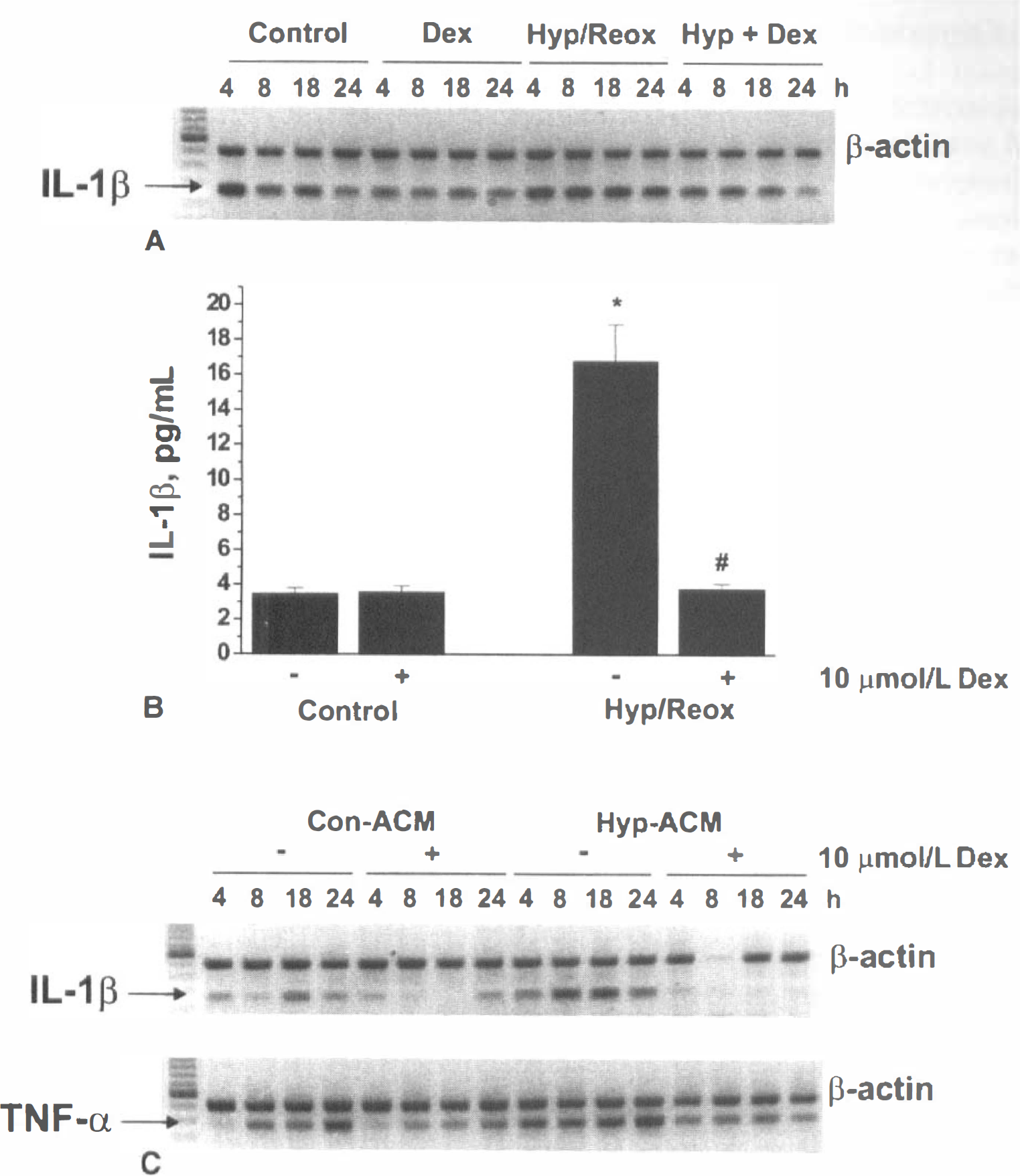
The effects of dexamethasone (Dex) on the hypoxia-induced IL-1β mRNA expression
The ability of ACM from DEX-treated FHAS to induce inflammatory genes in HCECs was examined. DEX was added to astrocytes 45 minutes before hypoxia, and media were changed after a 4-hour hypoxic period. Media were conditioned over a 24-hour reoxygenation period in the absence of DEX. HCECs exposed to ACM from DEX-treated control FHAS showed lower expression of both IL-1β and TNF-α mRNA (Fig. 7C) compared with control ACM. Furthermore, ACM from DEX-treated hypoxic FHAS failed to up-regulate either IL-1β or TNF-α mRNA in HCECs (Fig. 7C). ACM from DEX-treated hypoxic FHAS also failed to up-regulate IL-8, MCP-1, and ICAM-1 mRNA in HCECs (data not shown).
Effects of IL-1 receptor antagonist on HCEC activation induced by the hypoxic ACM
Additional evidence linking IL-1β to the ability of hypoxic ACM to trigger a proinflammatory HCEC activation was obtained by using IL-1β receptor antagonist (IL-1Ra) (R&D Systems, Minneapolis, MN, U.S.A.) to inhibit ACM-induced responses in HCECs. IL-1Ra binds to both IL-1RI and IL-RII receptors and has been shown to inhibit IL-1β-induced responses in various cell types (Arend et al., 1998; Dinarello, 1994, 1998).
HCECs were incubated with ACM from either control or hypoxic FHAS in the absence or presence of 50 ng/mL IL-1Ra. This concentration of IL-lRa was shown to inhibit 50 UNITS/mL IL-1β-induced IL-8 and ICAM-1 expression in HCECs (data not shown). IL-1Ra completely inhibited hypoxic ACM-induced ICAM-1, IL-8, and MCP-1 mRNA expression (data not shown). Hypoxic ACM induction of ICAM-1 protein in HCECs was completely prevented in the presence of IL-1Ra at 24 hours of exposure (Fig. 8A). In addition, ACM-pretreated with the neutralizing anti-IL-1β antibody (R&D Systems, Minneapolis, MN, U.S.A.), but not anti-TNF-α antibody (NeoMarkers, Union City, CA, U.S.A.), lost the ability to up-regulate ICAM-1 in HCECs (Fig. 8B). In a similar manner, the secretion of immunoreactive IL-8 (Fig. 8C) and MCP-1 (Fig. 8D) by HCECs was reduced to or below control levels after 4-hour or 24-hour exposure to hypoxic ACM in the presence of IL-1Ra. These experiments provide strong evidence that IL-1β released by hypoxic astrocytes is an important trigger of inflammatory HCEC phenotype.
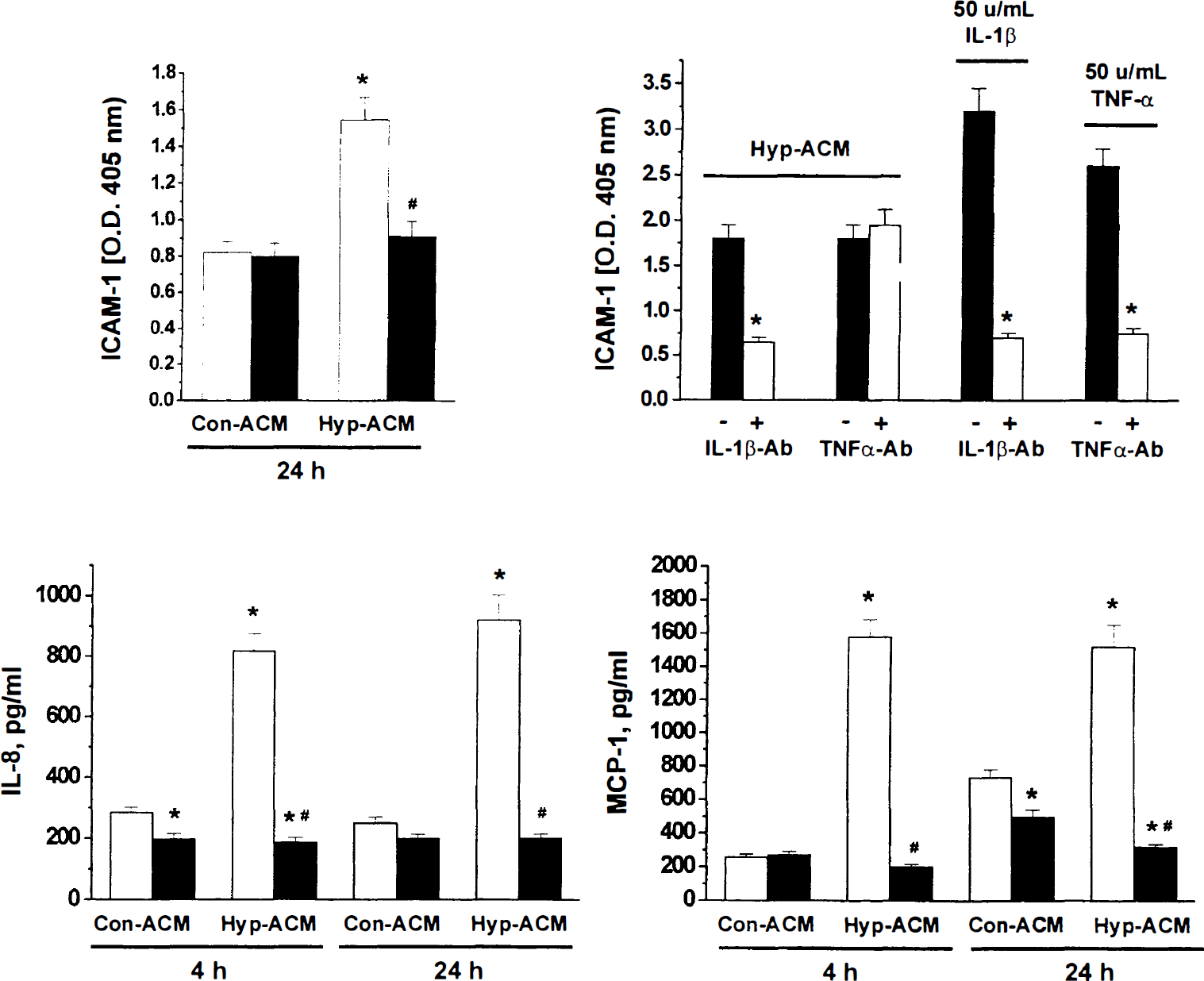
The effects of IL-1Ra and anti-IL-1β antibody on the ability of ACM from control (Con-ACM) and hypoxic (Hyp-ACM) FHAS to induce the expression of ICAM-1
DISCUSSION
This study demonstrates that hypoxic human astrocytes can evoke proinflammatory activation of human cerebral endothelial cells via mediator(s) secreted/released in cell media. Proinflammatory activation of HCECs by this paracrine pathway is manifested by the increased expression of the adhesion molecule ICAM-1, cytokines IL-1β and TNF-α, and the expression and secretion of chemokines IL-8 and MCP-1. Furthermore, the study provides strong evidence that IL-1β is a predominant HCEC-activating mediator secreted by hypoxic astrocytes. This conclusion is based on the following observations: (1) hypoxia up-regulated ICE and IL-1β mRNA expression in FHAS and increased secretion of IL-1β into FHAS media; (2) both hypoxic ACM and exogenous IL-1β downregulated the expression of IL-1RI in HCECs; (3) DEX simultaneously inhibited hypoxia-induced expression/release of IL-1 β from FHAS and the ability of hypoxic ACM to evoke proinflammatory activation of HCECs; and (4) anti-IL-1β antibody blocked ICAM-1 up-regulation and IL-1Ra inhibited proinflammatory activation of HCECs induced by hypoxic ACM. We have previously shown that IL-1β up-regulates ICAM-1 (Stanimirovic et al., 1997a), vascular cell adhesion molecule-1 and E-selectin (Stanimirovic et al., 1997b), and chemokines IL-8 and MCP-1 (Zhang et al., 1999) in HCECs. Taken together, these data suggest that IL-1β released from hypoxic astrocytes is a potent paracrine modulator of CEC activation/function in brain ischemia.
IL-1β has previously been identified as an important mediator of secondary ischemic brain damage in vivo (Dinarello, 1996; Feuerstein et al., 1997, 1998; Rothwell, 1999). An exogenous administration of IL-1β was found to exacerbate ischemic brain injury after MCAO in rat (Yamasaki et al., 1995). Animal studies have also shown that the expression of IL-1β is up-regulated between 24 hours and 7 days after focal brain ischemia (Davies et al., 1999) and that levels of IL-1β expression correlate well with the degree of ischemic brain damage (Feuerstein et al., 1998; Rothwell, 1999). IL-1β immunoreactivity in the ischemic brain has been localized to astroglia (Orzylowska et al., 1999), microglia, and scattered perivascular macrophages (Sairanen et al., 1997).
An active form of IL-1β is cleaved from the inactive, 269-amino-acid precursor pro-IL-1β by ICE/caspase I (Arend et al., 1998; Colotta et al., 1998). Increased levels of IL-1β (Orzylowska et al., 1999) and induced expression of ICE (Bhat et al., 1996) have been detected in animals after focal cerebral ischemia. The inhibition of ICE family proteases (Hara et al., 1997) as well as ICE gene knockout in mice resulted in the reduction of excitotoxic and ischemic brain injury (Schielke et al., 1998).
Several lines of evidence obtained in this in vitro study suggest that astrocytes exposed to hypoxia are a source of IL-1β. Hypoxia was found to up-regulate ICE mRNA expression in FHAS and this was accompanied by the secretion of immunoreactive IL-1β in astrocyte media. In addition, the ability of hypoxic astrocytes to induce proinflammatory genes in HCECs in a paracrine manner correlated well with the hypoxic stimulation of IL-1β expression and release by FHAS.
Both HCECs and FHAS were found to express a signaling receptor for IL-1β, i.e., IL-1RI, and IL-1RI downregulation was observed in response to exogenous addition of IL-1β and hypoxic ACM. IL-1RI is a receptor that mediates all the biological activities of IL-1β and α (Colotta et al., 1998; Dinarello, 1998; Rothwell, 1999), and IL-1RI downregulation by the ligand, IL-1β, may be a potentially important control mechanism that limits propagation of inflammatory response. The expression of IL-1RI has previously been demonstrated in rat cerebral endothelial cells (Van Dam et al., 1996). Recently, an increase in IL-1RI mRNA was detected in the hippocampus 8 to 24 hours after a transient global brain ischemia (Sairanen et al., 1997), and in the cerebral cortex 5 days after MCAO (Wang et al., 1997) in the rat, which suggests that neurons in discrete areas are target cells for IL-1β. IL-1RI expression by HCECs and FHAS shown in this study indicates that these cells are also likely targets of IL-1β in the brain.
DEX, a “classic” inhibitor of IL-1β in various cell types (Kern et al., 1988; Knudsen et al., 1987; Lee et al., 1988), was observed to simultaneously inhibit hypoxia-induced IL-1β expression/release in FHAS and the ability of hypoxic ACM to effect HCEC activation. This provides further evidence that IL-1β is an active component of ACM involved in stimulation of inflammatory genes in HCECs. Finally, IL-1Ra, a selective and specific endogenous receptor antagonist that binds IL-1RI and blocks actions of both IL-1α and IL-1β (Arend et al., 1998; Dinarello, 1998; Rothwell, 1999), eliminated the up-regulation of inflammatory genes and secretion of inflammatory mediators from HCECs induced by hypoxic ACM, which suggests that IL-1β released into hypoxic ACM triggers proinflammatory activation of HCECs. Blockade of IL-1 function in vivo by infusion of IL-1Ra rat (Garcia et al., 1995), or topical administration of anti-IL-1β antibody (Yamasaki et al., 1995), has been shown to significantly reduce infarct lesion, neuronal death, and neutrophil infiltration after MCAO. In a recent study, mice overexpressing IL-1Ra showed a reduction in ICAM-1–positive brain vessels after permanent focal ischemic insult (Yang et al., 1999). These studies and evidence presented in this study implicate IL-1β as an important mediator of both ischemic inflammatory changes in cerebral vasculature and ischemic brain damage.
In addition to IL-1β, in vitro hypoxia was also found to increase the expression of TNF-α mRNA in FHAS but no immunoreactive TNF-α was detected in ACM. Other studies have demonstrated that TNF-α is present as a biologically active membrane-bound form in many cell types and only a small amount of the membrane-bound TNF-α is cleaved by a metalloproteinase(s) into a free form (Decoster, et al., 1995; Grell et al., 1995). In a similar manner, IL-1α, in contrast to IL-1β, has been localized in cell cytosol and associated with cell membranes and is not released into cell media (Colotta et al., 1998). These observations suggest that TNF-α and IL-1α are not significant mediators of HCEC activation by hypoxic ACM. This is supported by the observation that the antibody against IL-1β effectively prevented hypoxic ACM-induced ICAM-1 expression in HCECs, whereas anti-TNF-α antibody was ineffective. However, we cannot completely exclude the possibility that proinflammatory activation of HCECs is aided by other factors secreted in hypoxic ACM, notably IL-8 (Zhang et al., 1999).
The in vitro experimental paradigm used in this study bears significant differences from in vivo models of stroke. Several important components of stroke in vivo, such as blood “flow,” acidosis, vascular reactivity, and changes in blood rheology, are absent in cell culture models. Furthermore, brain endothelial cells in vivo are exposed to various paracrine sources of “proinflammatory” mediators including those released by smooth muscle cells, pericytes, and perivascular macrophages. Whereas, in vivo, even small amounts of mediator released in intercellular spaces can reach very high local (i.e., physiologically relevant) concentrations, mediators released into ACM are substantially diluted when applied to HCECs. For example, in vivo, brain microvessels are enveloped by astrocytes and close proximity of the two cell types would allow for much higher local concentrations of IL-1β than those reached in conditioned media. This combination of factors may account for discrepancies in the magnitude of changes in any particular endothelial gene observed in this study and those seen in in vivo studies, notably multifold increases in ICAM-1 expression observed in microvessels in in vivo models of stroke (Wang et al., 1994).
Close apposition of CEC and astrocytes, which invest 99% of the abluminal surface of the capillary basement membrane in the brain in vivo, provides for the anatomic proximity necessary to exert paracrine interactions. Evidence from grafting experiments, developmental studies, and culture models demonstrate inductive influences of perivascular astrocytes on brain endothelium and vice versa (Abbott et al., 1992). Therefore, cooperation between two cell types is likely involved in initiating microvascular and inflammatory responses to ischemia. Peripheral leukocyte mobilization into the ischemic brain is critically dependent on the “inflammatory” status of CEC including CEC expression of adhesion molecules and chemokines. Our previous studies have shown that HCECs “activated” by simulated in vitro ischemia or exogenous IL-1β up-regulate the expression of ICAM-1, avidly bind freshly isolated neutrophils in an ICAM-1/CD18-dependent manner (Stanimirovic et al., 1997a), and release potent chemoattractants that stimulate neutrophil chemotaxis in vitro (Zhang et al., 1999). A maintenance/enhancement of this initial activation is likely mediated via paracrine cytokines released by neighboring cells. Based on the results of this study, it seems that cerebral endothelium is an important target for IL-1β produced by hypoxic perivascular astrocytes. The ability of IL-1Ra to reduce ischemic damage in vivo (Garcia et al., 1995) is probably, in part, the result of both attenuated CEC activation via IL-1β produced by perivascular astrocytes and reduced leukocyte infiltration into the brain. Therefore, therapies that target endothelial IL-1RI or reduce the production/release of IL-1β by glial cell compartment may be successful strategies to alleviate postischemic brain inflammation and secondary brain damage.
Footnotes
Acknowledgments
We thank Dr. Edith Hamel, and Dr. Jack Antel of Montreal Neurological Institute, for providing human temporal lobe biopsies and primary cultures of fetal human astrocytes, respectively, and Mrs. Rita Ball for preparation of human cerebromicrovascular endothelial cell cultures.