
Abstract
The pathogenesis of cerebral malaria (CM) remains largely unknown. There is growing evidence that combination of both parasite and host factors could be involved in blood–brain barrier (BBB) breakdown. However, lack of adequate
Keywords
Introduction
Malaria is a major public heath issue in developing countries with almost 1 million deaths per year (WHO, 2008).
Despite the effort of the scientific community, the pathogenesis of CM remains unknown. Several studies have pointed out that sequestration of parasitized red blood cells (PRBCs) is at the origin of this pathology, as it results in microcirculatory obstruction, decreased oxygen delivery, tissue hypoxia, metabolic acidosis, hyperlactatemia, and organ damage. This phenomenon has been reported to be higher in the brain of patients dying from CM than in other organs in the same patient and also higher than in patients without CM (MacPherson et al, 1985; Pongponratn et al, 1991). Therefore, PRBCs sequestration is necessary to the pathogenesis, but there is also evidence that it might not be sufficient by itself in initiating or maintaining the processes leading to CM and death, as demonstrated by the presence of sequestered PRBCs in non-CM cases (MacPherson et al, 1985; Montgomery et al, 2006).
There is growing evidence that a combination of both parasite and host environment could be implied in the blood–brain barrier (BBB) breakdown and in the pathogenesis of CM (Medana and Turner, 2006). Immunohistochemical and ultrastructural studies of postmortem brain tissue have shown brain endothelial cell (EC) activation, with formation of villous processes, which adhere to sequestered parasites (Brown et al, 1999a; MacPherson et al, 1985). It has also been described an upregulation of constitutively expressed molecules, such as human intercellular adhesion molecule 1 (hICAM-1) (Chakravorty and Craig, 2005; Tripathi et al, 2006), and reduction of junctional proteins expression at the BBB endothelium (Brown et al, 1999a; Pongponratn et al, 2003). Furthermore, the fact that plasma proteins leakage has been observed into the central nervous system (Boonpucknavig et al, 1990; Brown et al, 1999b) confirms that there is an alteration in the BBB integrity.
Until now, the hypothesis of the implication of PRBC interaction with the EC in BBB dysfunction has been difficult to study because of the lack of an adequate
New progresses have been recently achieved in the generation of a new human cerebral EC line named hCMEC/D3. These brain cells have been immortalized and have been shown to display a large number of human BBB characteristics, including tight junction proteins, adhesion molecules, chemokines receptors, and multidrug resistance proteins expression (Weksler et al, 2005). This cell line is widely used as a powerful tool to mimic BBB behavior in a large number of fields: studies of pathogens behavior, HIV (Afonso et al, 2008), meningoccocus (Coureuil et al, 2009), pathologies (prion protein) (Viegas et al, 2006), multiple sclerosis (Rampon et al, 2008), stroke (Cucullo et al, 2008), Alzheimer's disease (Vukic et al, 2009), inflammatory situation, and signaling pathway (tight junction regulation) (Eum et al, 2008; Sade et al, 2009).
We propose here the use of this well-established human cerebral EC line hCMEC/D3 to explore the implication of parasite factors and environmental conditions in the BBB breakdown and in the pathogenesis of CM.
Materials and methods
Cell Culture
The hCMEC/D3 cells were cultured on collagen type I (BD Biosciences, Bedford, MA, USA)-coated culture dishes at 37°C and 5% CO2 as described (Afonso et al, 2008). The EC culture medium (ECCm) contained EBM-2 (endothelial basal medium) basal medium (Lonza, Walkersville, MD, USA), supplemented with 5% heat-inactivated fetal bovine serum (Gibco), 10 mmol/L HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, Paisley, UK), 1.4 μmol/L hydrocortisone, 5 μg/mL ascorbic acid, and 1 ng/mL basic fibroblast growth factor (Sigma-Aldrich, Steinheim, Germany). Mouse brain EC bEnd5 cell line was a generous gift from Professor Engelhardt (Theodor Kocher Institut, University of Bern) and was cultured on gelatine (Sigma-Aldrich)-coated cell culture dishes in DMEM (Dulbecco's Modified Eagle Medium) (4.5 g/L glucose) supplemented with 10% fetal bovine serum, 2 mmol/L
P. falciparum Parasite Culture and Tryspin Treatment
For trypsin treatment, parasites were washed in Hank's balanced salt buffer, after gelatine enrichment, and incubated either in buffer or in 0.5 mg/mL trypsin/EDTA (ethylenediaminetetraacetic acid) at 10% final hematocrit for 5 minutes at room temperature. Reaction was stopped by adding RPMI 1640/5% fetal bovine serum medium.
Cytoadhesion Assay
The hCMEC/D3 cells were seeded onto collagen type I-coated eight-well Lab-Tek chamber slides (Nunc, Rochester, NY, USA), grown in ECCm to confluence and stimulated with 150 U/mL of tumor necrosis factor-α (TNFα) (Biosource, Camarillo, CA, USA) for 24 hours before the assay. Cells were fixed with 4% paraformaldehyde for 15 minutes and kept at 4°C until used. Mature forms (late trophozoites and schizonts) of
Alternatively, cells nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) after fixation. In parallel, gelatine-enriched parasites were stained with 2 mmol/L (((4-chloromethyl) benzoyl) amino) tetramethylrhodamine, orange cell tracking dye (Interchim, Montluçon, France), according to the manufacturer's instructions. Stained cells and PRBCs were incubated for 1 hour in cytoadherence culture medium at 37°C and 5% CO2, with gentle agitation every 15 minutes. Cocultures were washed in RPMI 1640 medium, air-dried, methanol fixed, and mounted for fluorescence microscopy analysis. For each well, 16 homogeneously distributed fields (magnification × 200) were imaged under the red filter (band pass, BP 610/75 nm) for PRBCs and under DAPI filter (BP 470/40 nm) for nuclei using a Leica epifluorescence microscope (Leica DMI 4000B, Leica Microsystems, Wetzlar, Germany) and a Zeiss camera (AxioCam MRc5, Oberkochen, Germany). In each wavelength, four images were assembled together using Zeiss software mounting tool and resulting TIFF images were analyzed using Image J software. The number of adherent PRBCs was counted on red filter images, and cells were counted on DAPI filter images by counting nuclei. The results were expressed as the number of adherent PRBCs per adherent cell.
Parasite Panning
Gelatine-enriched RAOL PRBCs were coincubated with TNFα-stimulated hCMEC/D3 cells in cytoadherence culture medium at 5% hematocrit for 1 hour at 37°C and 5% CO2, with gentle agitation every 15 minutes. Nonadherent parasites were removed by washing with RPMI 1640 medium. Adherent parasites were harvested by slight pipette flow, directly onto cells, with complete PCm at pH = 7.18, which facilitates PRBC detachment from cells. The obtained parasites were cultured, and the medium was changed the next day.
RNA Interference
The hCMEC/D3 cells were transfected with 20 nmol/L siRNA targeting either hICAM-1 or the corresponding siRNA scramble using Lipofectamine RNAimax (Invitrogen, Carlsbad, CA, USA). Transfected cells were allowed to settle down for 4 hours and then stimulated with 150 U/mL of TNFα for 24 hours. Transfected cells were analyzed for hICAM-1 expression by flow cytometry or used for PRBCs cytoadherence assay.
Flow Cytometry Analysis
The hCMEC/D3 or bEnd5 cells were harvested from culture dishes by treatment with 0.5 mg/L heated trypsin/EDTA (Gibco) for a very short period of time (3 minutes) and stained with anti-human ICAM-1 fluorescein-conjugated monoclonal antibody, or with the isotype control antibody (R and D system, Minneapolis, MN, USA). Short-time trypsin treatment did not alter surface antigens expression. Cells were washed with phosphate-buffered saline (PBS) and fixed with 1% formaldehyde and analyzed by flow cytometry using an EPICS XL cytometer (Beckman Coulter, Villepinte, France).
Lentiviral Production and Transduction
The lentiviral construct pTRIP-CMV-hICAM-1 ΔU3 was generated by gateway recombination cloning (Invitrogen). Briefly, the hICAM-1-coding sequence was PCR amplified from a plasmid using the following primers: hICAM-1-S 5′-ATGGCTCCCAGCAGCCCCCG-3′ and hICAM-1-AS 5′-TCAGGGAGGCGTGGCTTGTG-3′. The 1.6-kb resulting PCR product was cloned into the pENTR/D/TOPO plasmid (Invitrogen) to generate entry clone. The LR clonase II (Gateway Cloning System) recombination was performed using the entry clone and the pTRIP-CMV-rfa gateway ΔU3 destination vector as described previously (Russ et al, 2008). The pTRIP-CMV-GFP ΔU3 vector was used as control. Lentiviral vector stocks were produced by transient transfection of 293T cells with the p8.91 encapsidation plasmid, the VSV glycoprotein-G-encoding pHCMV-G plasmid, and the pTrip ΔU3 lentiviral vector as previously described (Zennou et al, 2000). Supernatants were treated with DNAse I (Roche Diagnostic, Mannheim, Germany) before ultracentrifugation, and the resulting pellet was resuspended in PBS, separated into aliquots and frozen at −80°C until use.
Mouse bEnd5 cells were transduced in suspension with the lentiviral vectors premixed for 15 minutes with 10 mg/mL DEAE-Dextran at 37°C. Transduced mouse cells were then seeded onto gelatine-coated culture dish in a minimal volume of bEnd5 culture medium. One hour on transduction, fresh medium was added. Two days after transduction, cells were either analyzed by flow cytometry for hICAM-1 expression or used for cytoadherence assays.
Confocal Microscopy Analysis
The hCMEC/D3 or bEnd5 cells were trypsinized and stained using an anti-hICAM-1 antibody (BioVision, Mountain View, CA, USA), followed by a phycoerythrin-conjugated secondary antibody (Sigma-Aldrich). Cells were fixed with 1% formaldehyde, mounted, and analyzed. For tight junction proteins expression, hCMEC/D3 cells were seeded onto collagen type I-coated Lab-Tek chamber slides, grown for 12 days and fixed with 4% paraformaldehyde for 10 minutes at room temperature. Cells were then permeabilized with 0.2% Triton X100 in PBS for 10 minutes, blocked in 3% bovine serum albumin/PBS for 30 minutes, and stained with an anti-
Permeability Assay
The hCMEC/D3 cells were seeded onto 12 mm-collagen/70% ethanol-coated Millicell cell culture inserts (0.4 μm; Millipore, Carrigtwohill, Ireland). To allow a better tightness of the junctions between cells, they were cultured in ECCm for 12 days. Cells were TNFα stimulated for 24 hours before the assay. Gelatine-enriched mature parasites, at late trophozoite and schizont stages, were added in the upper chamber (luminal compartment) at a given hematocrit and parasitemia (8 × 107/cm2 was the starting parasite concentration, equivalent to 50% parasitemia and 2.5% hematocrit). To carry out coculture experiment during 20 hours, a modified culture medium (MCm) was used. The MCm contained RPMI 1640 medium supplemented with
Lactate Dehydrogenase Assay
Parasite lactate dehydrogenase was measured following the manufacturer's instructions (DiaMed-France S.A., Chambly, France).
Merozoites Purification
The PRBCs infected with mature forms at 50% parasitemia were cultured at 2.5% hematocrit in MCm for 20 hours to obtain the equivalent number of merozoites obtained when PRBCs 8 × 107/cm2 density were added to the culture during 20 hours. After parasites burst, supernatants were harvested and centrifuged at 1800
Annexin V Staining
The PRBCs apoptosis was measured by annexin staining following the manufacturer's recommendation. The PRBCs were washed with ice-cold-binding buffer and stained with annexin V for 15 minutes in the dark. After washing, PRBCs were stained with propidium iodide. Samples were maintained on ice until analyzed by flow cytometry using an EPICS XL cytometer.
Statistical Analysis
Mean and standard deviation have been determined after 3 independent experiments. For multiple comparisons, analysis of variance followed by
Results
hCMEC/D3 Cells Allow Parasitized Red Blood Cell Cytoadherence
A cytoadherence assay was performed to determine whether the
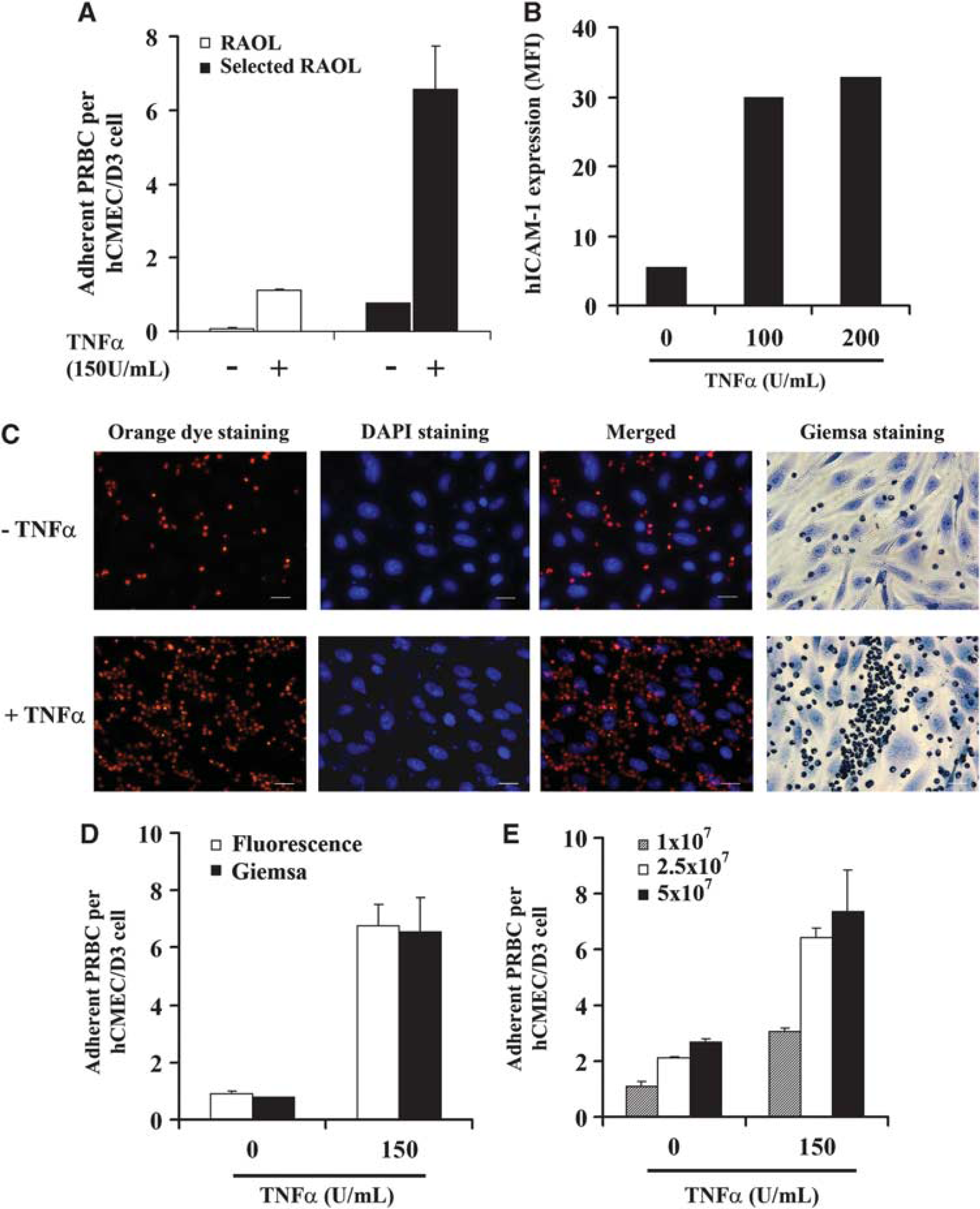
For a more rapid analysis of cytoadherence assay, we set up a new fluorescence-based detection protocol. The PRBCs were stained with the orange cell tracking dye and detected at 561 nm wavelength. hCMEC/D3 nuclei were stained with DAPI and detected at 460 nm wavelength (Figure 1C). The control Giemsa staining is also shown in Figure 1C. A semiautomatic quantification of the number of adherent PRBCs per hCMEC/D3 cell was obtained using Image J software. Figure 1D shows that both methods of cytoadherence quantification in nonstimulated or TNFa-stimulated cells yielded similar results. We further analyzed whether low doses of PRBCs are able to adhere to nonstimulated or stimulated hCMEC/D3 cells. As shown in Figure 1E, the lowest dose of PRBCs tested allows their adherence to nonstimulated hCMEC/D3 cells. The number of adherent PRBCs obtained using 2.5 × 107 PRBCs/cm2 increases by 2.5-fold compared with the adherent PRBCs observed using 1 × 107 PRBCs/cm2. Similar results were obtained using TNFa-stimulated hCMEC/D3 cells (Figure 1E). This result shows that hCMEC/D3 cells can support PRBCs cytoadherence even with low initial parasite load.
Human Intercellular Adhesion Molecule 1 Is Involved in Parasitized Red Blood Cells Cytoadherence to hCMEC/D3 Cells
To determine the adhesion molecule(s) responsible for
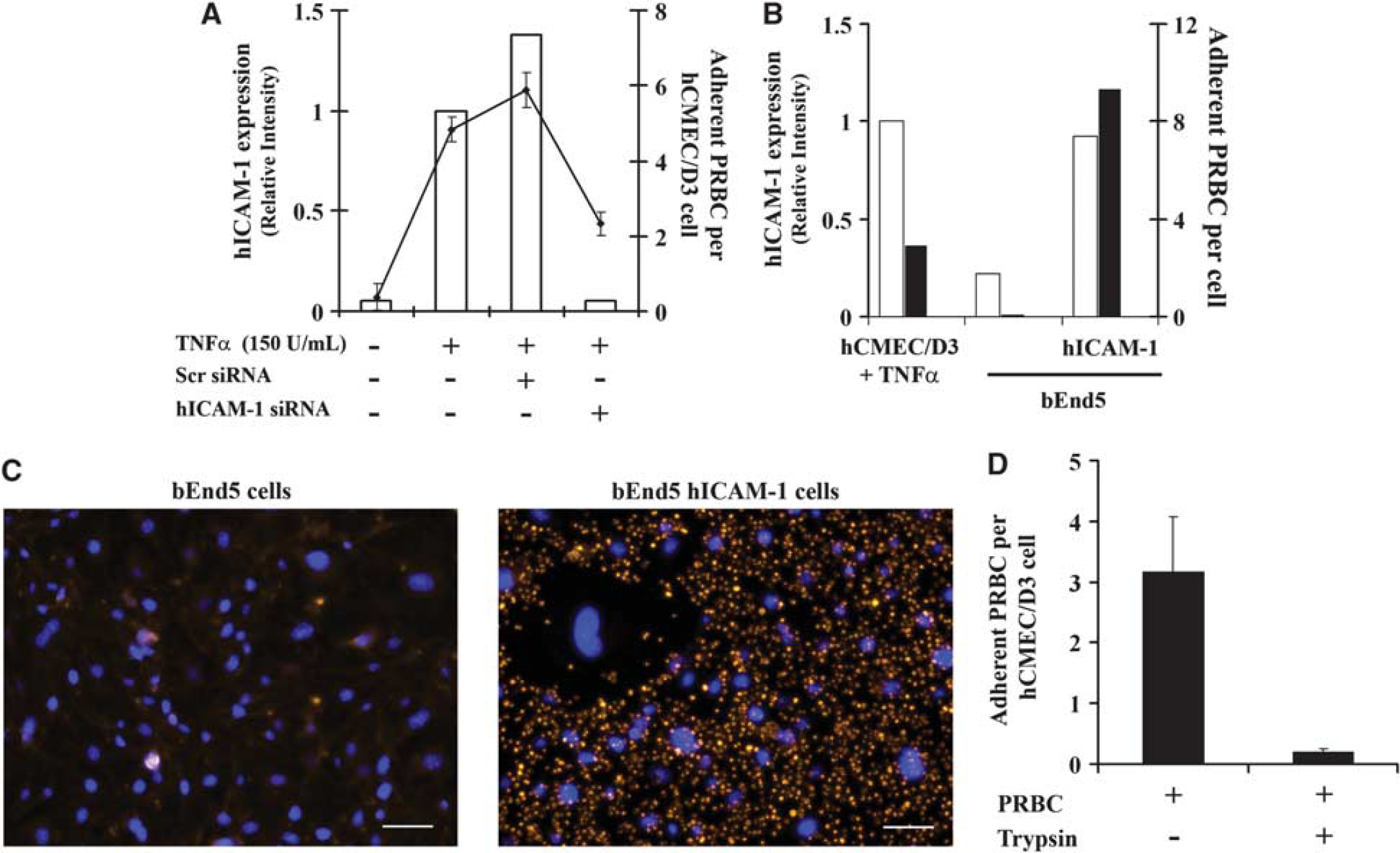
To further confirm these results, the mouse brain EC line bEnd5 was transduced using a lentiviral vector containing hICAM-1 sequence and assayed for
To go deeper into the mechanism of PRBCs cytoadherence, PRBCs were treated with trypsin to eliminate trypsin-sensitive
Parasitized Red Blood Cells Induce Endothelial Cell Permeability Enhancement and Modification of Tight Junction Proteins Expression
The following step of this study was to determine whether hCMEC/D3 cells could be used as a model to study the BBB breakdown triggered by PRBCs. The first parameter analyzed was the culture medium in which PRBCs and hCMEC/D3 cells could be cocultured. Cells were cultured alone or cocultured with RBCs or PRBCs in two-chamber culture inserts and the dye passing through was recovered in the lower chamber and analyzed 20 hours after coculture. We tested three different culture media: ECCm, PCm, and an MCm. Figure 3A shows that hCMEC/D3 cells form, as expected, a semipermeable barrier that reduces by 60% the amount of dye passing through the insert in all culture media analyzed. Coculture of hCMEC/D3 cells with RBCs does not modify significantly cell permeability compared with control cells. In contrast, coculture of cells with PRBCs induces a significant increase of the permeability, 43% in the MCm compared with cells cocultured with RBCs. No significant effect was observed when cells were cocultured in PCm or ECCm. As a consequence, MCm was used through the coculture experiments. We next analyzed the effect of parasite concentration on the permeability modification. Figure 3B shows that doses ranging from 1 to 4 × 107 parasites/cm2 have similar effect on the modification of the permeability. Ahigherdose (8 × 107 parasites/cm2) of parasite significantly increases cell permeability.
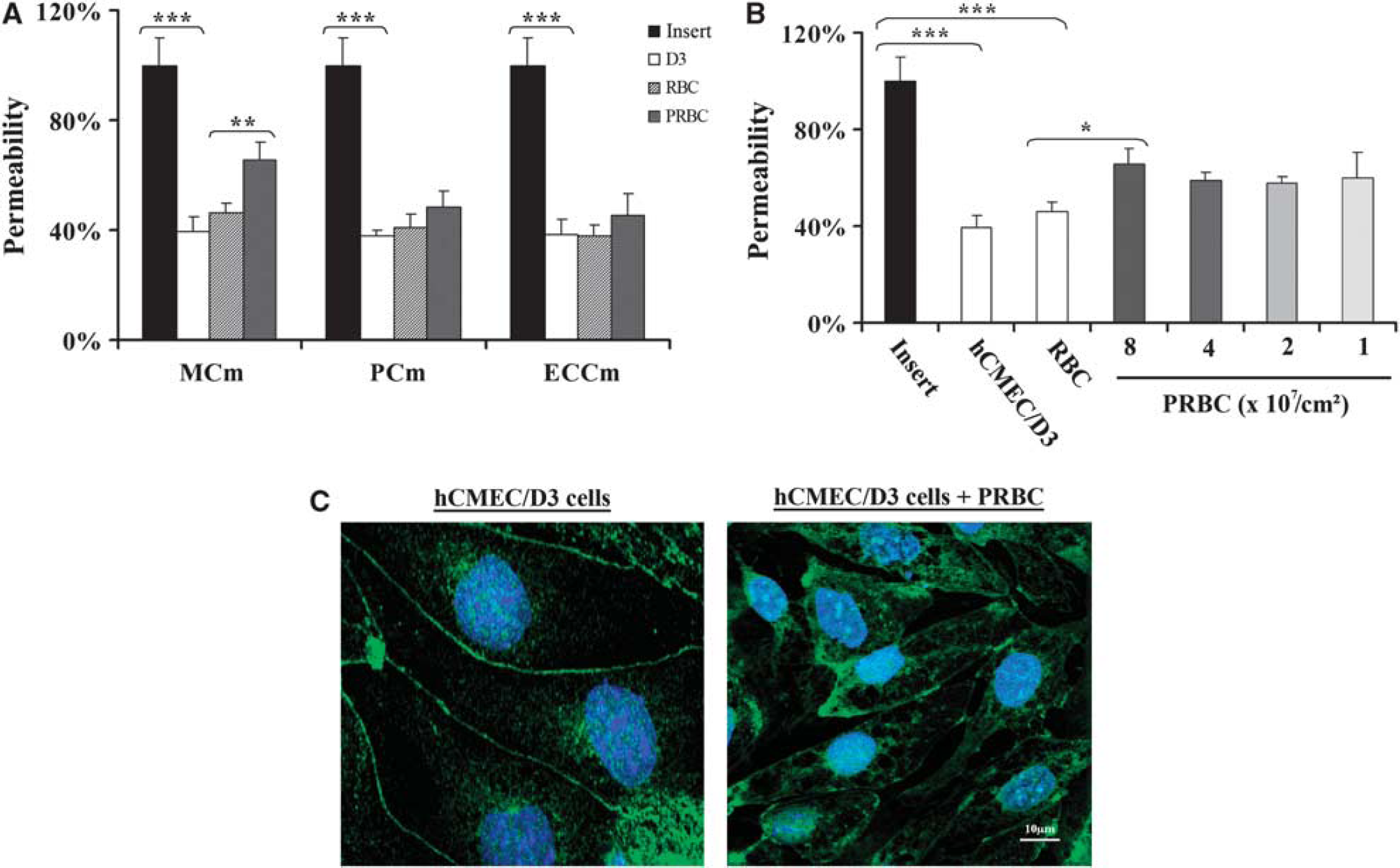
Effect of
We further analyzed whether the PRBCs-induced permeability modification could be associated with a disturbance of tight junction proteins of hCMEC/D3 cells. The expression of a tight junction protein, the submembranous tight junction-associated ZO1 protein, was determined (Figure 3C). Confocal microscopy analysis of immunostained hCMEC/D3 cells shows that ZO1 is expressed as a continuous line at the cell–cell contact. The TNFα stimulation of cells does not modify the staining of ZO1 protein (data not shown). In contrast, coculture of hCMEC/D3 and PRBCs shows a disorganization of ZO1 staining with gaps between cells and ZO1 relocalization in the intracellular compartment.
Parasitized Red Blood Cells Induced Permeability Modification Is Cytoadherence and Contact Independent
To analyze the role of cytoadherence in EC permeability, trypsin-treated and -untreated PRBCs were used in a permeability assay. Figure 4A shows that noncytoadherent trypsin-treated PRBCs are still able to induce an EC permeability increase similar to that observed in control nontrypsinized PRBCs. To exclude the hypothesis that other trypsin-resistant antigens could be involved in permeability, PRBCs were cultured in the lower chamber of the insert, preventing interaction with hCMEC/D3 cells. Figure 4B shows that independently of the contact, PRBCs could still induce permeability, compared with the control conditions.
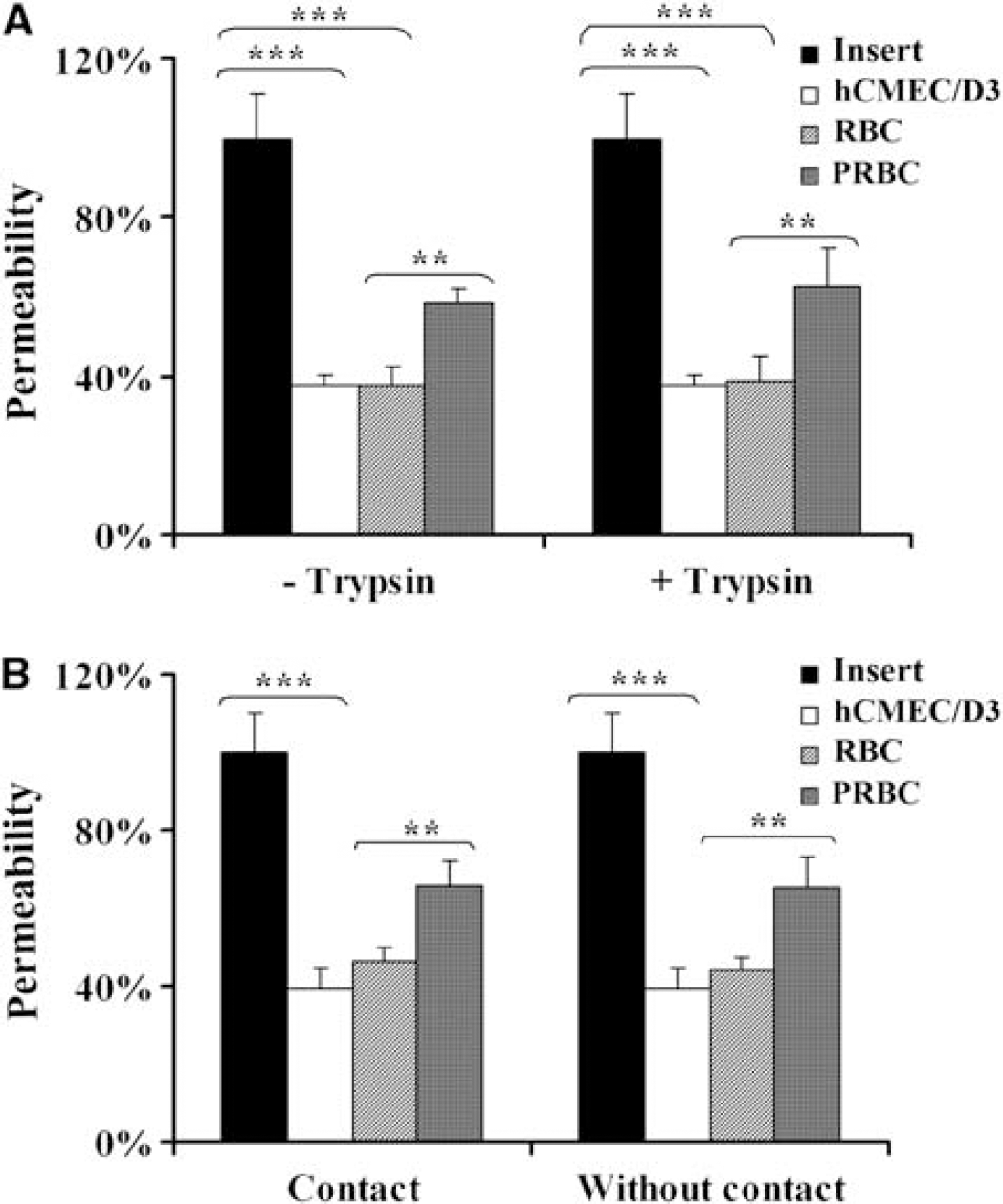
Evaluation of cytoadherence involvement in hCMEC/D3 cells permeability. (
Analysis of the parasite morphology after 20 hours of coculture showed that some PRBCs can mature to free merozoites (Figure 5A). Recent studies have shown that this parasite stage is implicated in the pulmonary endothelial barrier permeability (Gillrie et al, 2007). So the role of this parasite stage interacting with brain EC was also evaluated. Figure 5B shows that merozoites only induce an increase of 15% of permeability compared with cells cocultured with RBCs, whereas coculture of cells with PRBCs is able to induce an increase of 48% of the permeability compared with the control. All together, these results strongly suggest that the observed permeability is triggered through parasite cytoadherence and contact-independent mechanisms.
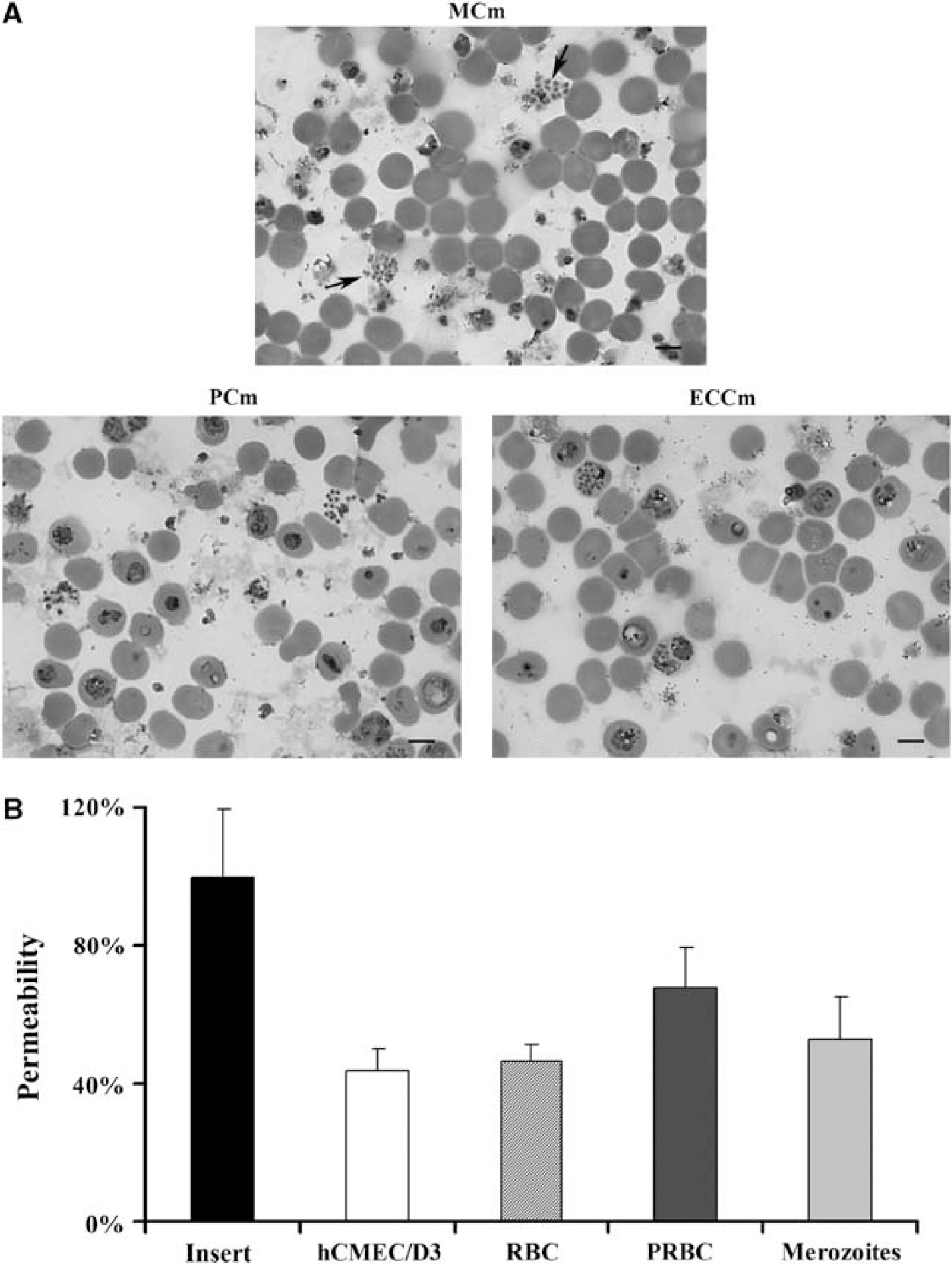
Effect of
The Role of Parasite Soluble Factors in the Endothelial Cell Permeability Enhancement
Previous studies have suggested that during parasite maturation, specific soluble factors released from PRBC mediate the decrease in human brain EC barrier resistance (Tripathi et al, 2007). Consequently, the parasite development in the different culture conditions was determined as an indirect measure of the release of parasite-specific soluble factors. Parasite development was evaluated through the measurement of the parasite lactate deshydrogenase (Figure 6A). Results show that parasite maturation, so release of specific soluble factors, was higher in the PCm and in the ECCm, both conditions, which failed to induce EC barrier permeability enhancement, than in the MCm. In parallel, PRBCs apoptosis was measured as a way to assess parasite viability in the different culture conditions. For this purpose, we performed annexin V and propidium iodide staining of PRBCs in different culture media. Figure 6B shows that PRBCs cultured in PCm and ECCm show very low level of late apoptosis, whereas PRBCs cultured in MCm show higher level of apoptosis. These results further point to a mechanism independent on a parasite-specific soluble factor.
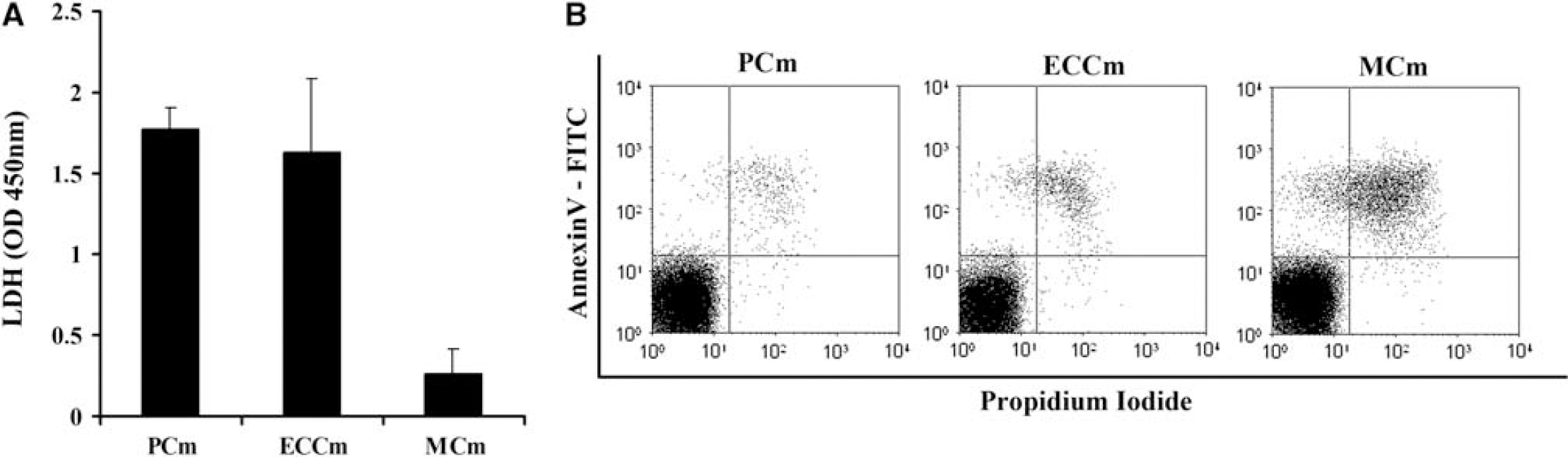
Evaluation of parasitized red blood cells (PRBCs) soluble factors release and viability on cell permeability. (
Parasitized Red Blood Cells Metabolism Induces Endothelial Cell Permeability Through the Decrease of the Environmental pH
As metabolic acidosis has been described as one of the strongest predictors of a fatal outcome in severe malaria (Dondorp et al, 2008), we decided to test whether the pH of the culture medium could be involved in the permeability enhancement observed. Results show that a decrease in the pH of the culture medium was correlated with an increase in the EC barrier permeability (Figure 7A). Consequently, we decided to study whether parasite metabolism could be responsible for an increase in the acidosis of the culture environment and associated with the permeability observed. Figure 7B shows the relationship between EC permeability and the culture medium pH measured after 20 hours of culture in the different conditions of the study. Results show a decrease in the culture medium pH because of the parasite metabolism when compared with the pH measured in the insert or in the EC conditions. Furthermore, this pH decrease was concomitant with the increase of the EC permeability. These results can be explained by the fact that MCm has a severe limitation in the buffer efficiency. This statement was determined comparing the final pH of the coculture in PCm and MCm. Difference between initial and final pH in PCm was 0.1 point, whereas in MCm it was 0.5 point. Consequently, in more alkaline media, PCm (pH = 7.18) and ECCm (pH = 7.31), parasite metabolism does not decrease enough the environmental pH to induce EC permeability. In contrast, in more acidic medium, MCm (pH = 6.78), parasite metabolism contributes to decrease the pH and to modify EC permeability compared with that observed in the control conditions. These results strongly suggest that a slightly acidic environment together with PRBC metabolism induces EC permeability through decrease of the environmental pH.
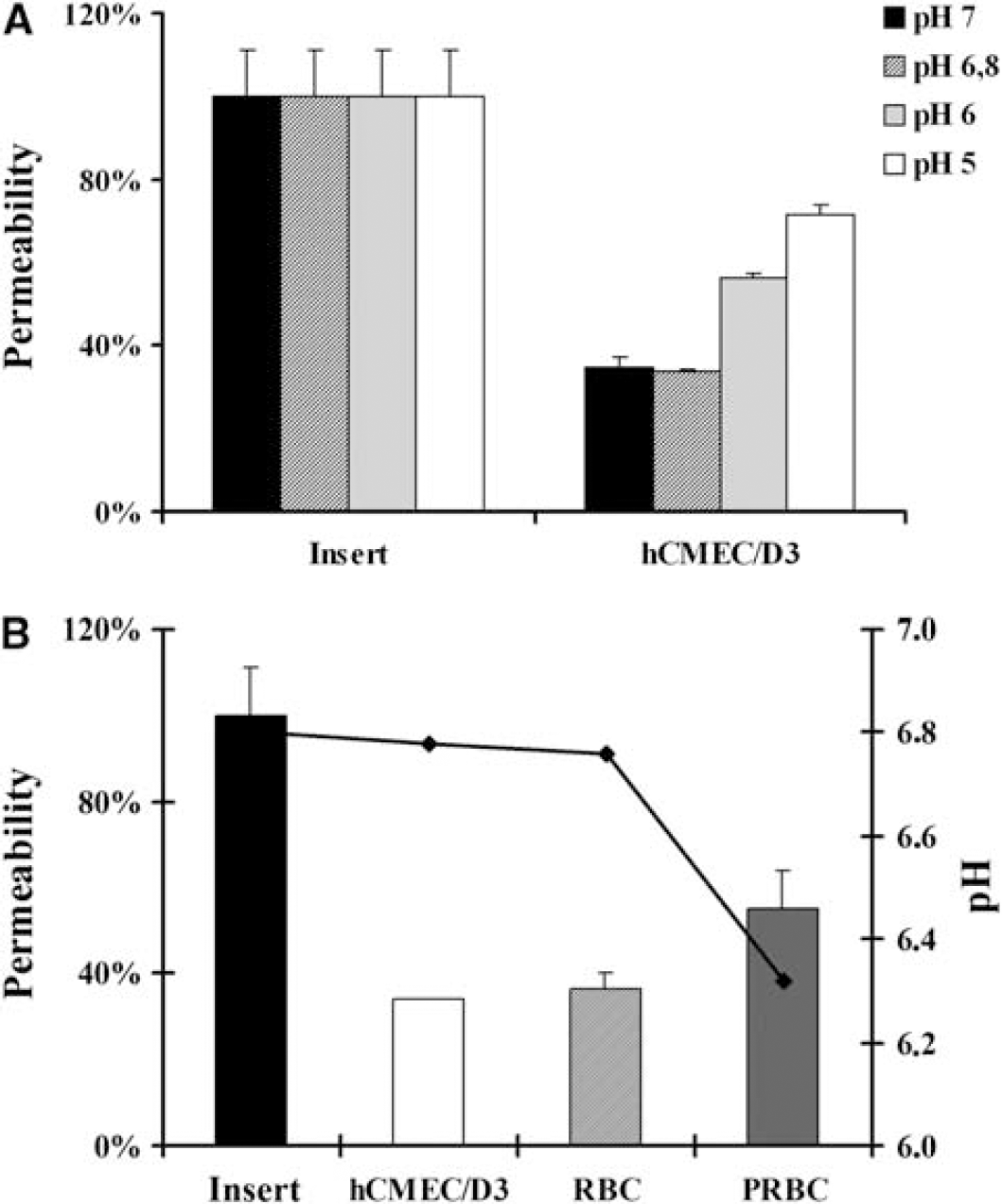
Effect of the environmental pH on endothelial cell (EC) permeability. (
Discussion
Cerebral malaria remains an important cause of death and therefore there is an urgent need of models to understand the pathophysiology of the disease. This study shows that hCMEC/D3 cells are a good model for studying
One interesting contribution of this study is that using hCMEC/D3 cell line, it has been possible to show that the selected strain, RAOL, isolated from a patient who suffered from severe malaria, induces an important permeability enhancement on the interendothelial permeability. This finding was consistent within different cell assays, suggesting that it is an intrinsic property of the parasite presence. Our results are in agreement with those previously reported, which showed the capacity of
To decipher the pathways implicated in the PRBCs-induced permeability enhancement, the role of the parasite cytoadherence phenomenon was explored. Previous studies, performed using cultures of human umbilical vein EC, human dermal microvascular EC, human pulmonary EC, or animal cerebral EC lines, have suggested that PRBCs cytoadherence to EC could induce a BBB alteration
Previous studies have proposed that parasite soluble factors such as proteins from parasite machinery released during parasite maturation and reinvasion could be at the origin of the EC barrier integrity breakdown (Gillrie et al, 2007; Tripathi et al, 2007). This hypothesis has been shown using artificial PRBCs lysate, which could introduce a bias in results obtained, as some parasite enzymes that are not naturally exposed to the EC will be released. In this study, we show that culture conditions, which allow a correct parasite maturation and reinvasion of new RBCs failed to induce a permeability enhancement, which suggests that parasite soluble factors related to the normal parasite maturation and reinvasion are not related with this phenomenon, even when working at high parasitemias such as 50% (8 × 107 PRBCs/cm2). In contrast, culture conditions, which reduce the parasite development and induce a PRBC apoptosis-like cell death trigger a permeability enhancement. Similar PRBC crisis forms have been observed in human CM (MacPherson et al, 1985), so we cannot discard the hypothesis that soluble factors produced during this apoptotic or death process can participate in the permeability enhancement observed in the hCMEC/D3 cell model. Moreover, these apoptotic structures can be more harmful as the reduced circulation in these obstructed microvascular vessels decreases the clearance of these parasitic wastes usually cleared by macrophages.
Finally, our results show a clear and expected relationship between pH of the culture medium and the integrity of the EC barrier. However, the more important and unexpected contribution of this study is to show up that, in a static condition, development of a high burden of parasites, in a slightly acidic environment with a limitation to control physiological pH, is enough to further decrease local environmental pH and alter the integrity of the EC barrier. One could conclude that culture conditions were not ideal for maintaining the integrity of the EC barrier; however, these results could have an important relevance in severe malaria. Indeed, recent published articles have shown a clear association between acidosis and the prognostic of the disease (Dondorp et al, 2008), mortality being greatest in children where acidosis coexists with impaired consciousness (Maitland and Newton, 2005). These clinical results, compared with the results obtained in our study, suggest that acidosis could be a main factor in BBB breakdown. This acidosis, because of systemic complications and reinforced by impaired blood perfusion because of parasites sequestration, leads to a deficiency to control the physiological pH (Sasi et al, 2007), which lets to think to the severe limitation in the buffer efficiency of MCm in our study. Finally, the high burden of sequestered parasites observed in the brain makes us thinking to the high parasite concentration needed in our experiments to decrease the environmental pH and to obtain the EC permeability enhancement.
The parallelism observed between our results and clinical data suggests that the culture conditions used in this study, even if they are not ideal for long-term parasite or EC culture, could be representative of the pathological situation observed in patients suffering from severe malaria. Thus, it is possible that in patients suffering from severe malaria, impaired perfusion of the brain, because of sequestered parasites, and massive parasite development are important factors in the etiology of acidosis increase in certain brain vessels and consequently in the BBB breakdown. However, the lack of metabolic disturbances or impaired perfusion of the brain could explain that infected human beings can support a high burden of sequestered parasites without undergoing CM.
In conclusion, our results establish that cytoadherence of mature parasites is not directly involved in permeability enhancement
Footnotes
Acknowledgements
The authors thank Catherine Blanc (flow cytometry core facility of Paris 6-Pitié-Salpêtrière) for flow cytometry assistance. Image acquisition was performed in the imaging facilities ‘Plate-forme d'Imagerie Cellulaire Pitié-Salpêtrière’ (Shared Resource Facilities of CRICM, IFR14, IFR113, INSERM, UPMC). The authors thank Claude-Marie Bachelet and Aurélien Dauphin for confocal microscopy assistance. The authors also thank Dr G Snounou for his scientific advices. SZ has a fellowship from La Fondation des Treilles.
The authors declare no conflict of interest.