
Abstract
Pneumococcal meningitis resulting from Streptococcus pneumoniae has a death rate of 28% in adults. In severe head injury and stroke, inflammatory changes and intracranial hypertension are improved by induced hypothermia, which also is neuroprotective. We hypothesized that moderate hypothermia ameliorates inflammatory changes in experimental pneumococcal meningitis. Wistar rats were cooled systemically, and meningitis was induced by pneumococcal cell wall components. The increase of regional cerebral blood flow in the meningitis animals was blocked by hypothermia at 6 hours. The reduction of intracranial pressure correlated with temperature. The influx of leukocytes into the cerebrospinal fluid and levels of tumor necrosis factor α in the cerebrospinal fluid were decreased. Cooling the animals 2 hours after meningitis induction to 30.5°C was also protective. We conclude that hypothermia is a new adjuvant approach to reduce meningitis-induced changes, in particular intracranial pressure, in the early phase of the disease.
Meningitis in adults caused by the most frequent pathogen, Streptococcus pneumoniae, still carries a death rate of 28% (Durand et al., 1993). The acute phase of meningitis is characterized by the most relevant factors for the disastrous outcome: influx of leukocytes into the cerebrospinal fluid (CSF), breakdown of the blood-brain and blood-CSF barrier, brain edema, increased CSF outflow resistance, and cerebrovascular complications, all of which contribute to intracranial hypertension (Quagliarello and Scheld, 1992). Besides the inflammatory response and possibly the effect of these alterations, bacterial factors may cause neuronal injury (Braun et al., 1999), which is responsible for the second clinical problem: permanent sequelae in up to 50% of the survivors (Bohr et al., 1984).
Induced hypothermia is an established tool used to protect the central nervous system during surgery (Sessler, 1995). Part of this protection is probably due to reduced brain metabolism and oxygen consumption (Croughwell et al., 1992; Marion et al., 1993; Metz et al., 1996). Further, hypothermia reduces cerebral blood flow in various models (Greeley et al., 1993; Marion et al., 1993).
Severe brain injury is complicated by a cascade involving similar inflammatory mediators and mechanisms like bacterial meningitis that may cause brain edema and increased intracranial pressure (ICP) (Goss et al., 1995). Induced hypothermia reduces increased ICP (Marion et al., 1993; Metz et al., 1996), neurologic deficits (Marion et al., 1997), elevated CSF cytokine levels (Goss et al., 1995), and histopathologic changes (Dietrich et al., 1994) in experimental brain injury. As a result, hypothermia improved the survival rate in an animal model of severe brain injury (Clark et al., 1996). Thus, hypothermia is widely accepted as an adjuvant therapy in patients with severe brain injury and otherwise uncontrollable intracranial hypertension (Marion et al., 1993; Metz et al., 1996).
In focal cerebral ischemia, there are extensive experimental data on the effect of induced hypothermia (Maher and Hachinski, 1993). In short, a reduction of brain temperature of 2°C or more results in markedly diminished neuronal damage and infarct size and in improved clinical outcome.
To date, there have been only two studies on induced hypothermia in meningitis. McCredie (1962) reported four children with meningococcal meningitis treated by hypothermia. A recent study showed a reduction of excitatory amino acids in bacterial meningitis under hypothermic conditions (Irazuzta et al., 1999).
Against this background, we hypothesized that induction of moderate hypothermia has a beneficial effect on increased regional cerebral blood flow (rCBF) and ICP, leukocyte influx to the CSF, and levels of tumor necrosis factor α (TNF-α) in the CSF during the early phase of pneumococcal meningitis.
METHODS
Animal preparation and experiments
We used a rat model of bacterial meningitis that has been described in detail (Weber et al., 1995). In short, male Wistar rats (280 to 300 g) were anesthetized using thiopental (Trapanal; Byk-Gulden, Konstanz, Germany) and mechanically ventilated with oxygen-enriched room air by a small-animal ventilator (AP-1; Effenberger, Pfaffing, Germany). The left femoral vein and artery were cannulated and 2 mL 0.9% saline was infused per hour. For meningitis induction and ICP measurement, a bur hole was drilled at the occipital pole and the cisterna cerebromedullaris was punctured.
Epidural temperature (ET) was used to determine the degree of hypothermia because it correlates closely with brain temperature (Miyazawa and Hossmann, 1992). For ET measurement, a craniotomy 2 mm in diameter was made 3 mm left of the sagittal suture and 2 mm caudal to the bregma. The underlying dura mater was left intact and under microscopic control a 0.2-mm temperature probe (T211; Physitemp Instruments, Clifton, NJ, U.S.A.) was carefully slid between bone and dura mater. Using a micromanipulator, a laser Doppler probe was placed on the right parietal bone in an area thinned and free of large vessels for rCBF measurement. During preparation, target temperature (ET 36.5°C, 34.5°C, 32.0°C, or 30.5°C) was reached and kept within a range of ±0.5°C. For hypothermia, a spontaneous reduction of body temperature was allowed; if necessary, a slow cooling of the body skin was performed by cool packs or warming using a heating pad. The target temperature was reached before rCBF recording. After a stable baseline of rCBF was recorded for at least 20 minutes, 75 mL CSF was withdrawn using the cisternal catheter and replaced by a sample of equal volume. In the posttreatment group, animals were cooled 2 hours after meningitis induction to ET = 30.5°C (during a period of 20 to 30 minutes). After 6 hours, the experiments were terminated by intravenous injection of KCl.
Continuous measurement was performed for end-expiratory pCO2 (ARTEMA MM 204; Heyer, Bad Ems, Germany), mean arterial blood pressure by the arterial line, ICP by a cisternal needle (Statham P10 EZ; Spectramed, Oxnard, CA, U.S.A.), rCBF by laser Doppler flowmetry (Periflux 4001 Master, Järfälla, Sweden), and ET and body temperature by a rectal probe (RET-2; Physitemp Instruments). All data were stored on a personal computer. Every 2 hours, arterial pH, pO2, and pCO2 were determined (Compact 1; AVL, Graz, Austria) for adjustment of ventilation. The white blood cell count in the CSF was obtained before and after the experiment.
Induction of meningitis
A preparation of pneumococcal cell walls was injected intracisternally (75 μL). The preparation has been described in detail (Weber et al., 1995). In brief, a nonencapsulated strain of S. pneumoniae (PnR-527; Jena, Germany) was heat-inactivated and disintegrated by ultrasound. Cell wall components were purified by centrifugation and resuspended in pyrogene-free saline. The preparation contained no detectable DNA/RNA (DNA DipStick; Invitrogen, Groningen, The Netherlands) or lipopolysaccharide (Chromogenic Limulus Amebocyte Lysate Tests; BioWhittaker, Walkersville, MD, U.S.A.). An equivalent of 107 cfu/mL S. pneumoniae was used. For control animals, saline was injected intracisternally.
TNF-α measurement
TNF-α bioactivity was measured by a modified L929 cytotoxicity assay. L929 cells were incubated with CSF in the presence of 1 μg/mL actinomycin D at 37°C for 20 hours. Cell viability was quantified by uptake of crystal violet in living cells, which was determined spectrophotometrically (595 nm) using an MR5000 ELISA Reader (Dynatech, Denkendorf, Germany). Equivalent concentrations of rat TNF-α (gift of Dr. P. Scholz, Schering AG, Berlin, Germany) were used for standards.
Experimental groups
The experimental groups were as follows:
Group 1: control animals receiving intracisternal saline injected at ET 36.5 ± 0.5°C (normothermic), n = 7 Group 2: control animals at ET 32.0 ± 0.5°C, n = 7 Group 3: meningitis animals at ET 36.5 ± 0.5°C, n = 7 Group 4: meningitis animals at ET 34.5 ± 0.5°C, n = 7 Group 5: meningitis animals at ET 32.0 ± 0.5°C, n = 7 Group 6: meningitis animals at ET 30.5 ± 0.5°C, n = 5 Group 7: meningitis animals at ET 30.5 ± 0.5°C, cooling started 2 hours after meningitis induction (posttreatment group), n = 5.
Statistical analysis
All data are expressed as mean ± standard deviation. Means of independent groups were compared by one-way analysis of variance and Student-Newman-Keuls multiple comparison; means of two independent groups were compared by the Mann-Whitney rank test. All statistical tests were done using SPSS for Windows 6.1 statistical software. P < 0.05 was considered significant.
RESULTS
At every time point, ET was lower than body temperature (mean Δ 1.6 ± 0.1°C). ET was different at different time points in different groups (Table 1).
Epidural temperature and physiologic parameters at 0, 2, 4, and 6 hours
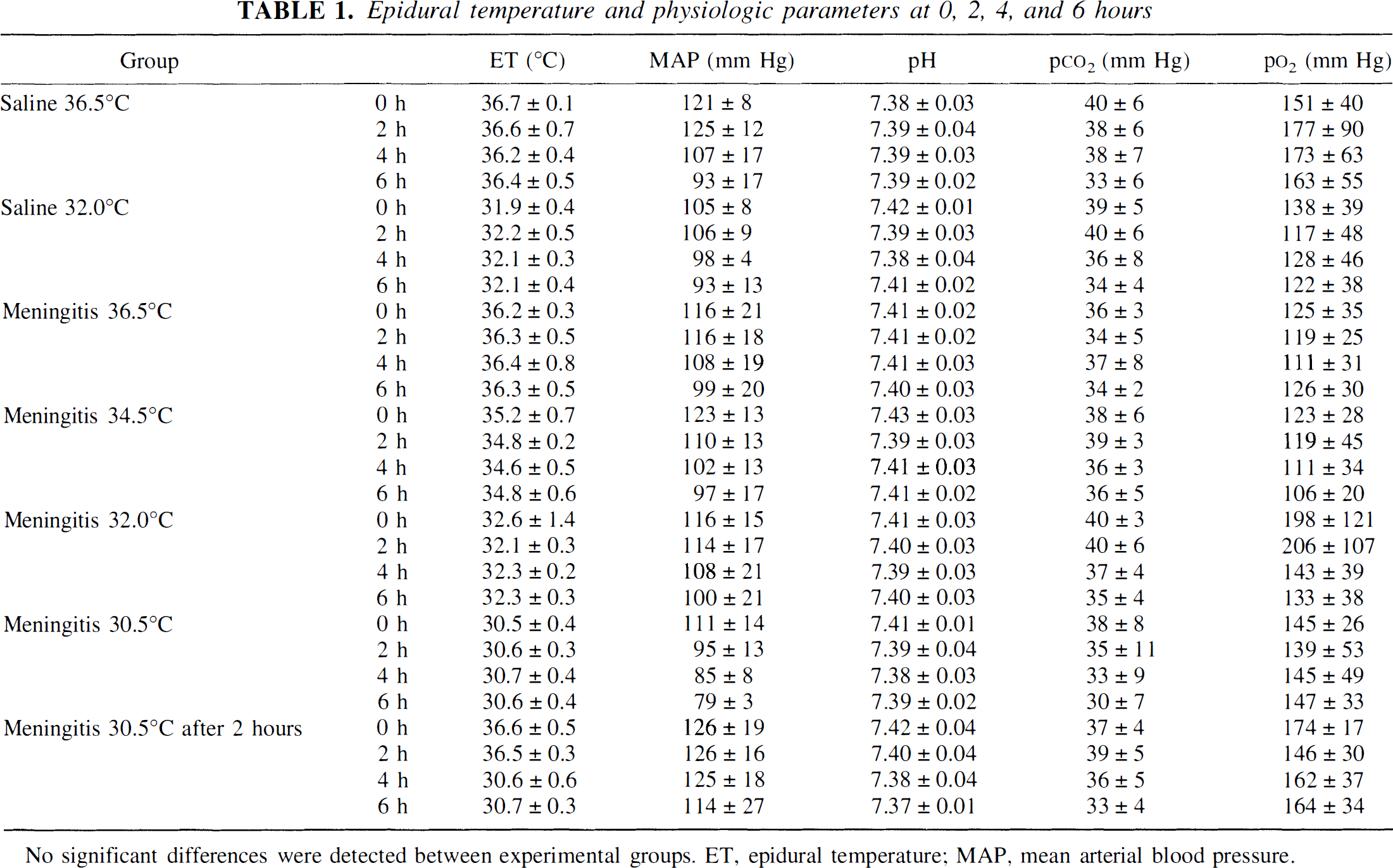
No significant differences were detected between experimental groups. ET, epidural temperature; MAP, mean arterial blood pressure.
In all groups, the mean arterial pressure, arterial pH, pCO2, and pO2 were in the normal range at any time (see Table 1). Between groups, there were no significant differences. Mean arterial pressure, however, tended to be lower at later time points in the 30.5°C meningitis group than in all other groups, but the difference was not significantly. In no animal did it fall to less than 70 mm Hg.
In control groups (normothermic and hypothermic), rCBF and ICP did not change over time (Figs. 1 and 2, Table 2), and the CSF white blood cell count was within normal limits (<5 per μL). These two control groups did not differ significantly. In normothermic animals, intracisternal injection of pneumococcal cell wall components resulted in a significant, slowly developing increase in rCBF (at 6 hours after meningitis induction, 212 ± 40% of baseline values) and in ICP (Δ 6 hours vs. baseline, 7.0 ± 2.8 mm Hg), as well as in marked CSF pleocytosis (5636 ± 1126 cells per μL). Hypothermia of 34.5°C or lower abolished the rCBF increase in the meningitis groups; the rCBF in all hypothermic meningitis groups was not significantly different from normothermic controls. ICP correlated with temperature (at 6 hours, r2 = 0.4128, P < 0.001), decreasing 1.1 mm Hg per degree; in the 30.5°C meningitis group, ICP did not differ from the control group. The CSF white cell count showed a statistically significant but rather low correlation with temperature (to mean ET: r2 = 0.2483, P < 0.05); hypothermic meningitis at all levels, however, showed significantly lower cell counts than normothermic meningitis. At 32.0°C, TNF-α levels in CSF were lower than under normothermic conditions (273 ± 256 vs. 927 ± 439 pg/mL, P < 0.05).
Analysis of rCBF, ICP, and CSF white blood cells
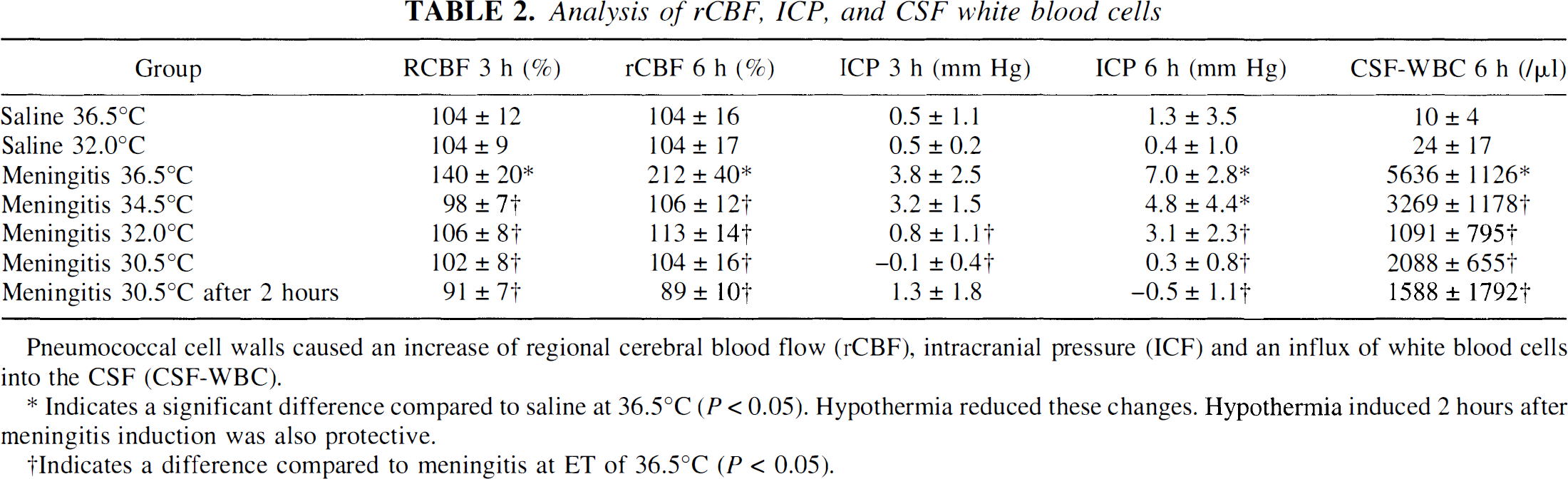
Pneumococcal cell walls caused an increase of regional cerebral blood flow (rCBF), intracranial pressure (ICF) and an influx of white blood cells into the CSF (CSF-WBC).
Indicates a significant difference compared to saline at 36.5°C (P < 0.05). Hypothermia reduced these changes. Hypothermia induced 2 hours after meningitis induction was also protective.
Indicates a difference compared to meningitis at ET of 36.5°C (P < 0.05).
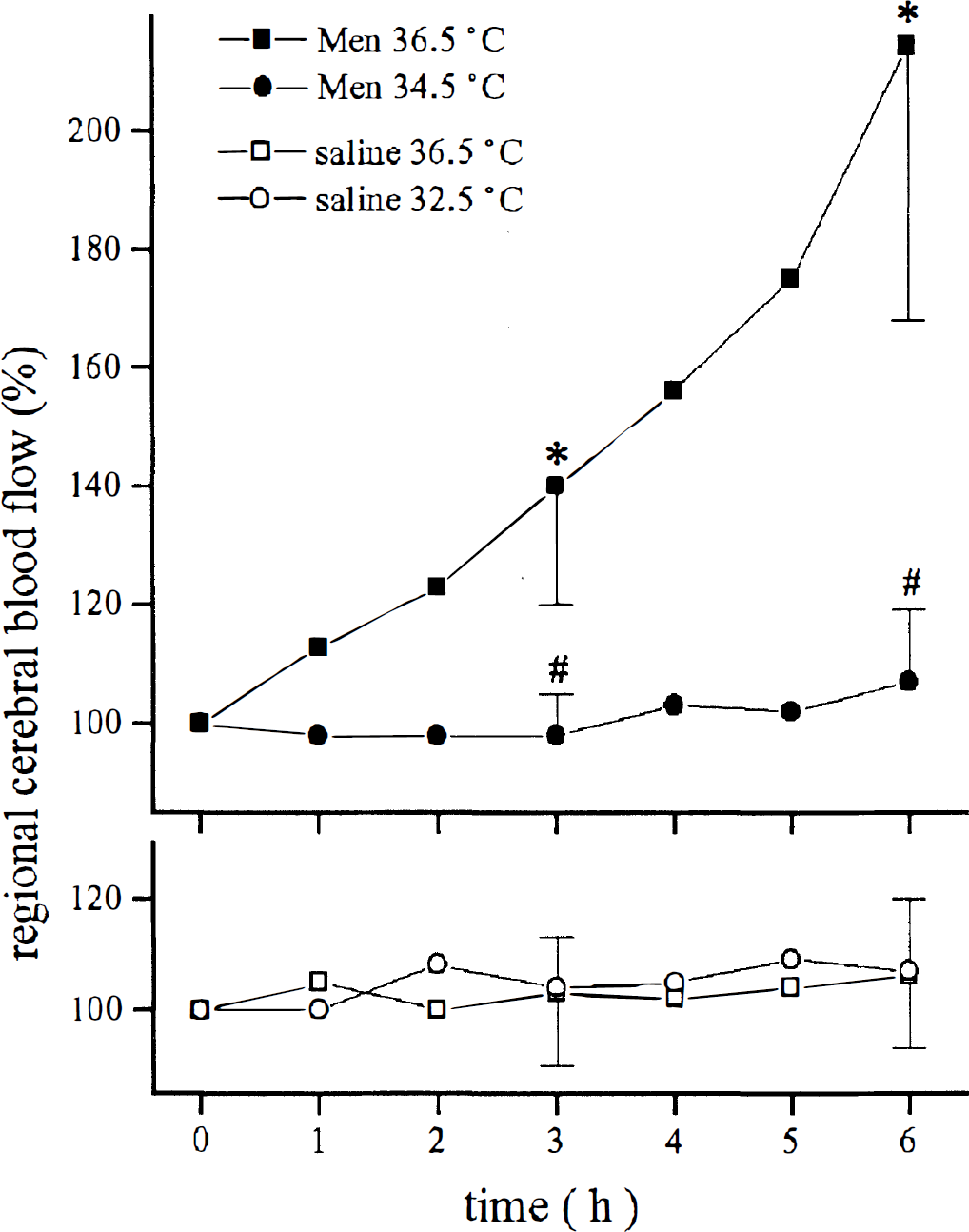
In normothermic animals, meningitis induced by pneumococcal cell wall components caused an increase in regional cerebral blood flow. At 34.5°C or lower, this increase was inhibited. In control animals, hypothermia did not change cerebral blood flow. * denotes a significant difference compared with all other groups (P < 0.05). # denotes a difference compared with meningitis at 36.5°C (P < 0.05).
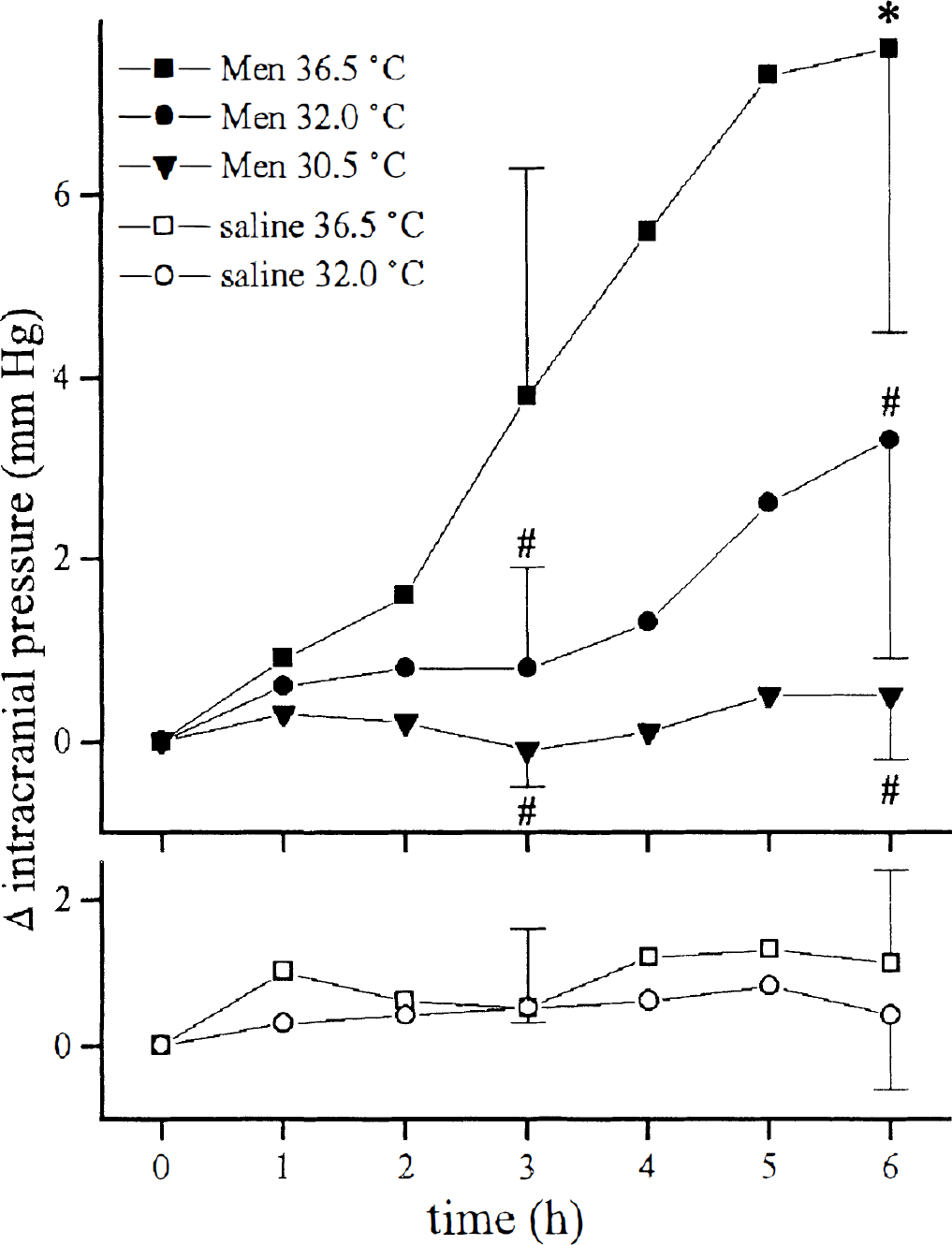
In normothermic animals, meningitis induced by pneumococcal cell wall components caused an increase in intracranial pressure. * indicates a difference compared with saline at 36.5°C (P < 0.05). # indicates a difference compared with meningitis at 36.5°C (P < 0.05). In control animals, hypothermia did not change intracranial pressure over time.
In the posttreatment group, rCBF (128 ± 13% of baseline values) and ICP (Δ: 1.6 ± 1.84 mm Hg) were slightly increased 2 hours after meningitis induction, similar to the normothermic meningitis group. Cooling during a period of 30 minutes caused a reduction of rCBF of 5.5 ± 1.4% per degree. At 6 hours after meningitis induction, rCBF was the same as at 2.5 hours (89 ± 10% vs. 91 ± 6% of baseline values) and ICP was significantly reduced compared with the normothermic meningitis group (Δ: −0.4 ± 0.7 vs. 7.0 ± 2.8 mm Hg). Further, in this group the CSF cell count was significantly decreased (1588 ± 1792 per μL).
DISCUSSION
Induced hypothermia attenuated the inflammatory response in a rat model of bacterial meningitis. In particular, a reduction of ET of 2°C significantly decreased the influx of leukocytes into the CSF and the increase in rCBF. ICP was correlated with ET, and the increase of ICP was reduced at lower temperatures. Further, induced hypothermia decreased TNF-α, a key cytokine, in the CSF.
Inhibition of leukocyte recruitment has been shown to reduce brain edema, ICP and rCBF increase, and neuronal damage and to improve survival in the rabbit and rat model of pneumococcal meningitis (Braun et al., 1999; Tuomanen et al., 1989; Weber et al., 1995). Hypothermia attenuated the influx of leukocytes into the CSF. Similar to our results, leukocyte migration was reduced by hypothermia in a rat model of chemical peritonitis (Balch et al., 1955). This may be secondary to a decreased production of proinflammatory cytokines, as demonstrated in experimental brain trauma (Goss et al., 1995). We measured significantly decreased levels of TNF-α in the CSF at ET = 32°C. Accordingly, isolated human monocytes produced less TNF-α, interleukin-1, and interleukin-6 at 30.0°C compared with 34.5°C after stimulation with an antigen of Staphylococcus aureus. In contrast, after lipopolysaccharide stimulation, levels of these cytokines were increased at the lower temperature (Luhm et al., 1998). A difference depending on the underlying pathogen must be considered. On human umbilical vein endothelial cells, expression of E-selectin and neutrophil adhesion was diminished at 25°C versus 37°C (Haddix et al., 1996). On leukocytes of patients undergoing coronary bypass surgery, up-regulation of adhesion molecules was delayed at lower temperatures (LeDeist et al., 1995).
A decrease of 2°C blocked the rCBF increase in our experiments. Cooling the animals 2 hours after intracisternal injection of pneumococcal cell wall components also prevented the rCBF increase; the difference was significant. Cooling animals 2 hours after meningitis induction to 30.5°C (as was done in the posttreatment group) reduced the rCBF, as in other studies (Croughwell et al., 1992; Greeley et al., 1993), whereas rCBF was stable over time in already cooled and sham-operated animals. Hypothermia reduced the rCBF increase also in severe experimental head injury (Marion et al., 1993). Cooling patients with severe brain injury to a core temperature of 32.5°C did not alter cerebral blood flow significantly but had a lowering effect on ICP and cerebral metabolic rates for oxygen and lactate (Metz et al., 1996). Hypothermia decreased nitric oxide levels in experimental brain trauma (Chatzipanteli et al., 1999). This effect should be considered because nitric oxide contributes to the rCBF and ICP increase in experimental meningitis (Koedel et al., 1995).
Clinically, the effect of hypothermia on ICP may be important because increased ICP is one of the most threatening complications in bacterial meningitis (Quagliarello and Scheld, 1992). In models of brain trauma and in patients with severe brain injury, induced hypothermia diminished intracranial hypertension (Marion et al., 1993; Metz et al., 1996). This may be due to protection of the blood-brain barrier, as reported in severe brain injury (Smith and Hall, 1996) as well as in stroke (Schwab et al., 1998).
Hypothermia in clinical use is considered to be safe (Marion et al., 1993; Metz et al., 1996). It transiently affects blood pressure, cardiac and renal function, and gastric motility (Metz et al., 1996). In our study we observed no relevant effects on the measured physiologic parameters (see Table 1) during a period of 6 hours. The slight decrease in mean arterial pressure should be considered carefully, because in bacterial meningitis cerebral autoregulation is impaired (Tureen et al., 1990), although hypothermia per se seems to have no effect on autoregulation (Greeley et al., 1993).
We conclude that cooling to moderate hypothermia is a new tool that can be used to attenuate the inflammatory response and to inhibit the development of life-threatening intracranial hypertension in the early phase of bacterial meningitis.