
Abstract
The heat shock response (HSR) provides protection against stress-induced damage, and also prevents initiation of inflammatory gene expression via inhibition of NFκB activation. This article describes experiments demonstrating that the HSR prevents induction of nitric oxide synthase type 2 (NOS2) in rat brain. Twenty four hours after intrastriatal injection of lipopolysaccharide (LPS), IL-1β, and IFN-γ, NOS2 immunoreactive cells were detected in striatum, corpus callosum, and to a lesser extent in cortex. Induction of a HSR by whole body warming to 41°C for 20 minutes, done 1 day before LPS plus cytokine injection, reduced the number of NOS2-positive staining cells to background levels. Staining for ED1 antigen revealed that the HSR also suppressed microglial/brain macrophage activation in the same areas. Striatal injection of LPS and cytokines induced the rapid activation of NFκB, and this activation was prevented by prior HS, which also increased brain IκB-α expression. These results suggest that establishment of a HSR can reduce inflammatory gene expression in brain, mediated by inhibition of NFκB activation, and may therefore offer a novel approach to treatment and prevention of neurological disease and trauma.
Inflammatory gene expression in brain contributes to pathogenesis of neurological disease and trauma, including Multiple Sclerosis, Alzheimer's disease, and stroke. Proinflammatory gene expression, including cytokines such as IL-1β or cell adhesion molecules like ICAM-1, and the production of reactive oxygen species like NO by the inducible enzyme NO synthase type 2 (NOS2, or iNOS), promote inflammatory reactions, cause cell damage, and initiate invasion of blood leukocytes into brain which can contribute to, and potentiate, ongoing inflammatory events. In cerebral ischemia, the intracerebroventricular injection of an IL-1β converting enzyme inhibitor reduced IL-1β expression and brain damage (Hara et al., 1997); NOS2 enzyme inhibitors reduced cell loss in focal ischemia (Iadecola et al., 1995); and stroke damage was less in homozygous ICAM-1 knockout mice than in control littermates (Connolly et al., 1996). In experimental autoimmune encephalomyelitis, the animal model for multiple sclerosis, NOS2 inhibitors can ameliorate the development of symptoms (Cross et al., 1994); and disease is more severe in mice lacking the anti-inflammatory cytokine IL-10 (Bettelli et al., 1998). The anti-inflammatory actions of nonsteroidal anti-inflammatory drugs reduce the risk of Alzheimer's disease, possibly because of reduction in NOS2 expression (Flynn and Theesen, 1999). These observations demonstrate that suppression of inflammatory activation in brain will provide therapeutically beneficial effects.
The expression of numerous inflammatory genes is under control of transcription factor NFκB (Baeuerle and Henkel, 1994), whose activation has been described during neurological disease and trauma. Levels of NFκB are increased in brains with Alzheimer's disease versus control brains (Kaltschmidt et al., 1997; Kitamura et al., 1997) during global (Clemens et al., 1997) and transient (Gabriel et al., 1999) forebrain ischemia in glial cells and neurons, in microglial cells after induction of experimental autoimmune encephalomyelitis (Kaltschmidt et al., 1994), in macrophages in human multiple sclerosis (Gveric et al., 1998), and in neurons as a consequence of excitotoxic damage (Qin et al., 1998). In some cases the prevention of NFκB activation (Clemens et al., 1998) was neuroprotective, suggesting that other methods to block NFκB activation in brain will also reduce inflammatory related damage.
It has been known for several years that the heat shock response (HSR) can protect cells and tissues from a variety of noxious stimuli, including thermal, chemical, and physical stress, possibly by facilitating renaturation of partially denatured proteins or by chaperoning nascent proteins to specific subcellular locations. The HSR can protect animals against endotoxin lethality (Chu et al., 1997; Hotchkiss et al., 1993; Ribeiro et al., 1994) and phospholipase-C-induced lung damage (Villar et al., 1994). Whether a HSR can protect the brain against inflammatory damage is not known. However, observations that HSPs are induced during stroke (Nowak and Jacewicz, 1994; Sharp et al., 1993; Sharp et al., 1999), and that ischemic damage is reduced in transgenic mice constitutively expressing HSP70 in neurons and glia (Mestril et al., 1994b; Plumier et al., 1995; reviewed in Yenari et al., 1999) suggests that a HSR will also be protective in the nervous system.
In addition to protecting against damage after insults, the HSR can prevent the initiation of inflammatory events themselves (Simon et al., 1995; Snyder et al., 1992) suggesting that in some cases the protective effects of HS are attributable to the prevention of proinflammatory gene activation. We demonstrated that the HSR prevents NOS2 induction by cytokines plus lipopolysaccharide (LPS) in rat primary astrocytes, C6 glioma cells, and Rat-1 fibroblasts (Feinstein et al., 1996), and similar results have been reported using rat pulmonary artery smooth muscle cells (Wong et al., 1995), human liver cells (de Vera et al., 1996b), rat hepatocytes (de Vera et al., 1996a), murine lung epithelial cells (Wong et al., 1997a), and cultured rat and human islets (Scarim et al., 1998). In whole animals, treatment with arsenite, which induces a HSR, blocked endotoxin-induced NOS2 expression in lung (Hauser et al., 1996). The HSR also prevents NFκB activation, due in part to the fact that the inhibitory IkB-α protein is a stress protein whose expression is increased by the HSR (Scarim et al., 1998; De-Meester et al., 1997; Feinstein et al., 1997; Meng et al., 1999; Thomas et al., 1998; Wong et al., 1997; Wong et al., 1999). We now report that the HSR is an equally effective inhibitor of NFκB dependent inflammatory events in vivo, and demonstrate that a brief period of hyperthermia prevents subsequent NFκB activation, NOS2 expression, and microglial/macrophage activation induced by injection of LPS and cytokines into brain.
MATERIALS AND METHODS
Animals
Female Sprague-Dawley rats (Charles River, MA, U.S.A.) weighing 250 to 300 g, were housed in groups of 4 under standard conditions at 22°C and a 12-hour light-dark cycle with free access to food and water.
Experimental protocol
Rats were anesthetized with pentobarbital (50 mg/kg, dissolved in 0.9% sodium chloride) and placed into a heating blanket. Body temperature was monitored by a rectal probe connected to the heating blanket, and was raised gradually to 41 ± 0.5°C over a period of 5 minutes, then maintained for 20 minutes. Control animals were anesthetized but not heated. Females rats were used for all experiments since even brief (10 minutes at 41°C) hyperthermia increased the mortality rate of male, but not female rats. Intrastriatal injections were performed as previously described (Heneka et al., 1999a). After 24 hours recovery, rats were again anesthetized as described above and placed in a stereotaxic frame (David KOPF, CA, U.S.A.). Two μL of a mixture containing recombinant rat IFN-γ (20 units/mL, Gibco, MD, U.S.A.), human IL-1β (10 ng/mL of 107 unit/mg, National Institutes of Health reagents program) and bacterial endotoxin LPS (1 μg/mL, Salmonella typhirium, Sigma, MO, U.S.A.) dissolved in phosphate buffered saline (PBS) (pH 7.4) was injected over a period of 120 seconds into the left striatum using a 5 μL Hamilton syringe, at AP 0.0, L −3.0, V 6.0 mm relative to Bregma and 2 μL of PBS were injected into the right striatum at AP 0.0, L+3.0, V 6.0. The needles were left in place for a further 5 minutes to prevent reflux up the needle tract. To maintain constant body temperature, animals were placed under a heating lamp until complete recovery from anesthesia. Either one or twenty-four hours after intrastriatal injection, animals were killed by an overdose of pentobarbital and then perfused transcardially with 200 mL heparinized sodium chloride (0.9%) and 200 mL fixative containing 10% formaldehyde, 10% acetic acid and 80% methanol. Brains were removed, immersed in fixative for 72 hours at room temperature, then paraffin embedded. In some cases brains were removed without perfusion and protein lysates prepared for immunoblot detection. All experiments were performed in accordance with the declaration of Helsinski, the animal welfare guidelines and laws of the United States of America, and were approved by the local ethical committee for animal experiments.
Processing of brain tissue and immunohistochemistry
Serial coronal sections of whole brain were cut 8 μm thick using a Leitz microtome and mounted on poly-L-lysine coated slides. Slides were immersed in 10 mmol/L citrate buffer (pH 6.0) and heated in a microwave oven, 4 cycles of 5 minutes each, to unmask antigen sites. Slides were cooled for 20 minutes at room temperature, then washed in PBS. Endogenous peroxidase activity was inhibited by rinsing slides in 0.1% hydrogen peroxide for 10 minutes. After washing in PBS, nonspecific binding was blocked by incubation in 10% normal goat serum in PBS for 1 hour at room temperature. After washing in PBS, sections were incubated overnight at 4°C with primary antibodies: (1) mAb N32020 directed against NOS2 (1:200 dilution, Transduction Laboratories, KY, U.S.A.); (2) ED1 mAb MCA 341 raised against rat lysosomal membrane antigen of activated macrophage/microglia (1:500 dilution, Serotec, Darmstadt, Germany); (3) rabbit polyclonal antibody SC-109 raised against NFκB subunit p65 (1:200 dilution, Santa Cruz, Heidelberg, Germany); (4) rabbit polyclonal antibody SC-1060 directed against HSP70 (1:300 dilution, Santa Cruz); or (5) rabbit polyclonal antibody SC-371 directed against IκB-α (1:1,500 dilution; Santa Cruz Biochemicals, Santa Cruz, CA, U.S.A.). The sections were washed extensively with PBS, then incubated with biotinylated anti-rabbit or anti-mouse IgG (1:200 dilution, Vector Laboratories, Burlingame, CA, U.S.A.) for 30 minutes at room temperature. Immunohistochemical localization was performed using the avidin-biotin peroxidase complex method (ABC-Kit, Vector Laboratories) with 3,3′-diaminobenzidine as chromogen.
Quantification of immunohistochemistry and statistical analysis
Quantitative analysis of NOS2 and ED1-antigen was performed on brain sections from animals (6 heat shocked and 6 control) injected with LPS, IFNγ, and IL-1β in saline, at 24 hours after injections. Antigens were detected in 7 sections having a defined distance relative to the level of intrastriatal injection. The sections were the middle section corresponding to the level of injection, with the injection site discernible, and the other 6 sections were taken at a distance of 25, 50, and 75 μm rostral and caudal of the injection site. Three different areas were defined and evaluated: striatum, corpus callosum, and cortex ipsilateral to the cytokine injection side. The number of cells within the respective fields was determined using a counting grid. Data are shown as mean ± SD of the number of positive cells per mm2. Differences between controls and heat shocked animals were assessed by one way analysis of variance followed by a Tukey test (SYSTAT, Evanston, IL, U.S.A.).
Western Blot Analysis
Brain region extracts were prepared by sonication of dissected brain regions (25 mg of wet weight tissue) in 10 volumes of 8 mol/L urea. Aliquots were immediately mixed with SDS sample buffer, boiled, and either used immediately or frozen at −80°C. Twenty μg protein was separated through 10% polyacrylamide sodium dodecyl sulfate gels and proteins transferred by semidry blotting to polyvinylidene difluoride membranes. The membranes were blocked in tris-buffered saline with 0.1% TWEEN 20 containing 0.5% bovine serum albumin, washed, incubated with primary antibody to HSP70 (mAb SPA-810, 1:1,000 dilution; Stressgen, Canada) or to the astrocyte-specific protein glial fibrillary acidic protein (mAb B2.210, Lee et al., 1984) overnight at 4°C, washed extensively, incubated with peroxidase conjugated goat anti-mouse IgG, then bands visualized with enhanced chemiluminescence reagents (Pierce, Rockford, IL, U.S.A.).
RESULTS
The efficacy of whole-body warming on inducing a HSR in brain was assessed by measuring levels of the inducible HSP70 protein 24 hours after increasing body temperature to 41°C for 20 minutes. Western blot analysis indicated that in control (anesthetized only) animals, HSP70 was easily detectable in cortex, and present but at much lower levels in striatum (Fig. 1, top panel). In both brain areas the antibody used, which is specific for the inducible HSP70 and does not cross react with constitutively expressed HSC70 (Manzerra et al., 1997), detected two bands of 68 to 70 kDa with comparable staining intensities. Heat shock greatly increased the levels of the slower migrating band in both brain areas, although higher levels were reached in cortex than striatum. The levels of the lower band were not significantly increased by HS. The levels of a third minor, faster migrating band (visible in cortex samples) was not effected by HS. Immunocytochemical staining for glial fibrillary acidic protein showed similar levels of this protein in control and heat shocked samples (Fig. 1, bottom panel). Time course studies (not shown) revealed higher expression of HSP70 protein between 18 and 24 hours after HS compared to either 4 hours or 48 hours after HS, and that a 20-minute HS was more effective than 10 minutes, but as effective as a 40-minute HS. We therefore chose 20 minutes of HS followed by 22 hours recovery for subsequent injection experiments.
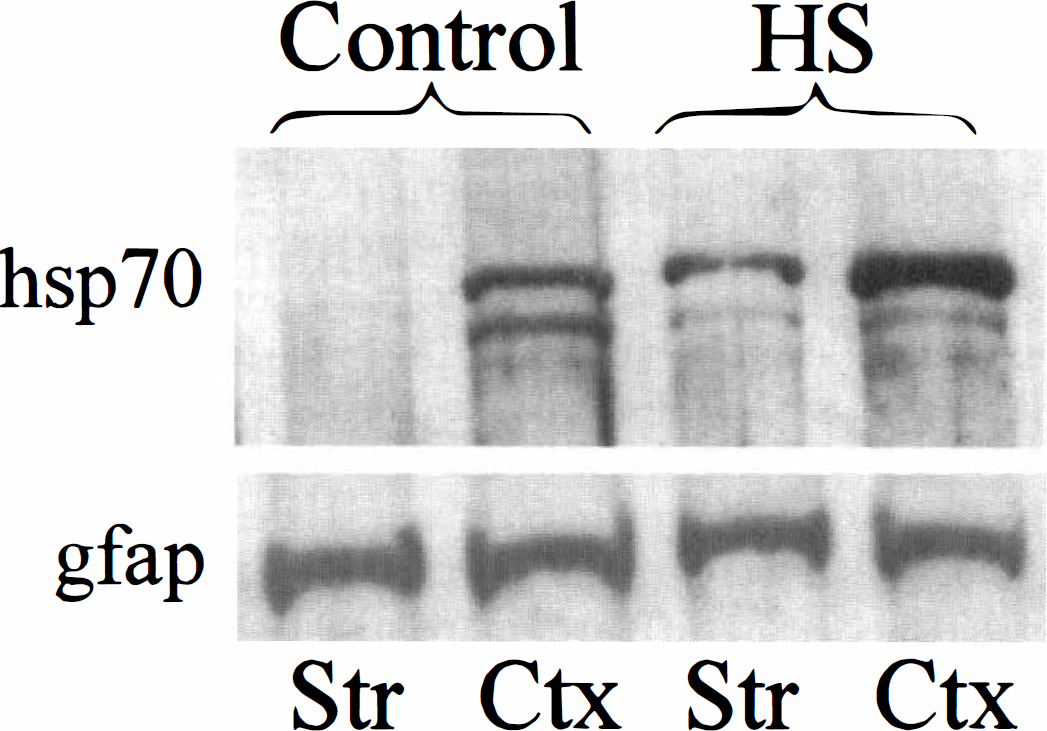
Heat shock induces HSP70 expression in rat brain. Adult female Sprague Dawley rats were anesthetized, then subjected to 20 minutes of whole-body heating at 41°C by warming in a heating pad. The next day, cell lysates were prepared from striatum (STR) and cortex (CTX) of control (anesthetized only) or heat-shocked animals, and aliquots (20 μg) examined by immunoblot for levels of HSP70 (top) or the astrocyte specific protein glial fibrillary acidic protein (gfap) (bottom).
Injection of a cytokine mixture (CM) containing bacterial endotoxin LPS together with cytokines (IFN-γ and IL-1β) induces inflammatory gene expression in brain (Kitamura et al., 1996; Garcion et al., 1998; Heneka et al., 1999a). Twenty four hours after intrastriatal injection of CM, numerous cells showed positive staining for NOS2 protein, with the highest number of positive cells observed in striatum, less in corpus callosum, and the fewest in cortex (Fig. 2). When equivalent injections were made into striatum of animals that had been heat shocked one day earlier, the number of NOS2 staining cells was greatly reduced (Fig. 2E). Contralateral injection of saline did not induce any positive staining in either control (Fig. 2C) or heat shocked animals (not shown). Injection of CM, but not PBS, into brain itself caused a slight increase in HSP70 expression that was restricted to the area around the needle tract (data not shown). Examination of serial sections through three brain regions (Fig. 3) revealed that HS reduced the number of NOS2 staining cells at all distances from the injection site, with an overall reduction of greater than 90% (Fig. 3B). The morphology of NOS2-positive cells was round to oval, resembling the typical morphology of brain macrophages and/or activated microglia. In addition, positive staining cells were seen within and surrounding brain microvessels. These results demonstrate that HS reduced CM-induced NOS2 staining in brain, and suggest that infiltrating monocytic cells constitute a large portion of the NOS2-positive cells.
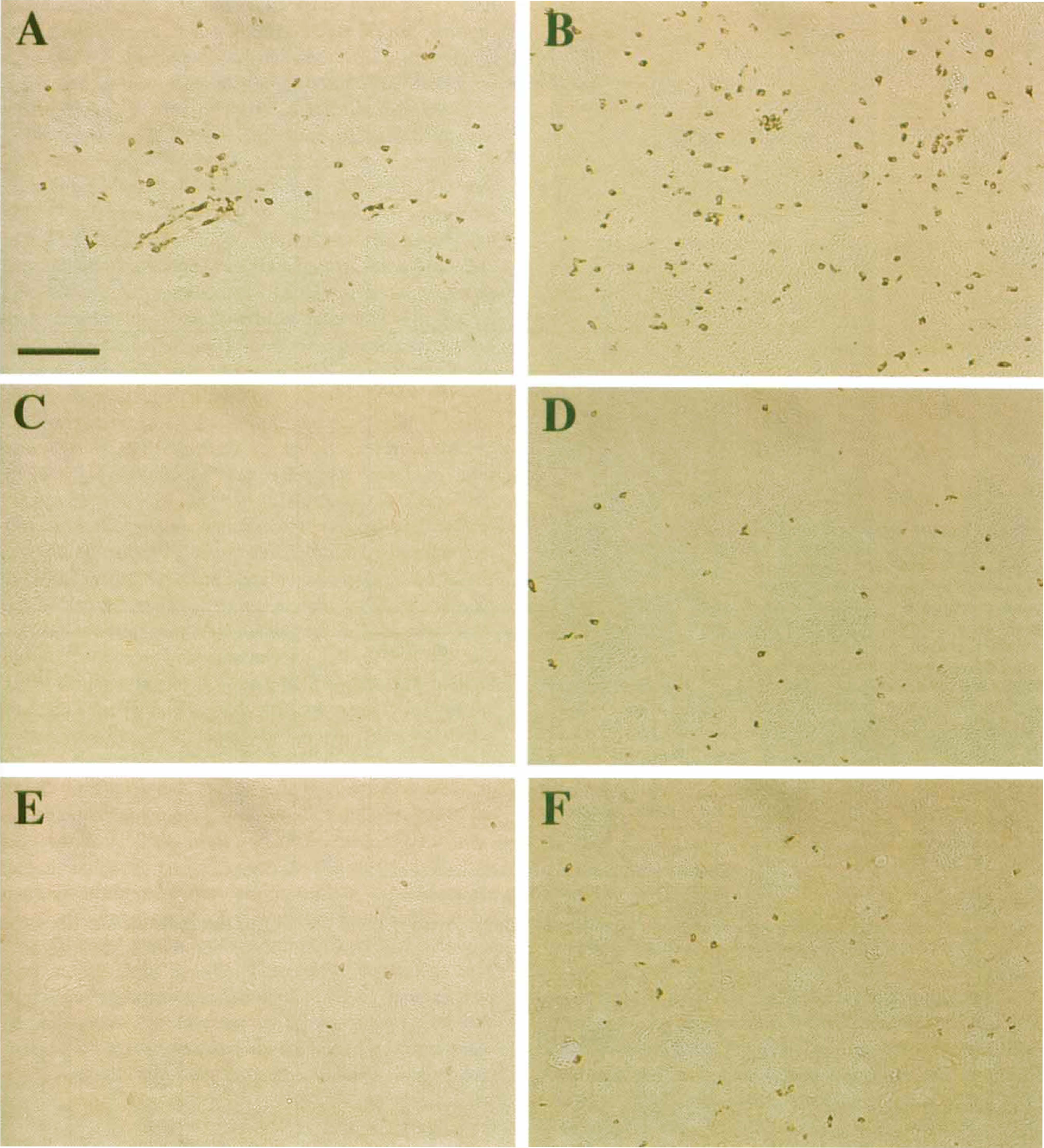
Heat shock prevents striatal nitric oxide synthase type 2 (NOS2) and ED1 expression. A mixture of lipopolysaccharide (LPS), IFN-γ, and IL-1β (“CM”) in 2 μL total volume in saline was injected into the left striatum of control or heat-shocked animals (n = 3 each). Saline alone was injected into the right striatum. After 24 hours, the animals were anesthetized, perfused, and sections stained for the presence of NOS2 (left panels) or ED1 (right panels).
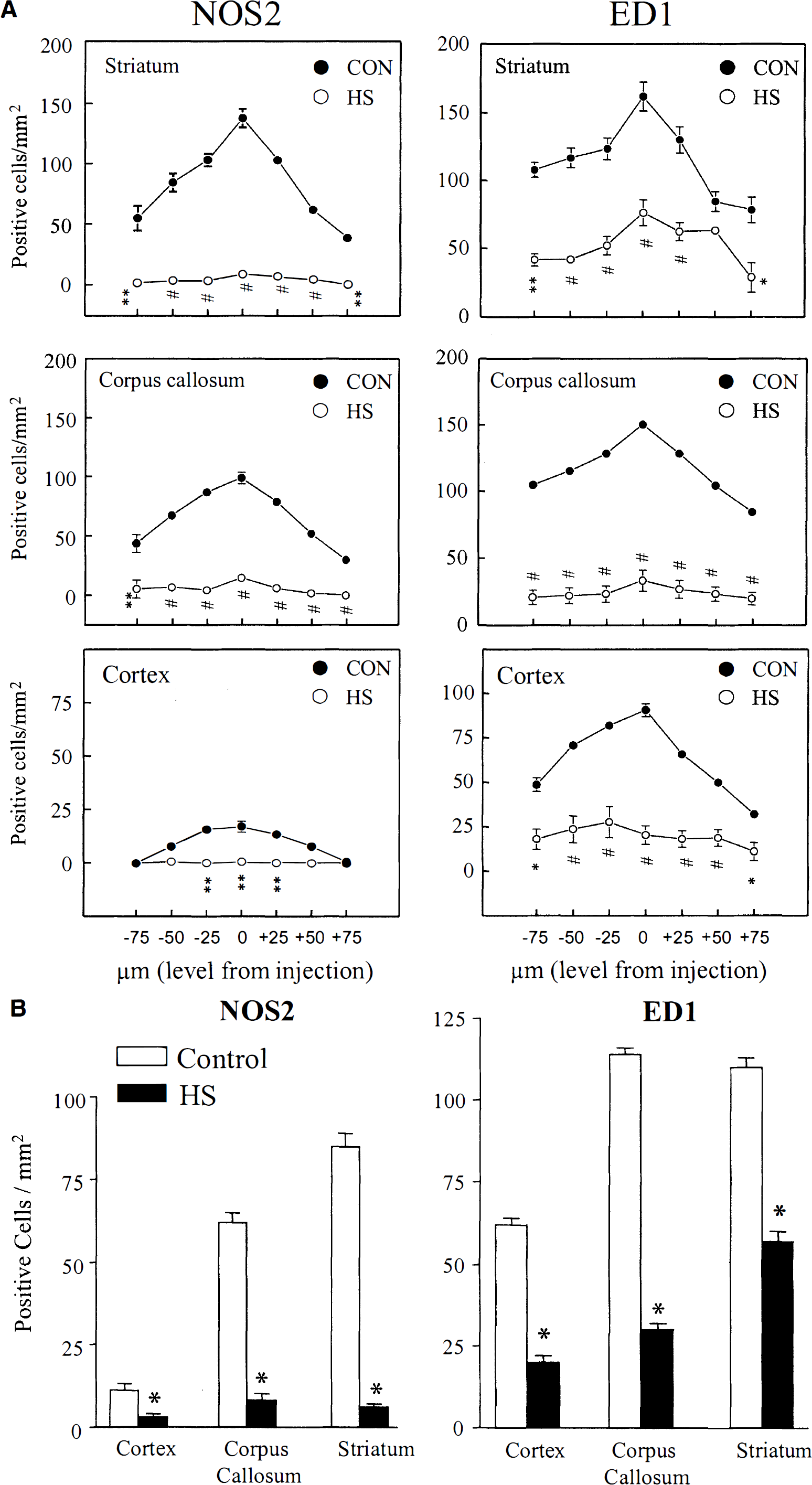
Heat shock prevents nitric oxide synthase type 2 (NOS2) and ED1 expression in different brain regions.
To determine if HS prevented macrophage or microglial activation, we stained sections for ED1 antigen 24 hours after intrastriatal CM injection (Figs. 2B, 2D, and 2F). As found for NOS2, LPS and cytokines induced a robust appearance of ED1, with most cells having the appearance of activated macrophages or microglia (Fig. 2B). The greatest numbers of ED1-positive staining cells were detected in striatum, with similar levels in corpus callosum, and lower levels in cortex. In all three brain regions, prior HS reduced staining for ED1 to background levels (Figs. 2F and 3).
Since activation of transcription factor NFκB is necessary for NOS2 induction, we tested the effects of HS on NFκB activation in brain (Fig. 4). One hour after saline injection into the right striatum, and CM injection into the left striatum in both heat-shocked and control rats, the NFκB p65 subunit was detected by immunohistochemistry. In the saline-injected hemisphere the NFκB p65 subunit was detected as a faint band of staining within the cytoplasm of parenchymal cells in both heat-shocked and non-heat-shocked animals (Figs. 4D and 4). In contrast, injection of CM resulted in a marked change of the pattern of p65 staining, namely loss of the light cytoplasmic staining, and appearance of dense staining in smaller cells (Fig. 4A to Fig. 4C). This change in staining pattern could be caused by increased p65 expression in a subpopulation of smaller cells, and/or to the movement of NFκB from a cytosolic to a nuclear location. There were two morphologically distinct labeling patterns observed after LPS/cytokine stimulation, a round to oval morphology with no visible cytoplasm (Fig. 4B), and a more uneven morphology with a lighter surrounding area (Fig. 4C). The round morphology may be infiltrating monocytes, while the uneven staining may be parenchymal glial cells. The injection of CM into striatum of heat-shocked animals yielded a staining pattern essentially the same as that following saline injection (Figs. 4F and 4G); i.e. a lighter staining restricted to the cytoplasm of parenchymal cells. Saline injection into heat-shocked brain gave the same results as into non-heat-shocked animals (data not shown). These results indicate that the HSR prevented the initial CM-dependent NFκB activation in brain, which could account for the subsequent decrease in NOS2 and ED1 staining observed.
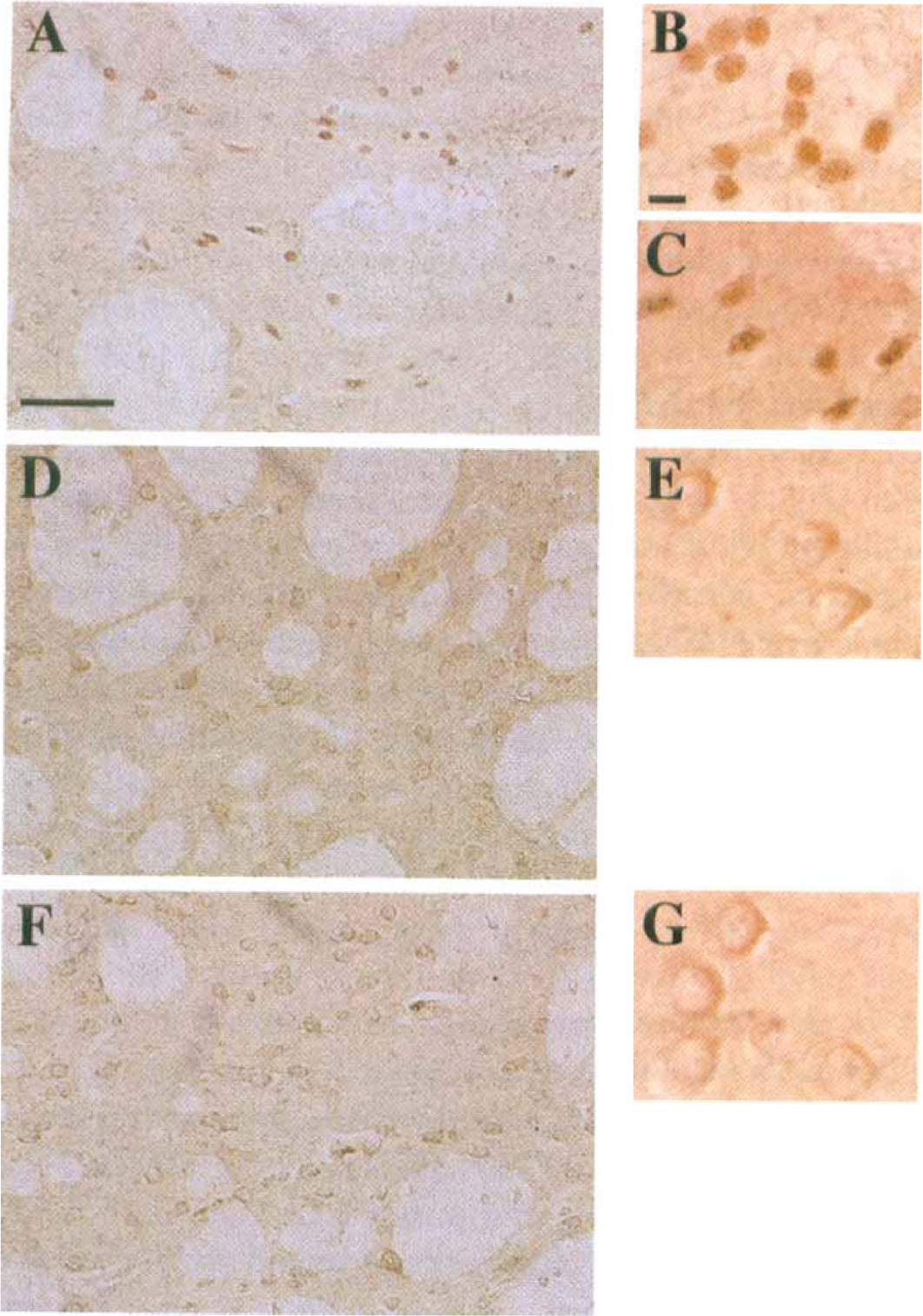
Heat shock prevents NFκB activation by lipopolysaccharide (LPS) and cytokines. A mixture of LPS, IFN-γ, and IL-1β (“CM”) in 2 μL total volume in saline was injected into the striatum of control or heat-shocked animals. Saline alone was injected into the contralateral striatum. After 1 hour, the animals were anesthetized, perfused, and sections stained for the presence of NFκB subunit p65.
The HSR has recently been shown to block NFκB activation because of increased expression of the inhibitory IκB-α protein (Feinstein et al., 1997; DeMeester et al., 1997; Thomas et al., 1998; Wong et al., 1999). We therefore tested if brain IκB-α protein levels were increased by HS. Immunocytochemical staining of striatum (Fig. 5) indicated low, basal levels of cytosolic IκB-α. Twenty four hours after HS, IκB-α levels were greatly increased in cells throughout the striatum, with morphologies similar to those identified by staining for NFκB. These results demonstrate that whole-body HS elevates brain IκB-α levels, and suggests that suppression of brain NFκB activation is mediated by increased IκB-α expression.
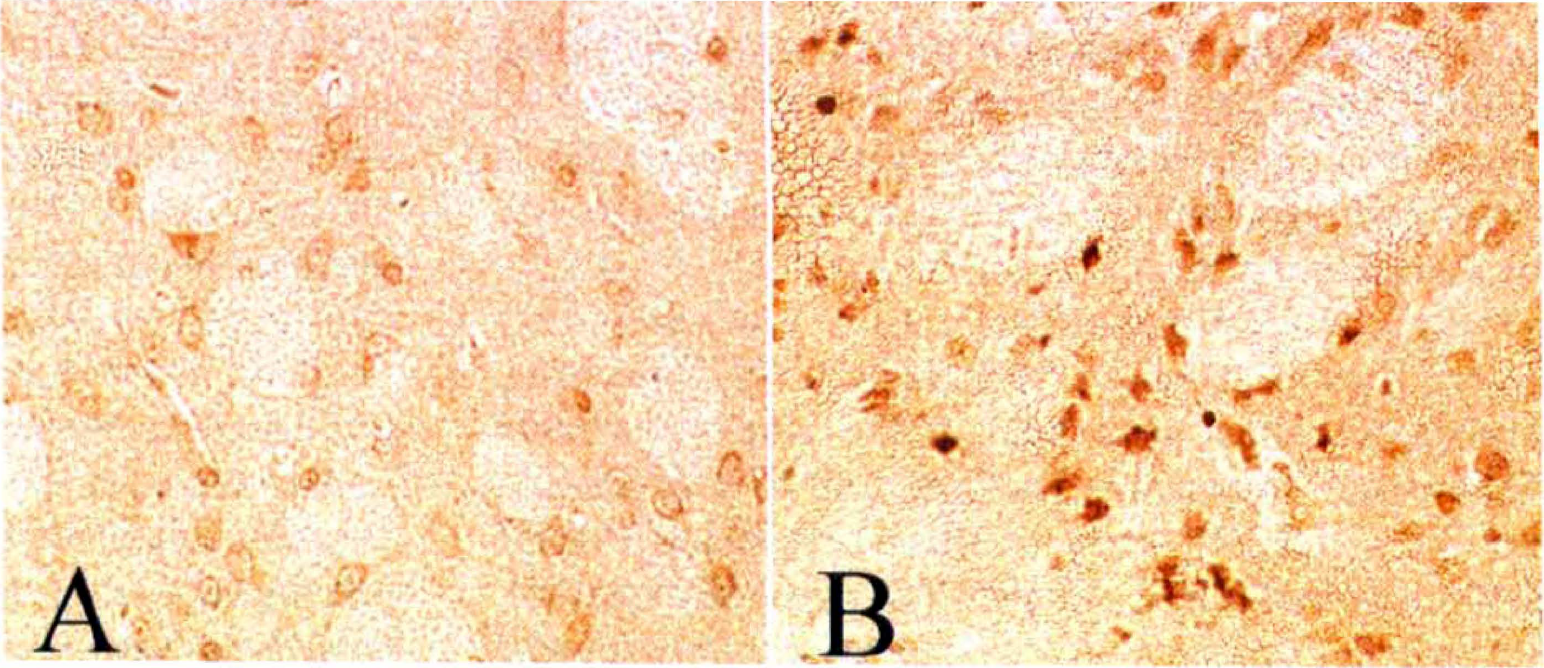
Heat shock increases IκB-α expression in brain. Adult female Sprague Dawley rats were anesthetized, then subjected to 20 minutes of whole-body heating at 41°C by warming in a heating pad. The next day, IκB-α expression was examined by immunocytochemical staining in striatal sections prepared from perfused control (anesthetized only, top panel) or heat-shocked (bottom panel) animals, using anti-IκB-α antibody SC-371.
DISCUSSION
In this report we demonstrate for the first time that prior HSR, induced by whole-body hyperthermia, prevents subsequent inflammatory reactions in rat brain caused by injection of bacterial endotoxin LPS and cytokines. At 24 hours after intrastriatal injection of LPS, IL-1β, and IFNγ (“CM”), we detected NOS2-positive cells in striatum, corpus callosum, and to a lesser extent in cortex. At the same time we also detected a large increase in ED1-positive staining cells, reflecting activation of resident microglial and/or infiltration of peripheral macrophages. Although injection of LPS alone (Garcion et al., 1998) or LPS plus IFNγ (Kitamura et al., 1996) can induce NOS2, we found that injection of a combination of LPS, IL-1β, and IFNγ to be a more reproducible stimulus. Prior HS reduced the CM-induced appearance of both NOS2- and ED1-positive staining cells in all three brain regions examined. Heat shock prevented the rapid activation of NFκB that was observed in non-heat-shocked animals at 1 hour after CM injection, supporting the idea that inhibition of brain NFκB activation contributes to the suppression of subsequent inflammatory gene expression or activation. Heat shock also increased brain IκB-α levels, which could account for reduced NFκB activation. These results suggest that a controlled induction of a HSR in brain could reduce inflammatory damage during disease or trauma.
In our studies, we assessed the efficacy of whole body warming on inducing a HSR in brain by measuring changes in expression of HSP70. We found by western blot analysis that greater levels of HSP70 were attained between 18 and 24 hours after HS compared to either 4 or 48 hours after HS, and that 20 minutes of hyperthermia was more effective than 10 minutes, but as effective as 40 minutes. Although we have not determined the identity of HSP70 expressing cells, previous studies have shown that hyperthermia results in primarily glial (both astroglial and oligodendroglial) HSP70 expression (McCabe et al., 1993; Foster et al., 1997), particularly at 24 hours after HS (Krueger et al., 1999; Marini et al., 1990). Our findings (Fig. 5) that whole body hyperthermia increased expression of IκB-α in brain indicate that, as expected, other HSPs were induced by the paradigm used. Although optimization of IκB-α expression may have been a better method to assess the anti-inflammatory role of the HSR, the anti-inflammatory effects we observed suggest that levels of HSP70 can be used as an effective index for studying anti-inflammatory effects of the HSR. Fortuitously, using a relatively short time of HS may have allowed for sufficient expression of antiinflammatory/ protective HSPs to occur while minimizing any damage due to excessive HS treatment.
Western blot analysis revealed two major 68 to 70 kDa HSP70 bands in control brain, of which one (of slower mobility) was greatly increased 24 hours after HS (Fig. 1). The two bands most likely correspond to HSP70, since (1) the antibody used (mAb SPA-810 from Stressgen) does not detect the highly related HSC70 proteins (Manzerra et al., 1997); (2) we did not observe strong staining in control striatum which is expected to express HSC70; and (3) basal expression of HSP70 in brain has previously been reported (Manzerra et al., 1997; Longo et al., 1993). The observed heterogeneity of HSP70 may be caused by distinct mRNA species (Lowe and Moran, 1986; Blake et al., 1990; Mestril et al., 1994), or multiple gene usage (see Longo et al., 1993). At least 4 distinct HSP70 proteins were detected in rabbit cerebellum, of which several, but not all, were greatly increased at 5 hours after HS (Manzerra et al., 1997). Our data similarly shows that whole body warming selectively increases some, but not all forms of HSP70 in rat brain.
Our results show that 24 hours after LPS and cytokine injection, numerous cells were stained for NOS2 protein. Strong NOS2 immunoreactivity was observed in areas around blood vessels, as well as in cells within vessel lumens, suggesting that at this time point the majority of NOS2 cells are infiltrating leukocytes, brain macrophages or activated microglia, as previously reported (Kitamura et al., 1996; Garcion et al., 1998; Heneka et al., 1999a). Heat shock performed 24 hours before injection of immunostimulants prevented the appearance of NOS2-positive staining cells in all brain regions. In vitro studies have shown that the HSR blocks cytokine mediated NOS2 expression in lung epithelial cells (Wong et al., 1997a); reduces IL1-β dependent nitrite accumulation and NOS2 mRNA expression in rat pulmonary artery smooth muscle cells (Wong et al., 1995); prevents IL-1β stimulation of islet cell NOS2 (Scarim et al., 1998); prevents cytokine-induced NOS2 expression in hepatocytes (de Vera et al., 1996a, 1996b); and prevents LPS- or cytokine-induced NOS2 expression in rat primary astrocytes, C6 glioma cells, and aortic smooth muscle cells (Feinstein et al., 1996). Our current studies therefore extend existing in vitro observations to an in vivo model of inflammation, and provide evidence that protection by HS in other animal models may be caused by reduction of NOS2 expression.
In parallel to reduced NOS2 expression, we also found that HS reduced the number of ED1-positive cells in brain induced by CM injection. The ED1 antibody recognizes cells of the macrophage/monocyte lineage in rat (Dijkstra et al., 1985) and in the CNS is specific for parenchymal microglia/brain macrophages and perivascular microglial cells in various states of activation (Graeber et al., 1990; Flaris et al., 1993; Streit, 1995). Because ED1 labels monocytes/macrophages and activated microglia to a similar extent, these two cell types cannot be distinguished by this antibody. Most parenchymal cells detected were oval to round in shape, the characteristic morphology of brain macrophages and activated microglia. Since cytokine injection causes recruitment of blood monocytes into brain, it is likely that both an infiltration of blood derived monocytes together with activated resident microglia account for NOS2 and ED1 positive staining in our model. In related studies (Heneka et al., 2000) we found that the majority of NOS2-positive cells were also ED1 positive, suggesting that CM injection does not induce significant astroglial NOS2 expression. Whether the HSR reduces levels of NOS2 and ED1 staining in infiltrating monocyte by reducing infiltration or by reducing activation once in the brain is not clear.
We have examined the effects of HS on brain NFκB activation after CM injection, since suppression of parenchymal NFκB activation would be expected to block parenchymal cell NOS2 and ED1 expression, as well as reduce blood monocyte infiltration caused by reduced expression of brain adhesion molecules and/or chemokines. In saline-injected animals, faint staining for the p65 subunit was observed in large cells, and appeared to be primarily cytosolic. Basal p65 expression has previously been detected in neurons and resting astrocytes (Gabriel et al., 1999; Kaltschmidt et al., 1994). One hour after injection of LPS and cytokines, the faint cytosolic staining was replaced by more intense p65 staining that was confined to smaller areas. This increased staining could be caused by movement of cytosolic p65 to the nucleus (thus accounting for loss of cytosolic immunoreactivity), and/or to increased p65 expression occurring in a second population of smaller cells (i.e., smaller glial versus larger neuronal cells, or infiltrating monocytes). The observation of two morphologically distinct staining patterns is consistent with activation of NFκB in multiple cell types. Prior HS prevented any changes in staining pattern observed for NFκB in brain, suggesting (1) that parenchymal cell NFκB activation was prevented; and (2) that NFκB activation in infiltrating cells was reduced, and/or there was suppression of monocyte infiltration into brain. These results are the first demonstration that the HSR is an effective inhibitor of NFκB activation in brain.
Whereas decreased NFκB activation in parenchymal glial cells could explain their lack of NOS2 (and ED1) expression, there are several ways by which reduced brain NFκB activation could suppression the inflammatory state of infiltrating monocytes in brain. In vitro, a HSR blocked cell adhesion molecule (ICAM-1) expression in monocytes (Housby et al., 1999), and chemokine expression in epithelial cells (Ayad et al., 1998). It is therefore possible that in vivo, the decrease of brain NOS2 and ED1 immunoreactivity is caused in part by reduced chemokine or adhesion molecule expression in resident cells, leading to a decrease in the number of invading cells. Alternatively, reduced proinflammatory cytokine expression from brain cells in response to the initial injection of immunostimulants, which normally activate and sustain NOS2 expression in infiltrating and resident cells, could contribute to reduced monocyte NOS2 and ED1 expression. Finally, the HSR could reduce the ability of peripheral cells themselves to become activated, and thus lose the capacity to migrate into brain.
Previously studies have shown that the HSR can provide protection against various forms of inflammatory damage both in vitro and in vivo. Thus, prior hyperthermia decreased lung damage and mortality caused by intratracheal instillation of phospholipase A2 (Villar et al., 1994); and greatly increased survival after intravenous injection of endotoxin (Chu et al., 1997; Hotchkiss et al., 1993). In vitro studies show that hyperthermia protects cultured cortical neurons from glutamate toxicity (Rordorf et al., 1991); increases hippocampal survival in primary cultures (Sato et al., 1996); and reduces cortical neuronal cell death from excitotoxic and apoptotic mechanisms (Snider et al., 1998). However, the protective role of the HSR, and HSP70 expression in ischemia is controversial. HSP70 mRNA is expressed in neurons and glia after ischemia (reviewed in Sharp et al., 1999); however, within the infarct area translation into protein is limited (Nowak and Jacewicz, 1994; Kinouchi et al., 1993). HSP70 is also expressed in cells outside the infarct area (Kinouchi et al., 1993), and most cells which showed HSP70 staining showed little evidence for cell damage as indicated by DNA fragmentation (States et al., 1998). This suggests that ischemic stress can activate HSP70 expression in neurons distal from the infarct site, and that this expression provides protection against ischemic damage. In several models of ischemic tolerance, increases in HSP70 expression were found to parallel the degree of tolerance to subsequent ischemic damage (Kirino et al., 1991; Glazier et al., 1994; Chen et al., 1996), although no correlation was observed between HSP70 expression and spreading depression induced tolerance (Kobayashi et al., 1995). More direct evidence that HSP70 expression provides protection against ischemic damage comes from transgenic studies in which constitutive HSP70 expression reduced ischemic damage, both in heart and in brain (Mestril et al., 1994; Plumier et al., 1995; reviewed in Yenari et al., 1999). Together with our current results, these findings suggest that prior establishment of a HSR, and/or HSP70 expression, will prove effective in reducing inflammatory as well as ischemic damage in brain.
The mechanisms by which HS prevents inflammatory gene activation are not completely understood. Observations that the HSR reduced cytokine (Snyder et al., 1992; Ayad et al., 1998) and NOS2 (Feinstein et al., 1996; Wong et al., 1995; de Vera et al., 1996a, 1996b; Scarim et al., 1998; Hauser et al., 1996) mRNA levels suggests that effects are mediated at the transcriptional level. This was confirmed in studies showing that the HSR reduced activation of the NOS2 promoter transfected into hepatocytes (de Vera et al., 1996a); C6 cells (Feinstein et al., 1996), and lung epithelial cells (Wong et al., 1997a). Because activation of transcription factor NFκB is a general activator of cytokines gene expression, the effects of HSR on NFκB activation have been studied. Heat shock prevented NFκB activation by IL-1β in islet cells (Scarim et al., 1998), by cytomix in hepatocytes (de Vera et al., 1996a, 1996b), by LPS and cytomix in astrocytes and C6 cells (Feinstein et al., 1996), by TNF-α in lung epithelial cells (Wong et al., 1997), and by LPS in endothelial cells (DeMeester et al., 1997a). It therefore appears that in vitro, the suppression of NOS2 by the HSR is mediated, at least in part, by inhibition of NFκB activation. However, additional mechanisms acting independent of NFκB inhibition could also be activated by the HSR and contribute to decreased NOS expression, for example leading to decreased NOS2 mRNA stability or translational efficiency.
It is not clear how the HSR reduces NFκB activation. As we previously suggested (Feinstein et al., 1997), the presence of a nuclear localization site in HSP70 could allow it to compete with NFκB for access to the nuclear pore complex (Feinstein et al., 1997) and thereby block NFκB nuclear uptake. Alternatively, cytosolic HSP70 could associate with the partially unfolded structure of the NFκB:IκB complex, thus preventing release of NFκB. The fact that HSP70 expression in transgenic mice reduced ischemic damage (Mestril et al., 1994; Plumier et al., 1995) suggests a direct effect of HSP70 on inflammatory damage. However, observations that the HSR prevented the cytokine-induced loss of IκB-α in C6 cells (Feinstein et al., 1997) and epithelial cells (Wong et al., 1997a, 1997b) provides a further explanation for inhibition of NFκB activation, since IκB-α degradation by the 26 proteasome is, in most cases, needed for release of NFκB and its migration to the nucleus. The HSR could increase IκB-α levels directly since it was recently shown that IκB-α is itself a stress protein whose promoter contains near-consensus binding sites for HS transcription factors (DeMeester et al., 1997; Thomas et al., 1998; Wong et al., 1999), or indirectly since the HSR can inhibit the activity of an IκB kinase (Curry et al., 1999). Our immunocytochemical data (Fig. 5) demonstrates that whole-body hyperthermia increases IκB-α expression in brain, suggesting that IκB-α functions as a stress protein both in vivo, as well as in vitro, and would therefore be expected to contribute to inhibition of NFκB activation.
Our results indicate that a brief period of prior hyperthermia can reduce subsequent inflammatory events in brain, suggesting a possible therapeutic application for the HSR in the prevention or reduction of inflammatory damage in brain. In a related set of studies (Heneka et al., unpublished observations, 1999) we found that intracerebellar injection of LPS, IL-1β, and IFN-γ, the same cytokine combination as used in the present study, induced both NOS2 expression as well as apoptotic cell death in cerebellar granule cells. Similarly, intrahippocampal injection of LPS (Garcion et al., 1998) or LPS plus IFN-γ (Matsuoka et al., 1999) caused neuronal apoptosis as assessed by TUNEL staining. Since in vitro, inhibition of NOS2 expression by selective NOS2 inhibitors can prevent neuronal apoptosis (Heneka et al., 1998) it is likely that inflammatory induction of NOS2 in brain contributes to eventual cell death. Our findings therefore suggest that prior establishment of a HSR in brain will prevent neuronal cell death, as well as acute inflammatory activation.
Because hyperthermia itself cannot be considered a practical therapeutic approach, alternative means to induce a HSR in brain must be considered. The observations that nonsteroidal anti-inflammatory drugs including indomethacin and ibuprofen, inhibit NOS2 expression (Aeberhard et al., 1995) and NFκB activation (D'Acquisto et al., 1998; Fiebich et al., 1999; Lee et al., 1995; Scheuren et al., 1998), together with findings that these same drugs induce a HSR (Housby et al., 1999; Lee et al., 1995; Fawcett et al., 1997) suggest that the mechanism of action of nonsteroidal anti-inflammatory drugs may be mediated, at least partly, through the HSR. Similarly, anti-inflammatory prostaglandins, including PGA1(Rossi et al., 1997) and PGJ2, an endogenous agonist of the peroxisome proliferator-activated receptor-γ (Colville-Nash et al., 1998; Petrova et al., 1999; Ricote et al., 1998; Jiang et al., 1998; Heneka et al., 1999b), block both NOS2 expression and NFκB activation, and also induce a HSR (Rossi et al., 1997; Colville-Nash et al., 1998; Amici et al., 1992; Kitamura et al., 1999). Finally, the knowledge that treatment with aspirin or indomethacin lowers the threshold for inducing a HSR (Lee et al., 1995) suggests that a combination of anti-inflammatory drug treatment with mildly elevated temperature (i.e., 39°C) could prove effective in reducing inflammatory damage. These findings raise the possibility that pharmacologic treatments can be used to induce a HSR under normothermic conditions.
Footnotes
Acknowledgment
The authors thank Patricia Murphy for assistance with sample preparation and animal handling.