
Abstract
Although the thrombolytic activity of tissue-type plasminogen activator (t-PA) may be beneficial in the acute treatment of stroke, recent studies have suggested that this serine protease could also play a critical role in determining the extent of neuronal death after injury to the central nervous system (CNS). This hypothesis is based on several experimental results: t-PA-deficient mice are resistant to excitotoxic neuronal death induced by the intrahippocampal injection of kainate; the infarct volume induced by occlusion of the middle cerebral artery is reduced in t-PA knockout mice; and the intravenous injection of t-PA can under certain circumstances potentiate the infarct volume in animals subjected to middle cerebral artery occlusion.
In the CNS, the serine proteases have been identified to occur both in neurons and glial cells. Their enzymatic activity regulates the balance between the accumulation and the degradation of the extracellular matrix. They are involved in many physiologic functions, ranging from synaptic outgrowth during perinatal development to plasticity in adults. For instance, thrombin and t-PA are known to modulate neurite outgrowth and tissue remodeling in the early stages of development. In the adult brain, t-PA may contribute to the late phase of long-term potentiation and to the subsequent synaptic growth in the hippocampal mossy fiber pathway.
This balance between the degradation and accumulation of the extracellular matrix may also be integral to various pathologic processes involved in acute brain injury. For example, compounds that modulate the activity of serine proteases exhibit neuroprotective activity. Based on the above, numerous studies have focused on the production and modulation of the endogenously produced serine protease inhibitors, termed serpins, such as type 1 plasminogen activator inhibitor, neuroserpin, and protease nexin-1.
In the present review, we will discuss the need to distinguish between the potentially neurotoxic effects of t-PA and its beneficial effect on reperfusion. We will present data supporting the idea that the modulation of serine protease activity may represent a novel and efficient strategy for the treatment of acute cerebral injury in humans.
Keywords
Because the usual causes of stroke are related to thromboembolic processes in cerebral arteries, the idea that the prevention of clot formation might be helpful for patients was understood as early as the 1940s. After unsuccessful clinical trials using different pharmacologic agents (e.g., streptokinase or urokinase) directed toward the pathway for lysis of clots, the National Institutes of Health recommended in the 1980s the use of tissue-type plasminogen activator (t-PA) for the treatment of thromboembolic diseases. In 1991, an advanced study was initiated to study the effects of t-PA (Hacke et al., 1995). Although the clinical results were not conclusive, the authors suggested that recombinant t-PA (rt-PA) treatment (at 1.1 mg/kg) after the onset of stroke speeded neurologic recovery. In 1995 (Adams et al., 1995), the National Institute of Neurological Disorders rt-PA Stroke Study Group conducted a randomized, double-blind, placebo-controlled study of the effect of the intravenous injection of t-PA within 3 hours after the onset of symptoms in 624 patients. This time, the results of the clinical study were more conclusive: t-PA administration produced an absolute increase of 11% to 13% in the proportion of patients who regained normal neurologic function without increasing the fraction that had a poor outcome. Although t-PA treatment induced a 10-fold increase in the rate of symptomatic intracerebral hemorrhage, ischemic brain damage was reduced. Notwithstanding these secondary effects, the intravenous injection of t-PA was approved by the Food and Drug Administration as the first agent acknowledged for the treatment of focal cerebral infarction. The same year, a report that described the participation of t-PA in excitotoxic neuronal death was published (Tsirka et al., 1995). Since then, numerous studies have been conducted to determine the role of serine proteases and their natural inhibitors (serpins) in physiologic and pathologic conditions.
In this brief review we will first discuss the expression pattern of serine proteases and their inhibitors in the healthy and injured brain. Then we will present experimental data to demonstrate the involvement of serine proteases in various types of neuronal injury. Finally, we will advance the hypothesis that although the thrombolysis induced by t-PA may exert a beneficial activity in the treatment of acute stroke, t-PA may play other roles as well, some of which might be deleterious to brain tissue. Based on the literature reviewed, we suggest that the modulation of the activity of serine proteases may represent a novel therapeutic target for limiting the extent of acute and neurodegenerative brain damage.
EXPRESSION OF SERINE PROTEASES AND THEIR COGNATE INHIBITORS
The serine proteases are a superfamily of proteins in which most members exert their proteolytic activity by cleavage after the amino acid arginine (Table 1). These proteins have been studied extensively in relation to coagulation and thrombolysis (Carmeliet and Collen, 1997). In addition to their existence in the blood compartment, several of these proteases have been localized in the central nervous system (CNS). The CNS serine proteases most studied are the plasminogen activators (t-PA and urokinase-type plasminogen activator [u-PA]) and thrombin, which are thought to play a critical role in CNS homeostasis.
Serine proteases and serpins location

t-PA, tissue-type plasminogen activator; u-PA, urokinase-type plasminogen activator; PAI-1, type 1 inhibitor of plasminogen activator; PN-1, protease nexin-1.
The maintenance of vascular patency and the dissolution of fibrin clots are key functions of plasminogen. Plasminogen is a proenzyme that can be converted to the active enzyme, plasmin, by different types of activators. Two physiologic plasminogen activators, t-PA and u-PA, have been identified, initially based on their immunologic relations with the plasminogen found in urine.
t-PA, the predominant plasminogen activator in blood, is a serine protease with a molecular weight of 60 to 70 kDa and is synthesized and secreted by endothelial cells in a single-chain form. This form is cleaved by the plasmin into the double-chain form held together by a disulfite bond (Lijnen et al., 1980). In the brain, t-PA is involved in the catabolism of the extracellular matrix through the conversion of the inactive plasminogen to the protease plasmin (Fig. 1). Although there is no clear evidence of an obligatory interaction between t-PA and a membrane-bound receptor to initiate proteolysis, t-PA can interact with the cell surface by means of the calcium-regulated phospholipid-binding protein annexin II (Hajjar et al., 1998). The expression of annexin II has been detected in the developing murine CNS (Hamre et al., 1995), but its distribution in the adult brain remains to be established. During embryogenesis, increased t-PA expression coincides with extensive cell migration, proliferation, and tissue remodeling in the CNS (Friedman and Seeds, 1995). The first report of the implication of t-PA in cell migration was obtained from differentiated lines of neuroblastoma cells. A fibrin overlay assay revealed that the predominant site of t-PA activity was on the growth cones (Krystosek and Seeds, 1981). Cerebral t-PA mRNA levels are modulated according to the stage of development. Northern blot analyses performed on mouse brain revealed that t-PA mRNA levels are low from embryonic day 1 to day 7, increase from postnatal day 1 to day 7, and thereafter decline about threefold in the adult (Friedman and Seeds, 1994). In the mature brain, t-PA is synthesized by neurons in most regions, with the most prominent expression in the hippocampus and hypothalamus (Sappino et al., 1993). In vitro, t-PA mRNAs are detected in both cultured cortical neurons and astrocytes (Buisson et al., 1998). The earliest data on the physiologic role of t-PA were obtained from mice in which the t-PA gene was rendered nonfunctional (t-PA −/−) (Carmeliet et al., 1994). These knockout mice displayed a selective reduction in the late phase of the phenomenon of long-term potentiation (Huang et al., 1996). Similarly, the administration of inhibitors of t-PA proteolytic activity such as endogenous plasminogen activator inhibitor, type 1 plasminogen activator inhibitor (PAI-1), or a synthetic t-PA inhibitor, t-PA stop, inhibit the long-term potentiation induced in the mossy fibers of the hippocampus (Baranes et al., 1998). In summary, the available evidence indicates that t-PA is central to the regulation of neuroplasticity in both in the developing and mature brain.
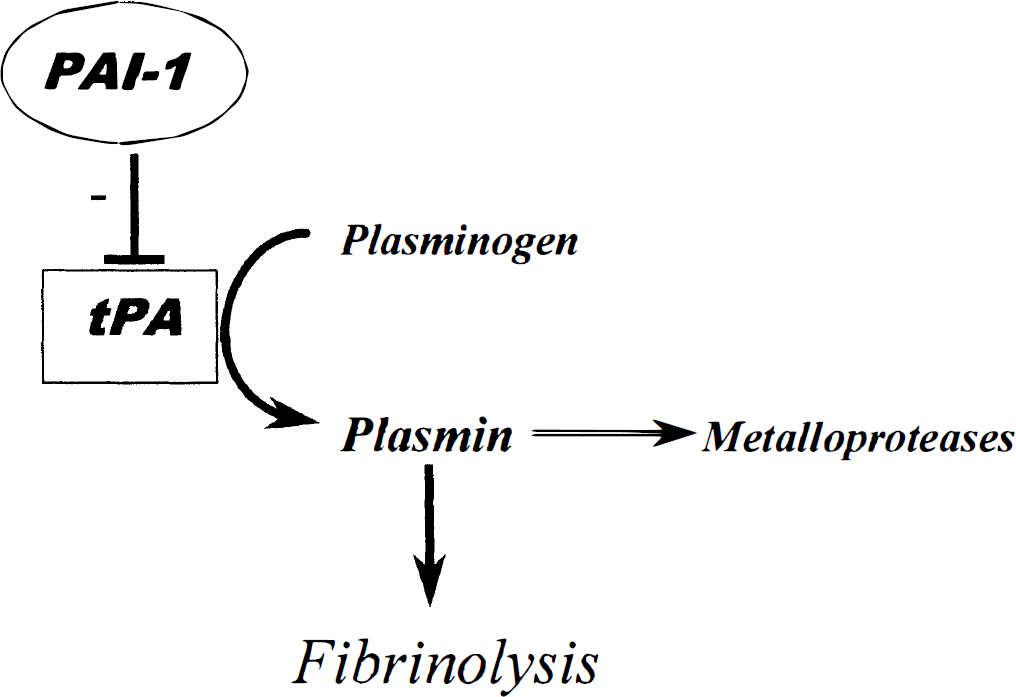
The tissue-type plasminogen activator (t-PA) proteolytic cascade. t-PA cleaves the proenzyme plasminogen in the active serine protease plasmin. Plasmin activation facilitates extracellular proteolysis either directly or by the activation of metalloproteases.
u-PA, a second serine protease involved in the activation of plasminogen, has a molecular weight of 54 kDa. To be activated, u-PA requires conversion by plasmin from an inactive single chain to a catalytically active double-chain derivative (Lijnen et al., 1980). The major difference between t-PA and u-PA is that u-PA needs to be bound to a specific cell surface receptor, the urokinase plasminogen activator receptor. This feature gives u-PA a precise localization for its proteolytic activity that affects the degradation of the extracellular matrix (Yamamoto et al., 1994). During embryonic development, u-PA is expressed throughout the growth process of neurons and astrocytes in the cerebellum, hippocampus, and cerebral cortex. These data suggest that the function of u-PA is related to cell proliferation, migration, and differentiation during brain development (Tranque et al., 1992, 1994). In the adult, in situ hybridization in mice has shown that u-PA is predominantly expressed in the subiculum, the entorhinal and parietal cortices, and in the hippocampus (Masos and Miskin, 1996). The physiologic role of u-PA in the CNS should be further investigated.
The serine protease thrombin (38 kDa) is a key component in the coagulation cascade and is also localized in the CNS. Thrombin exerts its effects via a G-protein-coupled protease-activated receptor (PAR-1) that is found on the cell surface. These receptors are cleaved by thrombin and then undergo a conformational change that leads to their activation. Both neuronal and glial cells express the thrombin precursor prothrombin and the PAR-1 transcript (Niclou et al., 1994). In situ hybridization experiments have revealed a ubiquitous but lowintensity signal for PAR-1 mRNA in the late embryonic and early postnatal nervous systems. In the adult, PAR-1 expression is more pronounced but is confined to specific neurons such as mesencephalic dopaminergic and cerebellar Purkinje cells (Niclou et al., 1998). In summary, the role of thrombin in the CNS seems distinct from its function in the coagulation system, but its physiologic importance remains to be established.
A novel and brain-specific serine protease, neuropsin (55 kDa), has been recently identified (Chen et al., 1995; Scarisbrick et al., 1997). This protease is markedly expressed in the neurons of the murine limbic system and in the pyramidal neurons of the hippocampus. This restricted localization could imply involvement in neural plasticity (Tomizawa et al., 1999).
In biologic systems, the activity of serine proteases depends on their equilibrium with their serine protease inhibitors, termed serpins. These serpins act as a pseudosubstrate for their target protease(s) and establish a tight bond with the catalytic site. By attenuating extracellular proteolysis, serpins modify the processes that involve the proteolysis of the extracellular matrix such as neuronal migration, axogenesis, or the formation of mature synaptic connections. Two major inhibitors of t-PA have been identified in the brain: PAI-1 (50 kDa) (see Fig. 1) and the neuroserpin (55 kDa). PAI-1 is a fastacting inhibitor of t-PA and u-PA and has been shown to be present in the rodent cerebral cortex. In vitro studies have revealed that the PAI-1 mRNA and the protein are exclusively restricted to astrocytes. PAI-1 mRNA and protein are not detected in cultured cortical neurons (Docagne et al., 1999).
Initially identified as an axonally secreted protein in cultured chicken neurons taken from the dorsal root ganglion, neuroserpin is a serine protease inhibitor present both in the developing and in the adult murine nervous system (Berger et al., 1998; Hastings et al., 1997; Krueger et al., 1997). In the adult CNS, neuroserpin is expressed predominantly in the neocortex, the hippocampal formation, the olfactory bulb, and the amygdala (Hastings et al., 1997; Krueger et al., 1997). In vitro, murine cortical neurons and astrocytes both express neuroserpin mRNA (Docagne et al., 1999).
Protease nexin 1 (PN-1) (44 kDa) is the most potent tissue inhibitor of thrombin known (Smirnova et al., 1996). In situ hybridization performed during embryonic CNS development has shown expression of PN-1 in the mesencephalon (Kury et al., 1997). In the developing postnatal brain, PN-1 expression appears transiently in many neuronal populations (Mansuy et al., 1993). Further, in vivo studies have shown that the expression of PN-1 is abundant not only in the astrocytes of the parenchyma but also in the perivascular astroglial end feet (Choi et al., 1990). In vitro, there is evidence of expression of PN-1 mRNA in both cortical neurons and astrocytes (Docagne et al., 1999). The presence of PN-1 has also been detected in Schwann cell cultures (Mulligan et al., 1991). Studies performed in the adult human cerebral cortex have revealed a pronounced immunoreactivity for PN-1 in capillaries and in the smooth muscle cells of arteries and arterioles (Choi et al., 1990).
INVOLVEMENT OF SERINE PROTEASES AND INHIBITORS DURING NEURODEGENERATIVE DISEASE
Various studies have shown that the expression of serine proteases is modulated during brain injury (Qian et al., 1993). However, their involvement in the consequent neuronal damage remains controversial (Table 2).
Beneficial and deleterious effects of serine proteases and their inhabitors

t-PA, tissue type plasminogen activator; u-PA, urokinase type plasminogen activator; PAI-1, type 1 inhibitor of plasminogen activator; PN-1, protease nexin-1.
In 1995, it was reported for the first time that t-PA mediates neuronal cell death induced by the intrahippocampal injection of kainic acid in mice (Tsirka et al., 1995). Explicitly, these authors observed that t-PA-deficient mice display a dramatic resistance to the excitotoxic injury induced by this glutamate agonist. To analyze the mechanism by which t-PA-deficient mice were resistant to the neurotoxin, the same group subjected mice deficient in plasminogen, the zymogen substrate of t-PA, to the same type of injury. They showed that the intrahippocampal injection of kainic acid induced a minimal degeneration in the plasminogen-deficient mice, whereas fibrin-deficient mice displayed a vulnerability to the excitotoxin similar to that seen with the wild-type animals (Tsirka et al., 1997a). These results were further strengthened by the observation that the infusion of α2-antiplasmin, the physiologic plasmin inhibitor, into the hippocampus of wild-type mice confers neuroprotection against excitotoxic damage (Tsirka et al., 1997b). Moreover, intrahippocampal delivery of plasminogen to plasminogen-deficient mice restored degeneration induced by glutamate analogue injection to wild-type levels, but intravenous delivery did not (Chen et al., 1999). These results suggest that excitotoxic neuronal death and blood-brain barrier breakdown are independent processes. The same group (Chen and Strickland, 1997) clarified the potential role of plasmin in neuronal excitotoxic mechanisms by identifying a target protein, laminin, cleaved by plasmin in response to a kainate infusion. These authors showed that the intrahippocampal injection of kainic acid induces a loss of the laminin immunoreactivity that precedes neuronal death. This laminin cleavage and the neuronal death subsequent to the kainate injection were abolished in t-PA-deficient mice or when kainate was coinjected with α2-antiplasmin into the hippocampus of wild-type mice. These results indicate that the cleavage of plasminogen into plasmin by t-PA is a key step in the excitotoxic cascade.
Because excitotoxic neuronal injury is thought to participate in ischemia-induced brain damage, it was important to test the implication of a t-PA/plasmin pathway in an animal model of cerebral ischemia. This step was achieved in 1998 by Wang et al., who demonstrated that t-PA-deficient mice subjected to 3 hours of middle cerebral artery occlusion (MCAO) exhibited a 50% reduction in the infarct volume compared with wild-type mice. These data have been confirmed by Nagai et al. (1999), who showed that the infarct size induced by a permanent MCAO is threefold smaller in t-PA-deficient mice than in wild-type animals. The implication of t-PA in ischemia-induced neuronal cell death was strengthened by the results obtained in PAI-1-deficient mice. When subjected to permanent MCAO, PAI-1-deficient mice exhibited a twofold increase in infarct volume. No modification of the infarct size was observed in u-PA-deficient mice (Nagai et al., 1999). Taken together, these data suggest that t-PA is involved in the deleterious cascade of events leading to ischemia-induced neuronal death. In these latter studies, the involvement of plasmin in the t-PA deleterious effect during cerebral ischemia remains controversial. In contrast to the previous investigations, in which a lesion was induced by the intrahippocampal injection of an excitotoxin, Nagai et al. (1999) reported that plasminogen-deficient mice subjected to MCAO exhibited a larger infarct volume compared with wild-type animals. Similarly, mice with an increased plasmin activity (α2-antiplasmin-deficient mice) were resistant to an ischemic insult.
The cellular origin of the t-PA present in brain tissues after CNS injury is not well characterized. It has been suggested that t-PA is produced by activated microglial cells in response to the neuronal injury induced by an intrahippocampal injection of kainic acid (Rogove and Tsirka, 1998; Tsirka, 1997). However, experiments performed with the “neuronal-like” cell line PC-12 demonstrate an activity-dependent secretory mechanism that can rapidly increase the amount of t-PA (Gualaudris et al., 1996). These authors suggested that t-PA could be stored in vesicles and released along with neurotransmitters. Thus, during hypoxic depolarization, t-PA could be released with glutamate into the extracellular space and participate in the excitotoxic pathway leading to ischemic neuronal death.
The resistance of t-PA-deficient mice to ischemic insults raises the question of whether the potentially deleterious effect of rt-PA is independent of its beneficial thrombolytic activity. To test this hypothesis, it was considered necessary to use a treatment with rt-PA in an animal model of cerebral ischemia that did not involve the formation of a thrombus. Wang et al. (1998) demonstrated that the intravenous injection of rt-PA (1 mg/kg) 20 minutes after MCAO with an intravascular filament increased the lesion induced by 120 minutes of MCAO. These data support the idea that the intravenous injection of rt-PA promotes undesirable effects by potentiating the extent of the lesion. However, this result remains controversial: two other groups have reported conflicting results. Kilic et al. (1999) showed that the intravenous injection of rt-PA (10 mg/kg) 15 minutes after the onset of a 90-minute transient MCAO in mice reduces the ischemic injury. Klein et al. (1999) showed that the intravenous injection of rt-PA does not exacerbate ischemia-induced lesions in models of global and focal cerebral injury in rats.
Recently, other workers have suggested that t-PA could also exert a neuroprotective role independent from its catalytic activity. Kim et al. (1999) have described a neuroprotective effect of t-PA against zinc-induced neuronal death in vitro as well as in a model of free radicalmediated neuronal injury in murine cortical cell cultures. They strengthened their preliminary observations by injecting t-PA into the cerebrospinal fluid (CSF) of animals subjected to kainate-induced seizures. This treatment reduced both zinc translocation into neurons and the subsequent kainate-induced neuronal death in the hippocampus. They postulated that the neuroprotective effect of the t-PA treatment does not involve its proteolytic activity.
The role of other serine proteases in acute brain injury or neurodegenerative diseases has not been extensively studied. However, thrombin has been shown to exert deleterious effects on both neurons and astrocytes in vitro. These effects, ranging from neurite retraction to induction of apoptotic cell death, appear to be mediated through an interaction with thrombin cell surface receptors and are prevented by PN-1 (Turgeon and Houenou, 1997; Turgeon et al., 1998). For example, synthetic peptides that directly activate the thrombin receptor have been shown to induce apoptosis (Donovan et al., 1997). The neurotoxic effects of the β-amyloid protein on cultured neurons can be potentiated by the activation of the thrombin receptor (Pike et al., 1996). Taken together, these data suggest the participation of thrombin in certain cell death pathways that are involved in neurodegenerative diseases. However, thrombin has been shown to exert a neuroprotective role in various types of injury. It has been reported that thrombin protects rat primary hippocampal neurons from cell death induced by hypoglycemia or growth supplement deprivation (Vaughan et al., 1995) and to reduce rat astrocytic death induced by hypoglycemia or oxidative stress (Vaughan et al., 1995). Overall, these data underline the potential participation of thrombin in various neurologic disorders.
With respect to the potential involvement of serine proteases in neurodegenerative diseases, an impairment in the balance between serine proteases and their cognate inhibitors in the CNS may lead to a pathologic state. Accordingly, it is important to study the level of expression of serpins in various neurodegenerative conditions. In humans, it has been reported that the expression of the most potent inhibitor of t-PA, PAI-1, is increased in the CSF from patients with neurologic disorders such as Alzheimer disease, cerebral infarction, CNS infection, and CNS neoplasia. Overall, there was no correlation between plasma and CSF levels of PAI-1. These observations suggest that CSF PAI-1 levels may represent a nonspecific marker of CNS disease (Sutton et al., 1994). Similar results were obtained in experimental models of cerebral ischemia in rodents. After permanent focal cerebral ischemia in mice, semiquantitative RT-PCR (Fig. 2A) and in situ hybridization studies revealed that PAI-1 is overexpressed 24 hours to 3 days later at the border of the ischemic core and in tissues that matched GFAP immunostaining (Docagne et al., 1999). This upregulation of PAI-1 expression has also been observed as early as 6 hours after the onset of focal ischemia in rats (Docagne et al., unpublished results).
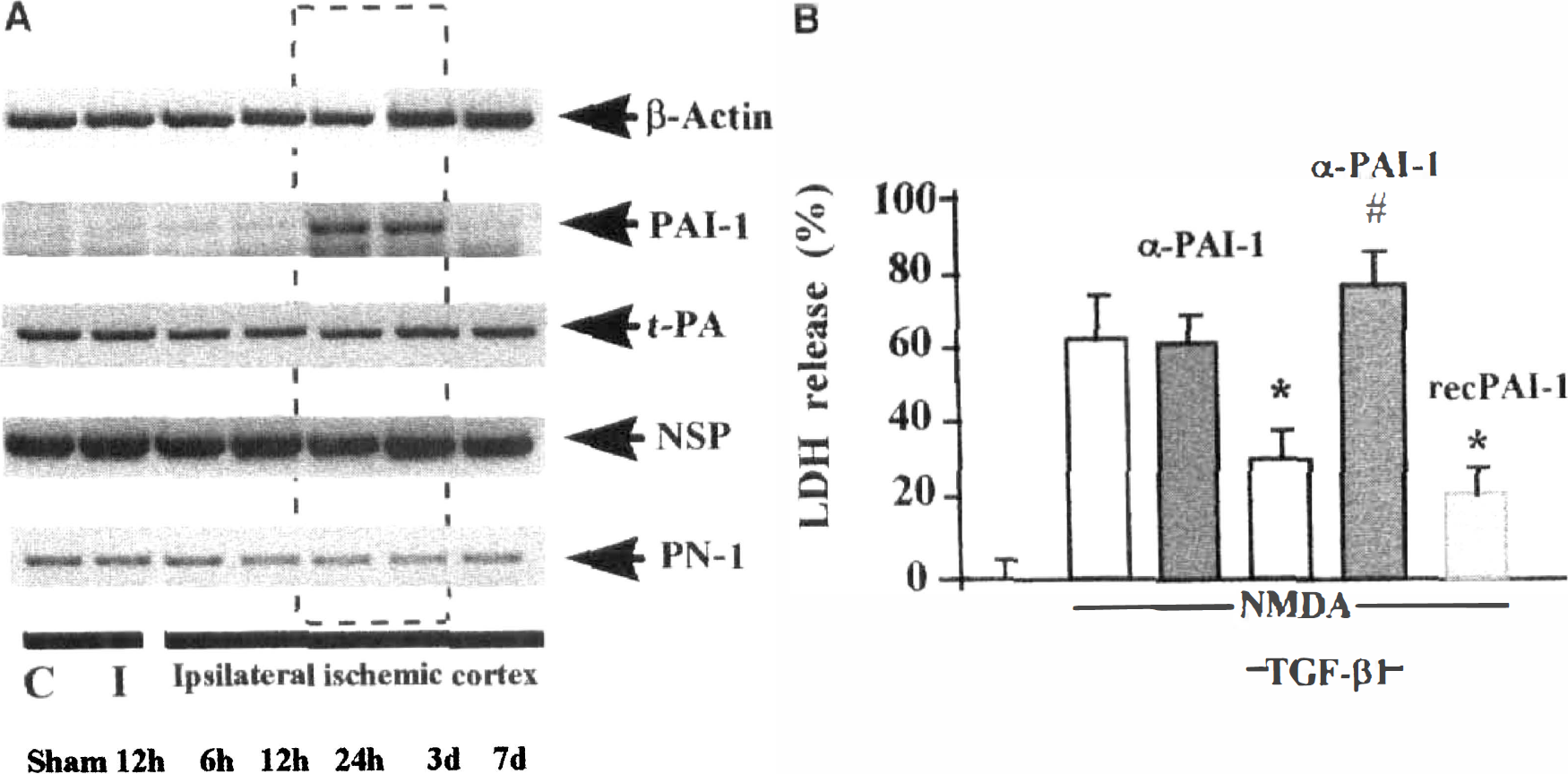
(
Other serine protease inhibitors may also be modulated after brain injury. PN-1 expression is up-regulated in glial cells after different types of CNS lesions that are associated with a disruption of the blood-brain barrier (Erno et al., 1996). In some cases, this expression persists 1 year after the induction of transient global ischemia in rat brain (Nitsch et al., 1993). In contrast, a marked reduction of PN-1 and PN-2/β-amyloid protein precursor is observed in the brain of patients with Alzheimer disease (Choi et al., 1995).
Overall, these observations suggest that serpins may influence the outcome of brain injury. Experimental data support this postulate. Tsirka et al. (1997b) have mentioned that PAI-1 confers extensive neuroprotection after a hippocampal excitotoxic lesion. In vitro, PAI-1 produced by astrocytes is neuroprotective against necrosis mediated by N-methyl-D-aspartate (NMDA). This neuroprotective activity of PAI-1 is restricted to the necrotic cell death mediated by NMDA, because no effect has been observed against either kainate-mediated necrosis or apoptotic paradigms (Buisson et al., 1998). It has also been reported that PN-1 protects cultured rat hippocampal neurons from glucose deprivation-induced damage through attenuation of the increase in intracellular calcium levels associated with such damage. This protection is abolished by an equimolar concentration of thrombin (Smith-Swintowsky et al., 1995). PN-1 also can rescue spinal motoneurons during the period of programmed cell death that naturally occurs during ontogenesis (Houenou et al., 1995).
MODULATION OF SERINE PROTEASE/SERPIN AXIS BY INJURY-RELATED CYTOKINES IN THE CNS
In many biologic systems, the expression of the serine protease/serpin axis is modulated by various cytokines. In the CNS, this control by cytokines has not been extensively studied and remains speculative. Nevertheless, recent data have described a control of serine protease expression by neurotrophins. It has been demonstrated that brain-derived neurotrophic factor, neurotrophins 4 and 3, and nerve growth factor increase the secretion of plasminogen and u-PA from microglia (Nakajima et al., 1998). Thus, neurotrophic factors may contribute to changes associated with synaptic plasticity by means of a modulation of t-PA expression (Fiumelli et al., 1999). Other cytokines have been shown to decrease t-PA activity by up-regulating the expression of PAI-1. Interleukin-1β induces a dose-dependent decrease in t-PA activity in cultured astrocytes through an overexpression of PAI-1 (Rogister et al., 1990). Basic fibroblast growth factor (b-FGF) also can induce an accumulation of PAI-1 mRNA in astrocytes in vitro(Rogister et al., 1988). In a comparable study, both transforming growth factor-β 1 (TGF-β1) and b-FGF stimulated PAI-1 mRNA expression in astrocytes with a maximal 5-fold increase for b-FGF and a 30-fold increase for TGF-β1 (Treichel et al., 1998). The expression of the thrombin inhibitor PN-1 is also under the control of cytokines. For example, injury-related cytokines such as interleukin-1, tumor necrosis factor-α, and TGF-β can stimulate the secretion of PN-1 by the neuronal and glial cells from a neuroblastoma cell line (Vaughan et al., 1993).
Among the cytokines involved in the control of the expression of both serine proteases and serpins, TGF-β1 has been described as an important regulator of PAI-1 expression in the vascular system. In the CNS, TGF-β1 is overexpressed after various types of injury (Krieglstein and Kreiglstein, 1998). In vitro, TGF-β1 exerts a neuroprotective activity against NMDA-induced neuronal death (see Fig. 2B) but only in mixed cultures of neurons and astrocytes, which would suggest that astrocytes mediate this effect. These data have been confirmed by the blockade of TGF-β's neuroprotective activity after a selective transfection of astrocytes with a dominant negative receptor for TGF-β. Moreover, the neuroprotection induced by TGF-β1 is prevented by the addition of an antibody raised against PAI-1 and is mimicked by the coapplication of the recombinant form of PAI-1 (see Fig. 2B). These data suggest that the neuroprotective activity of TGF-β1 occurs by means of upregulation of PAI-1 in astrocytes (Buisson et al., 1998). This study has given evidence for a tight link between TGF-β1 and the serpins in the CNS. TGF-β1 belongs to a large family of peptides including activin and bone morphogenetic proteins, which are known to be expressed in the CNS (Dewulf et al., 1995; Ebendal et al., 1998). Recently, two other members of the same family, termed the glial cell line-derived neurotrophic factor (Lin et al., 1993) and neurturin (Kotzbauer et al., 1996), have been identified in brain tissues. Although TGF-β1 and activin can enhance the expression of PAI-1 in astrocytes (Docagne et al., 1999), bone morphogenetic proteins, glial cell line-derived neurotrophic factor, and neurturin are devoid of this activity.
These results provide novel insights into the regulation of the serpin/t-PA axis and the mechanisms by which an injury-related cytokine, such as TGF-β, may be neuroprotective in a situation of induced excitotoxicity.
SERINE PROTEASE INHIBITORS: A THERAPEUTIC TARGET FOR LIMITING ISCHEMIC BRAIN DAMAGE?
The idea that serine proteases-or more precisely the balance between serine proteases and serpins-may be involved in regulating neuronal fate during CNS development is now widely accepted. In the adult brain, serine proteases have been extensively studied for their role in synaptic plasticity. In pathologic conditions, there is increasing evidence to suggest an involvement of serine proteases and their inhibitors in neurodegenerative diseases.
Data obtained using transgenic mice indicate that serine proteases participate in the neuronal death observed in acute brain injury. Most studies have focused on the involvement of t-PA in acute brain injury because of its clinical relevance. The experimental data obtained from studies with t-PA-deficient mice have led to the same conclusion: t-PA participates in excitotoxic neuronal death (Chen and Strickland, 1997; Tsirka et al., 1995, 1997a, 1997b) and ischemia-induced neuronal death (Nagai et al., 1999; Wang et al., 1998). This conclusion is reinforced by the results obtained in PAI-1-deficient mice (Nagai et al., 1999). The involvement of t-PA in the excitotoxic pathway is still not well understood. Does it involve the activation of plasminogen into plasmin, or is it a direct action of t-PA? The answer remains unclear. Studies based on in vitro models of excitotoxicity may help to elucidate the cellular mechanisms through which t-PA participates in excitotoxic cell death. t-PA is not the only serine protease believed to be involved in the control of neuronal death. Thrombin seems to be implicated in the control of the viability of both astrocytes and neurons after different types of stimuli in vitro. Overall, these data suggest that the blockade of the activity of serine proteases might be beneficial in various types of CNS injury.
These experimental results have raised the question of the possible side effects of t-PA in stroke treatment in humans. Clinical studies have revealed that t-PA treatment is clearly beneficial in the treatment of acute stroke. However, because the ischemic lesion is thought to progress largely by an excitotoxic pathway, the data that demonstrate that serine proteases may potentiate excitotoxin-induced neurodegeneration raise the question of potential risks in the use of t-PA for the treatment of acute stroke. The disruption of the blood-brain barrier that occurs in cerebral ischemia may facilitate the access of systemically administered t-PA to neuronal tissues, thus potentiating neuronal death. As discussed above, the results obtained from rodents treated with intravenous t-PA and subjected to focal cerebral ischemia remain controversial. These discrepancies could be the result of differences in the t-PA regimen, such as the dose of t-PA or the type of paradigm used to induce neuronal death. Because of its clinical relevance, researchers should make a concerted effort to clarify this issue. The idea that thrombolytic agents may be used in combination with potent neuroprotective agents merits further attention. Because serine protease inhibitors specific for t-PA are neuroprotective, it is possible to suggest novel ways to counteract the neuronal death observed in acute stroke. The inhibition of the proteolytic activity of serine proteases on the cerebral parenchyma or the potentiation of serpin expression in brain cells could be a potential therapeutic intervention for disorders in which excitotoxicity is thought to play a critical role. These observations raise the possibility of distinguishing potentially neurotoxic effects of t-PA from its beneficial effect on reperfusion; the expectation would be that a combination of serine proteases and their inhibitors may usefully complement each other in the treatment of cerebral ischemia in the clinical setting (Fig. 3). Because PAI-1 would also block the beneficial effect of t-PA on the original clot, one could envisage initial treatment with t-PA followed by a second application of serpin. According to the data presented in this review, the roles and the mechanisms of action of serine proteases and their inhibitors in the CNS deserve greater consideration by the scientific and medical communities.
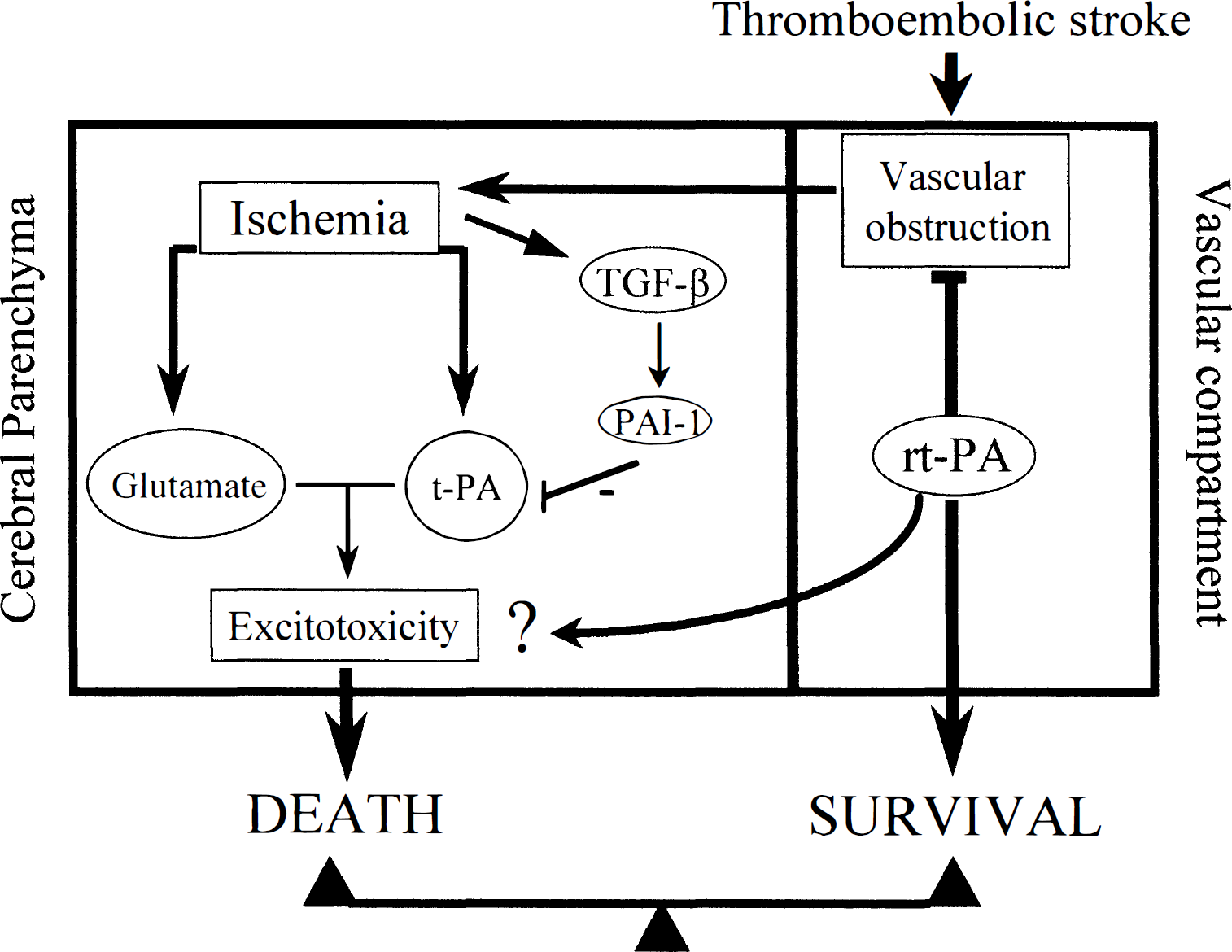
Proposed mechanism for the involvement of the tissue-type plasminogen activator (t-PA)/type 1 plasminogen activator inhibitor (PAI-1) axis in the central nervous system during acute stroke. Intravenous injection of t-PA is used for the treatment of focal cerebral infarction in humans (thrombolysis). In the cerebral parenchyma, t-PA could be released from stressed neurons (Gualandris et al., 1996) and could potentiate excitotoxin-mediated neuronal cell death (Tsirka et al., 1995). The expression of transforming growth factor-β 1 (TGF-β1) is up-regulated after either permanent or transient cerebral ischemia in rodents (Vivien et al., 1998). Then, TGF-β1 induces an overexpression of PAI-1 in astrocytes, a mechanism leading to the neuroprotective activity of TGF-β against N-methyl-D-aspartate (NMDA)-mediated excitotoxicity (Buisson et al., 1998). Concerning these results, we can postulate that the neuroprotective activity of TGF-β1 and PAI-1 could be related to their inhibitory effects on t-PA. All these results suggest that both endogenous t-PA and recombinant t-PA used for thrombolysis may interfere in the control of the neuronal outcome after stroke in humans.
Footnotes
Acknowledgments
The authors thank Dr. E.T. MacKenzie for helpful discussions and comments, and the other members of our team for their help during the preparation of this review.