
Abstract
In the immature brain, postischemic metabolism may be influenced beneficially by the effect of inducing hypercarbia or hypothermia. With use of 31P nuclear magnetic resonance spectroscopy, intracellular pH (pHi) and cellular energy metabolites in ex vivo neonatal rat cerebral cortex were measured before, during, and after substrate and oxygen deprivation in in vitro ischemia. Early postischemic hypothermia (fall in temperature −3.2 ± 1.0°C) delayed the normalization of pHi after ischemia by inducing an acid shift in pHi (P < 0.01). Postischemic hypercarbia (Krebs—Henseleit bicarbonate buffer equilibrated with 10% carbon dioxide in oxygen) and hypothermia induced separate, but potentially additive, reversible decreases in pHi, each of approximately −0.16 pH unit (P < 0.05). When these postischemic perturbations were applied in isolation, there was significant improvement of ∼20% in the recovery of β-ATP (P < 0.05). In combination, however, hypercarbia and hypothermia worsened recovery in ATP by ∼20% (P < 0.05). In control tissue, which had not been exposed to ischemia, ATP content was also significantly reduced by co-administration of the two treatments (P < 0.05), an effect that persisted even after discontinuing the perturbing conditions. Therefore, in this vascular-independent neonatal preparation, early postischemic modulation of metabolism by hypercarbia or hypothermia appears to confer improved bioenergetic recovery, but only if they are not administered together.
Keywords
In infants and young children with critical illness, treatment of acute cerebral ischemic insult, in many instances, is practical only after the event (Tasker et al., 1991). There has therefore been an understandable emphasis on the clinical translation of experimental effects such as the modulation of carbon dioxide (CO2) (Vannucci et al., 1995) or mild to moderate hypothermia (Edwards et al., 1995; Thoresen et al., 1995; Yager and Asselin, 1996; Bona et al., 1998), which can be implemented easily in the postischemic period in severely ill, mechanically ventilated patients.
Carbon dioxide readily crosses cell membranes and may protect brain tissue by improving intracellular buffering capacity (Hurn et al., 1991; Katsura et al., 1992). By contrast, lowering body temperature 2 to 5°C (mild to moderate hypothermia) may protect by attenuating the periischemic release of excitatory neurotransmitters (Busto et al., 1989). Despite these very different effects, however, there may be a common mechanism underlying the neuroprotection mediated by each of these physio-chemical perturbations. For example, many studies of postischemic hypothermia in immature animals do not report a specific acid-base management during the period of hypothermia and, in the main, comparative blood gas measurements are not given. During hypothermia, if blood gases are corrected to actual body temperature, more severe hypocarbia, relative to normothermic blood gas values, develops with deepening hypothermia. Failure to control or increase minute ventilation during hypothermia will, relative to the optimal adaptive response, be the same as imposing an hypercapnia challenge, or respiratory “acidosis.” One plausible mechanism of protection, therefore, is the postischemic modulation of intracellular pH (pHi) (Tombaugh and Sapolsky, 1993; Sapolsky et al., 1996; Vornov et al., 1996).
We have previously undertaken studies using phosphorus (31P) nuclear magnetic resonance (NMR) spectroscopy in the superfused brain slice preparation to look at energy metabolism after substrate and oxygen deprivation, so-called in vitro “ischemia.” In a vascular-independent neonatal preparation, an insult known to deplete all high-energy phosphates was followed by reproducible bioenergetic recovery with secondary energy failure and chemical evidence of apoptosis within 2 hours of reperfusion with oxygen and glucose (Tasker et al., 1998). We now describe further infant brain slice studies that examine the hypothesis that postischemic bioenergetic recovery is influenced by changes in pHi. To this end, our aims were first to confirm whether there are similarities in the benefit induced by postischemic hypercarbia and hypothermia and second to assess how these effects may interact.
MATERIALS AND METHODS
Preparation of cortical brain slices and incubations
Cerebral cortical slices of 350-μm thickness from 7-day-old rats (three to seven animals per experiment) were prepared using a McIlwain tissue chopper. Each brain was removed from the cranial cavity within 30 seconds, and slices were rinsed in oxygenated Krebs—Henseleit bicarbonate (KHB) buffer before transferring them to a 20-mm NMR tube. Once in the tube, slices were perfused with KHB buffer containing (in mmol/L) 124 NaCl, 5 KCl, 1.2 MgSO4, 1.2 CaCl2, 26 NaHCO3, 1.2 KH2PO4, and 10
Nuclear magnetic resonance techniques
In these experiments, the metabolic status of the tissue was assessed from determinations of pHi and the relative concentrations of ATP and phosphocreatine (PCr). 31P NMR spectroscopy was used to follow changes in pHi and metabolic recovery of high-energy phosphates. Spectra were recorded using a 20-mm NMR probe in an 8.5 T vertical magnet interfaced with an SMIS console operating at 145.7 MHz. Magnetic field homogeneity was optimized by adjusting room temperature shim currents until the water proton linewidth from the preparation was < 30 Hz. Throughout the experiments, 31P spectra were acquired (45° pulse; relaxation delay, 1.1 seconds; sweep width, 10 kHz; 4, 000 points; 280 or 500 scans) and processed as described previously (Kauppinen and Williams, 1990) with a 1/4 sine filter over the first 2 milliseconds of the free induction decay (to suppress broad phospholipid resonances) followed by 15-Hz line broadening. These pulsing conditions do not allow for full relaxation of all signals (particularly PCr) between pulses; therefore, peak area ratios do not correspond directly to concentration ratios. The data have not been corrected for this effect, but from a series of experiments previously reported (Tasker et al., 1996) comparing the above acquisition to spectra acquired with an 8-second relaxation delay, the β-ATP signal is fully relaxed for all conditions. The saturation factor for the PCr signal was 64.4 ± 3.2% (mean ± SD, n = 12), and it was not influenced by condition or age. Therefore, because the acquisition conditions in the experiments described were the same for all 31P spectra and we were interested in time-dependent changes in individual metabolites during and after ischemia, the changes in peak heights of PCr and β-ATP relative to their control values were used (Kauppinen and Williams, 1990).
Calculation of intracellular pH
Absolute pHi determinations by NMR are accurate to within ± 0.10 pH unit, but changes can be measured with a precision of ± 0.03 pH unit (Taylor et al., 1983). In the present experiments, during normothermic (36.7 ± 0.6°C) conditions, the pHi was calculated from the chemical shift (σ) of Pi relative to PCr at 0 ppm using the following titration curve (Taylor et al., 1983): pHi = 6.75 + log [(σ − 3.27)/(5.69 – σ)]. We also checked the validity of this calibration under mild hypothermic conditions (32.3 ± 0.8°C) over a pH range of 6.5 to 7.6 pH units by using a solution containing (in mmol/L) 1 MgCl2, 5 NaCl, 130 KCl, 5 KH2PO4, 5 K2HPO4, and 10 PCr. We found that the above titration curve, when used during mild hypothermia, overestimated pH with a mean bias of 0.10 ± 0.02 pH unit. Because this bias was consistent (95% confidence interval for the bias was 0.09 to 0.11 pH unit) over the range studied, we elected to subtract 0.10 pH unit from the above NMR-derived calculation of pHi to temperature-correct the value rather than use a completely different titration equation at different times in the experiment.
Application of hypothermia and hypercarbia
A period of hypercarbia was induced by changing the superfusion medium from KHB equilibrated with 5% CO2 to KHB equilibrated with 10% CO2, that is, normocarbia and hypercarbia, respectively. To study this effect, independently of changes in oxygen, specially prepared gases were used: O2/N2/CO2 mixture, 16:3:1 for 5% CO2 and 8:1:1 for 10% CO2. Previous studies by ourselves (Tasker et al., 1996, 1998) and others (Espanol et al., 1992) indicate that this partial pressure of O2 (∼600 mm Hg) and CO2 (∼40 and 60 mm Hg at 5 and 10%, respectively) is not detrimental to ex vivo normothermic cortical brain slices.
A period of hypothermia was induced by altering the temperature of the superfusion medium. The temperature of the buffer within the 20-mm NMR tube was measured and monitored during each experiment by using a calibrated NMR-compatible thermocouple connected to a medical precision thermometer (DM852; Ellab, Copenhagen, Denmark). To mimic the experimental protocol of many in vivo studies of postischemic hypothermia, prehypothermic CO2-gassing conditions (5%) of the superfusion medium were left unchanged, which at normothermia maintains medium pH at 7.41 ± 0.03 pH units. The time course and duration of these changes, although clearly different, were selected specifically so that steady-state levels of metabolites could be expected at specific a priori chosen timepoints in the course of the experiments (40, 100, and 130 minutes after a 40-minute control or ischemic period). In this regard, although pHi changes rapidly attain steady-state values after making changes in CO2 concentration, ∼30 minutes is required in vivo to achieve steady-state levels of other metabolites (Cohen et al., 1990).
In vitro ischemia studies
After recovery from dissection, perfusion of slices with well oxygenated KHB buffer was continued for 45 to 60 minutes. After this period, the perfusion medium was changed to KHB buffer containing a reduced concentration of phosphate (100 μmol/L). The washout period for medium changes in our apparatus is < 5 minutes. Over the next 50 minutes, six control 31P spectra were acquired (3 × 500 scans, 3 × 280 scans), and the mean values of peak heights were used as intraexperiment controls for the two methods of acquiring 31P spectra. Next followed two 20-minute intervals, during each of which three 31P spectra were acquired (3 × 280 scans). The slices were perfused with either KHB buffer lacking oxygen and glucose or normal control buffer. Next, during recovery with oxygen and glucose, over the following 100 minutes, 12 31P spectra were acquired (6 × 280 scans, 6 × 500 scans) while hypothermia and hypercarbia were applied, either individually or in combination. Last, all brain slices were perfused with normal control (normothermic, normocarbic) KHB buffer, during which time three further 31P spectra were acquired (3 × 500 scans).
Experimental design and statistical analysis
Twenty-four separate brain slice NMR spectroscopy experiments were conducted: three experiments in each of two conditions (control or ischemia 40 minutes) undergoing four permutations of two postischemic perturbations (hypercarbia with normothermia, hypothermia with normocarbia, hypercarbia with hypothermia, and normocarbia with normothermia). This balanced design enables the use of multiway analysis of variance to examine for significant main effects and interactions between factors at a priori chosen timepoints in the protocol: end of insult, 40 minutes; recovery, 40, 100, and 130 minutes (Minitab Statistical Software, State College, PA, U.S.A.).
Significant differences in NMR metabolites and change in pHi among groups were determined with analysis of variance followed by the Tukey-Kramer HSD multiple comparison test for differences between mean values. Significance was accepted with P < 0.05. Analysis of variance was performed on the logarithms of percentage changes to overcome the nonnormal distribution of ratios. Throughout this article, quantitative results are presented as means ± SD.
RESULTS
Each experiment was conducted over a 170-minute time course. Cortical brain slices, whether undergoing control, in vitro ischemia, or perturbing conditions, were returned to control, normocarbic, and normothermic superfusion KHB by the end of the experiment (that is, after 100 to 130 minutes of recovery). In comparing the observations made during the preexperiment control period and at the end of the study, there was no significant difference in the pHi of the cortical brain slices (7.19 ± 0.02 and 7.18 ± 0.03, start and end of experiment, respectively). Neither was there any difference in the temperature in the 20-mm NMR tube containing the brain slices (36.5 ± 0.6 and 36.8 ± 0.6°C, start and end of experiment, respectively). In vitro ischemia consisted of a 40-minute period during which time the brain slices were superfused with KHB lacking oxygen and glucose. This insult caused a complete loss of NMR-observable energy metabolites. There was also a significant fall in pHi (−0.01 ± 0.04 and −0.19 ± 0.03, end of control and end of ischemia, respectively; P < 0.0001).
The following results describe the acutely induced changes in pHi and any consequent effects on energy metabolites (PCr and β-ATP) resulting from hypercarbia or hypothermia introduced after a 40-minute period of in vitro ischemia.
Effect of hypercarbia and hypothermia on intracellular pH
The data in Fig. 1 and Table 1 summarize the changes in pHi during the different experimental conditions: at the end of a 40-minute control or ischemic insult period and after 40, 100, and 130 minutes of recovery, that is, respectively, 40, 140, and 170 minutes into the time course of the experiment. Because of the balanced design of the experiment, there is an experimental group for each condition. By carrying out analysis of variance on these data, we are able to evaluate independently the effects of ischemia (none or 40-minute insult), CO2 (5 or 10%), and temperature (normothermia or hypothermia) on the overall change in pHi (before ischemia to recovery, ΔpHi) and also to determine whether these factors act independently or synergistically.
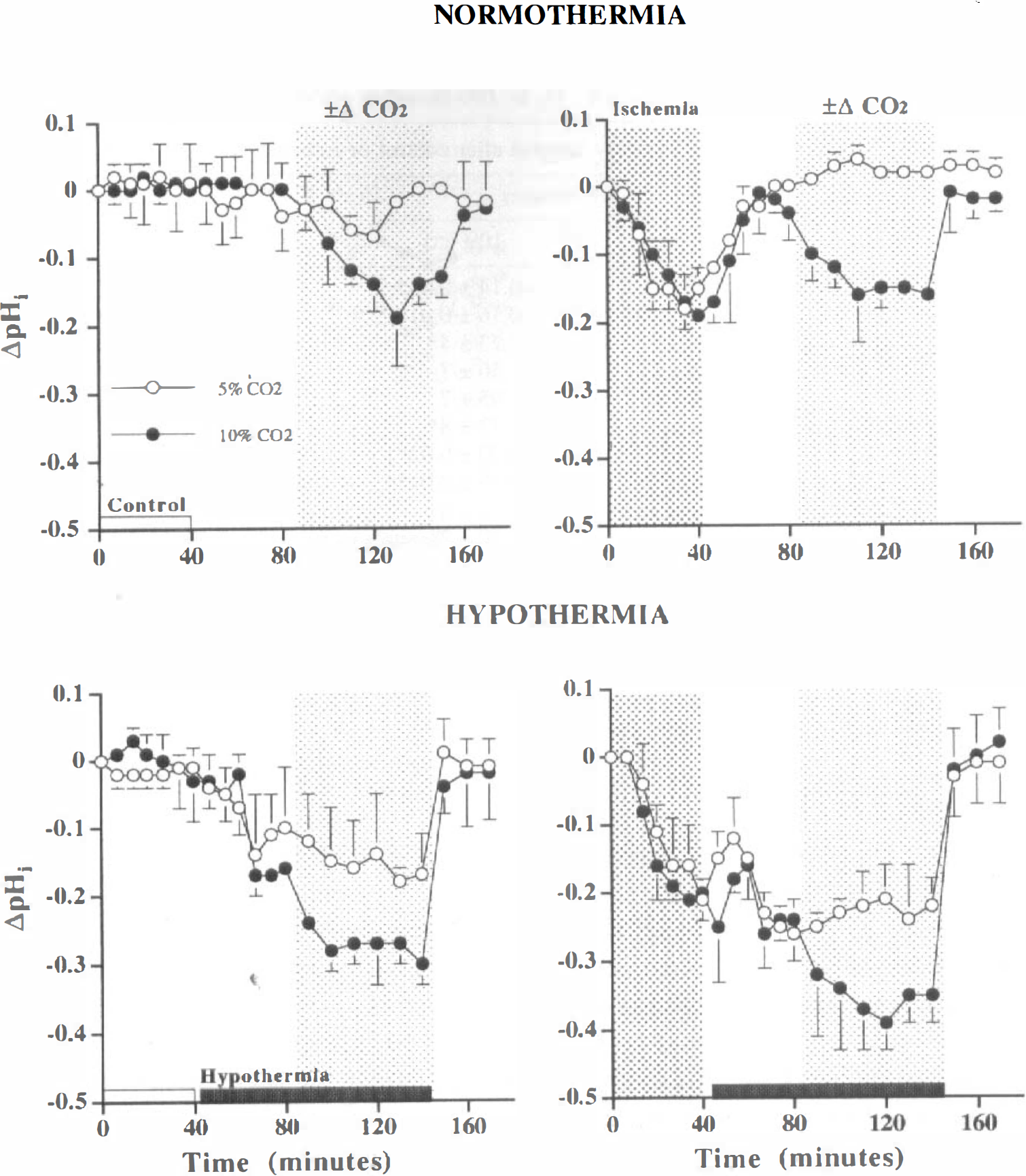
Change in intracellular pH (ΔpHi relative to preinsult control) during a 40-minute control (indicated by open bar on abscissa, “Control”) or in vitro ischemic insult (indicated by dark hatched background area, “Ischemia”) and during 130 minutes of recovery (timepoints 40 to 170 minutes).
A summary of changes relative to preinsult values induced by substrate and oxygen deprivation for 40-minutes (Ischemia) observed after 40-, 100- and 130-minutes recovery in the presence or absence of hypercarbia (5% or 10% CO2, 40-to 100-minutes of recovery) or hypothermia (36°C or 33°C, 0- to 100-minutes of recovery)
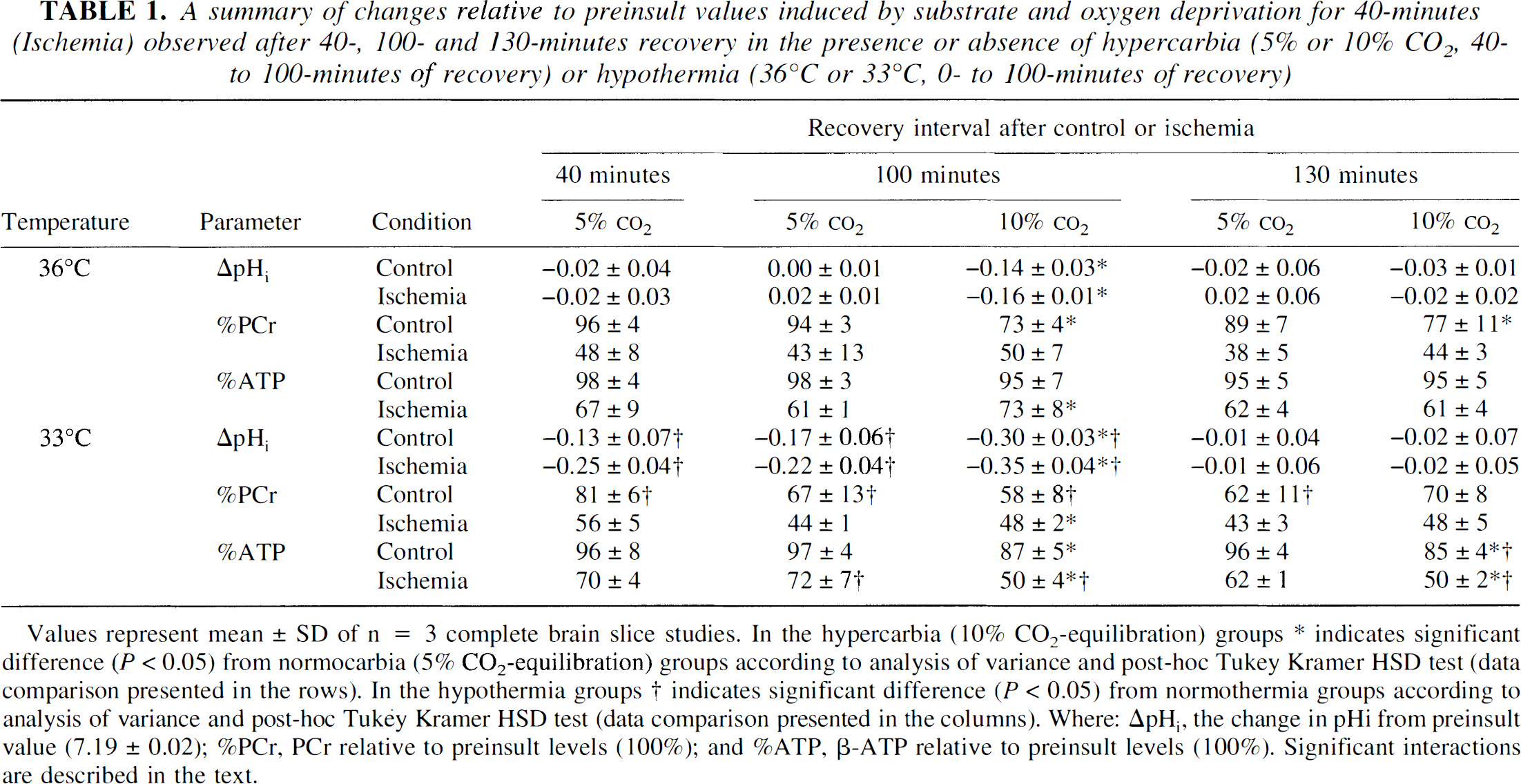
Values represent mean ± SD of n = 3 complete brain slice studies.
In the hypercarbia (10% CO2-equilibration) groups * indicates significant difference (P < 0.05) from normocarbia (5% CO2-equilibration) groups according to analysis of variance and post-hoc Tukey Kramer HSD test (data comparison presented in the rows).
In the hypothermia groups † indicates significant difference (P < 0.05) from normothermia groups according to analysis of variance and post-hoc Tukey Kramer HSD test (data comparison presented in the columns). Where: ΔpHi, the change in pHi from preinsult value (7.19 ± 0.02); %PCr, PCr relative to preinsult levels (100%); and %ATP, β-ATP relative to preinsult levels (100%). Significant interactions are described in the text.
First, with respect to ΔpHi after 40 minutes of recovery (80-minute timepoint), there are significant main effects for both variables tested at this timepoint (P < 0.01). Taken together with an inspection of the data, the findings indicate that cortical brain slice pHi is significantly more acidotic during hypothermia. The interaction between temperature and ischemia (P < 0.01) shows that these factors do not act independently. Rather, after ischemia, hypothermia not only delays the expected recovery in pHi (see above) but also exacerbates further the acidosis.
Second, after 100 minutes of recovery (140-minute timepoint)—when the effects of CO2 and temperature were tested together—there are significant main effects for CO2 and temperature (P < 0.0001) with no interactions. That is, hypercarbia and hypothermia induced, independently, an acidotic change irrespective of whether the cortical brain slices had been exposed to an ischemic insult. Furthermore, these effects were additive, with each causing a ΔpHi of approximately −0.16 pH unit. For example, the ΔpHi induced in control tissue was −0.14 ± 0.03 by 10% CO2 alone, −0.17 ± 0.06 by hypothermia, and −0.30 ± 0.03 by combination treatment.
Effect of hypercarbia and hypothermia on bioenergetic recovery from in vitro ischemia
Phosphocreatine. Via the creatine kinase equilibrium, an acid shift in pHi will result in a decrease in the amount of PCr. The acid shifts described for hypercapnia and hypothermia should therefore be associated with a decrease in the amount of PCr (relative to the value during the preexperiment control period; %PCr) after 40 and 100 minutes of recovery. At the earlier timepoint, the effect of temperature was tested, and at the later time-point, the effects of temperature and CO2 were tested, either alone or in combination. The data in Fig. 2 and Table 1 summarize the changes observed.
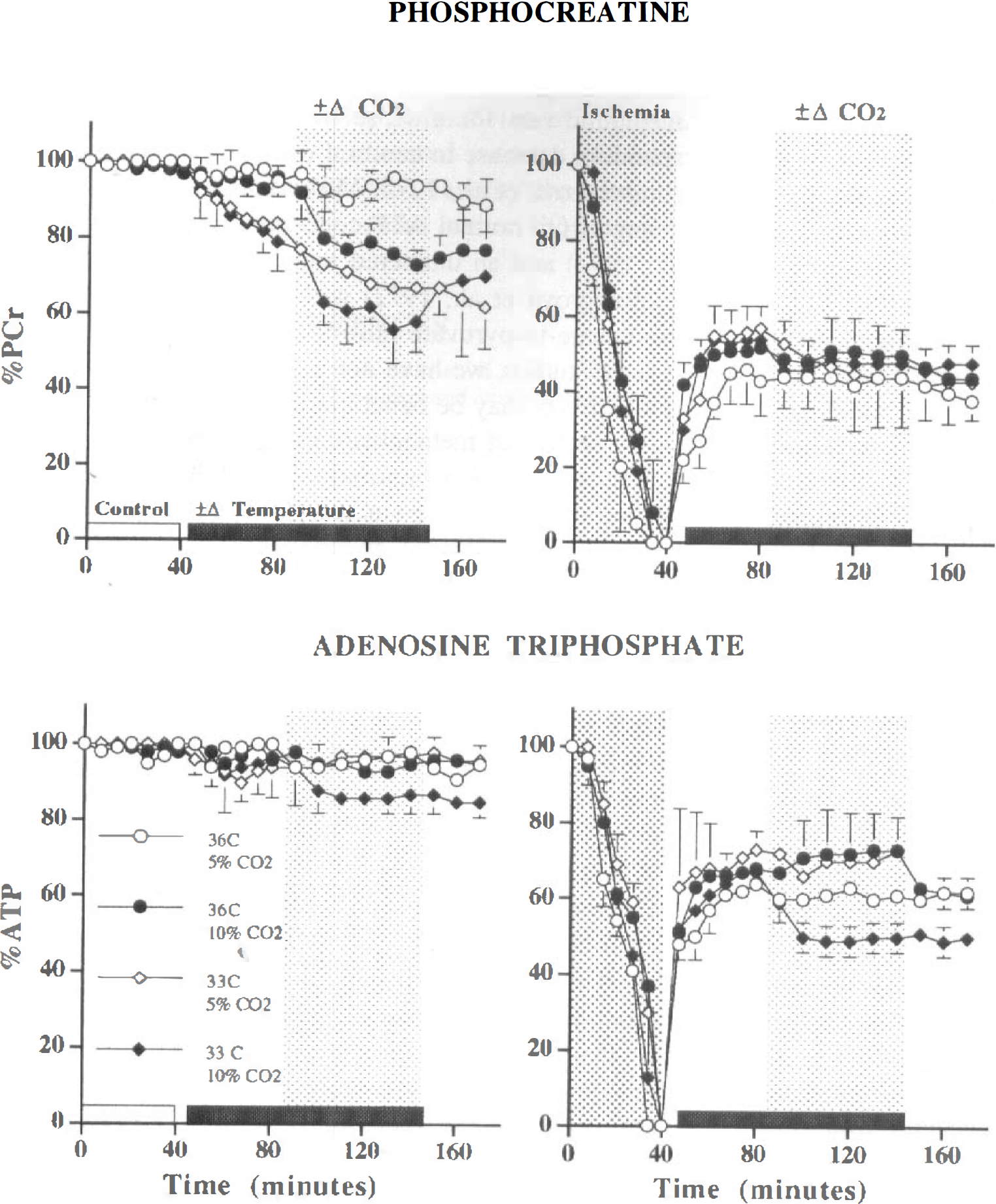
Change in high-energy phosphates during a 40-minute control (indicated by open bar on abscissa, “Control”) or in vitro ischemic insult (indicated by dark hatched background area, “Ischemia”) and during 130 minutes of recovery (timepoints 40 to 170 minutes). The dark bar on the abscissa (±Δ Temperature) indicates the interval when hypothermia (33°C) was applied. The light hatched background area (±Δ CO2) indicates the interval when hypercarbia (10% CO2) was applied. Eight groups exposed to four separate permutations of these two perturbations after control or ischemia are indicated. Values represent means ± SD. (
After 40 minutes of recovery (80-minute timepoint), as expected, there is a significant interaction between ischemia and temperature (P < 0.001). On inspection of the data, it is evident that these findings demonstrate first the expected acidosis-related hypothermia-induced fall in PCr in control tissue. Second, the significant interaction implies that the effect of ischemia is modulated by temperature in such a way as to ameliorate recovery of PCr under hypothermic conditions.
After 100 minutes of recovery (140-minute timepoint), there are significant main effects for ischemia (P < 0.0001) and temperature (P < 0.05) as well as significant interactions between temperature and ischemia (P < 0.05) and between CO2 and temperature (P < 0.05). In regard to the interactions, similar to the opposing effects in control and postischemic tissue after 40 minutes of recovery, the data again indicate that there are the expected acidosis-related decreases in percent PCr in control tissue when under the effect of hypothermia alone, or hypercarbia alone, or when these two perturbations are combined. Also, there is an absence of an acidosis-related fall in percent PCr in postischemic tissue.
After 130 minutes of recovery (by which time all the perturbed preparations had been returned to normal conditions of temperature and CO2 for 30 minutes), the ΔpHi was not significantly different from preexperiment control values. At this timepoint, therefore, changes in PCr would not be influenced by changes in the creatine kinase equilibrium. As expected, there is a main effect of ischemia on recovery in PCr (P < 0.0001). There is also an interaction between temperature and ischemia (P < 0.01). Inspection of the data indicates that the level of PCr in control slices has been detrimentally affected by the preceding period of hypothermia and that this effect is common to both CO2 regimens. For example, percent PCr in normocarbic-normothermic tissue was 89 ± 7%, whereas in postichemic tissue, it was 62 ± 11 and 70 ± 8% in tissue exposed to 5 and 10% CO2, respectively (P < 0.05, Tukey HSD). It is notable that PCr recovery after ischemia is not improved by hypothermia or hypercarbia, alone or in combination.
Adenosine triphosphate. After 40 minutes of recovery (80-minute timepoint), the only significant effect on β-ATP was that induced by ischemia (P < 0.0001; Fig. 2; Table 1). After 100 minutes of recovery (140-minute timepoint), however, there are significant main effects for all three variables (ischemia, P < 0.0001; temperature and CO2, P < 0.05), with a three-way interaction between them (P < 0.01). Inspection of the data indicates the following. First, in postischemic tissue, there is a significant amelioration in percent ATP when either hypercarbia or hypothermia is applied in isolation. These increases in β-ATP represent a 20% relative improvement from the untreated postischemic bioenergetic state. Second, when these perturbations are combined in postischemic tissue, in comparison with the above values, there is a significant worsening of percent ATP. Last, the detrimental effect on percent ATP, of combining hypercarbia and hypothermia, was not confined solely to postischemic tissue, as evidenced by the decrease in percent ATP in control tissue (98 + 3 and 87 ± 5%, normothermia with normocarbia and hypothermia with hypercarbia respectively; P < 0.05, Tukey HSD).
After 130 minutes of recovery (170-minute timepoint), there are significant main effects for all three variables (ischemia and CO2, P < 0.0001; temperature, P < 0.05), with a two-way interaction between temperature and CO2 (P < 0.0001). The absence of any significant interactions with ischemia implies that the treatments are ineffective in ATP recovery after ischemia. Evidently, the beneficial effects of hypothermia or hypercarbia, as applied in this experimental paradigm, are temporary and not preserved when the preparations are returned to normal conditions after 100 minutes of treatment. In contrast, the detrimental effect on ATP of applying both perturbations together persisted. These decreases in (3-ATP represent about a 10 and 20% relative worsening from the untreated control and postischemic state, respectively.
DISCUSSION
The results of these studies show that, in the developing brain, postischemic perturbation with hypercarbia or mild hypothermia has significant bearing on pHi and cell bioenergetics. Our three principal findings were first that early postischemic hypothermia delayed the normalization of pHi after ischemia-related acidosis; second, that hypercarbia and hypothermia induced separate, but potentially additive, reversible decreases in pHi (∼0.16 pH unit); and third, that when these perturbations were applied in isolation, in association with the acid shift in pHi, there was significant recovery in ATP (relatively ∼20%). However, in combination, rather than having an additive effect (like those observed in the pHi), hypercarbia and mild hypothermia together were detrimental and worsened recovery of ATP. Importantly, this negative response was not confined to postischemic cortical tissue but also applied to brain slices not exposed to in vitro ischemia. Furthermore, the compromised bioenergetic state persisted even after discontinuing these briefly administered perturbing conditions (representing a relative worsening in ATP of ∼10 and ∼20% in control and post-ischemic tissue, respectively).
Before a discussion of these findings, it should, however, be noted that there are both advantages and limitations of studying energy failure in the brain slice model. This preparation is commonly used to study the pharmacology, physiology, and biochemistry of the brain, where it offers the benefits of a model with relatively preserved cell-to-cell interaction and microanatomical architecture as well as the ease of undertaking experimental manipulations. Nevertheless, extrapolation of results of an in vitro test of perfusing buffer aglycemic-hypoxia to an in vivo state of ischemia must take into account certain unavoidable facts, not least the inevitable tissue injury occurring during preparation, the differences in the experimental insult, and the physical isolation of the brain slice from systemic effects and the influence of other brain regions.
Brain tissue intracellular pH, acidosis, and the pH paradox
In these studies, the NMR-detected changes in pHi in brain slices represent at best a tissue-averaged pHi and indicate that more extreme, or even opposite, changes—acid, alkali, or both—may be occurring in specific cell types within the preparation. Nevertheless, we observed that hypercarbia and hypothermia each induced separate, but potentially additive, decreases in pHi (∼0.16 pH unit). This magnitude of change after hypercarbia was as would be expected for the change in extracellular pHo of ∼0.30 pH unit. However, in regard to mild hypothermia, by a similar calculation, the expected fall in pHi would have been ∼0.06 pH unit (allowing for a relative difference in CO2 at the lower temperature of ∼5 to 7 mm Hg and a 3°C drop in temperature), which is only one-third of the change actually observed. These findings suggest that hypercarbia and hypothermia induce brain tissue acidosis by independent mechanisms, a conclusion that is also supported by their equally additive effects when co-administered. In this context, it is of note that a similar degree of change in pHi has been reported in adult rats undergoing both deep hypothermia (∼28.5°C) and hypercarbia (10% CO2) during control conditions (Johnson et al., 1989) and in rats undergoing hypothermia after fore-brain ischemia (Chen et al., 1992).
A hallmark of ischemia is intracellular acidosis. Paradoxically, during reperfusion after anoxia in neonatal rat cardiac myocytes, it is the normalization of acidotic to physiological pH that triggers lethal injury, rather than reoxygenation (Bond et al., 1991, 1993). A similar phenomenon has also been observed in neurons where acidosis has the potential to be neuroprotective during certain circumstances (Sapolsky et al., 1996; Vornov, 1995; Vornov et al., 1996). Also, in vivo, brief postischemic hypothermia for 2 hours induces an intracellular acidosis, but this perturbation does not modify the expected histological findings 7 days after forebrain ischemia (Chen et al., 1992). This failure of benefit, however, may be because of an inadequate duration of postischemic hypothermia (Dietrich et al., 1996; Colbourne et al., 1997), which needs to be longer than 3 hours (Dietrich et al., 1993) and possibly at least 12 hours (Colbourne and Corbett, 1994) for chronic neuroprotection. Taken together with the current report, therefore, there remains the question of whether the so-called postischemic pH paradox (Bond et al., 1991, 1993) contributes to hypothermia-related neuroprotection.
pH and bioenergetic effect of hypercarbia and mild hypothermia
The effect of hypothermia in delaying the postischemic normalization of pHi is similar to the postischemic change in pHi reported in our previous studies where glyceryl trinitrate, depolarization, and dantrolene afforded significant bioenergetic protection (Tasker et al., 1996, 1998). The central question, therefore, is whether there is any causality underlying such a consistent association between sustained maintenance of pHi below 7.0, during the early postischemic period, and subsequent improved bioenergetic recovery. For example, when postischemic hypercarbia or mild hypothermia is applied in isolation, in association with an acid shift in pHi there is, relatively, about a 20% improvement in ATP recovery. The magnitude of these changes may be small, numerically, but in other brain slice studies, similar effects signify the early prevention of in vitro ischemia-induced poly(ADP-ribose) polymerase-related bioenergetic failure (Tasker et al., 1998) and limitation of histological injury (Espanol et al., 1998).
Induction of hypercarbia in the absence of hypoxia is probably the best model available for studying the effects of acidosis on energy metabolism, as CO2 acidifies both intracellular and extracellular fluids. In vivo, acidosis due to hypercarbia is accompanied by distinct change in brain energy metabolism, such as a reduced cerebral metabolic rate for glucose in the absence of a corresponding decrease in cerebral metabolic rate for oxygen (Folbergrová et al., 1975; Miller and Veech, 1975) or a change from normal ATP concentration (Folbergrová et al., 1992) and an unaltered reduction to oxidation state (Folbergrová et al., 1972) with a pH-dependent rise in the lactate-to-pyruvate ratio (Siesjö et al., 1972). In the current studies, we have not examined why these metabolic effects may be beneficial in the cortical brain slice in the setting of metabolism after ischemia.
Hypothermia has minimal effect on extracellular pH, but, as shown in these studies, it induces an acid shift in pHi and, after ischemia, an amelioration in level of ATP. This protection may be due in part to a depression of cellular metabolism, as demonstrated by a substantially lowered cerebral metabolic rate of oxygen (Yager and Asselin, 1996). However, recent in vivo evidence indicates that, in addition to a 30 to 40% decrease in metabolism, a temperature of 31°C is associated with an increased fraction of glucose metabolism shunted through the pentose phosphate pathway (Kaibara et al., 1999). In this regard, it is of interest that, in neutrophils, stimulation of the pentose phosphate pathway (or hexose monophosphate shunt) generates sufficient protons for lowering pHi (Grinstein and Furuya, 1986): a mechanism that may account for the hypothermia-related changes in pHi described in the current report. The ameliorating effect of hypothermia may be related, therefore, to the tissue-protective effects of NADPH on maintaining membrane potential and ion transport and on removing membrane-toxic oxygen radicals (Baquer et al., 1988).
Taken together, this discussion suggests that, after ischemia, the change in pHi during hypercarbia affects, directly, glucose metabolism and the improved bioenergetic state. In contrast, during mild hypothermia, the change in pHi may be an epiphenomenon of increased metabolic shunt through the pentose phosphate pathway. Such a proposal, based on two different effects on glycolysis, would be consistent with all of the pH and bioenergetic observations in the current report and may account for the detrimental interaction (which occurs when hypercarbia and mild hypothermia are combined) in terms of impaired or disturbed glucose metabolism. In particular, a possible explanation is proffered for the anomaly that even though hypercarbia-induced changes in pHi (as large as −0.63 pH unit) do not lead to detrimental changes in ATP in vivo (Litt et al., 1985) or in vitro (Espanol et al., 1992; Pirttila and Kauppinen, 1993), when half such pH changes are induced by combining hypercarbia with mild hypothermia, there is an interaction that results in a persistent and detrimental effect on bioenergetic state.
Finally, in regard to the clinical translation of these ex vivo data, further in vivo investigation is warranted, not least because they suggest that the clinical utility and trials of mild hypothermia may be fundamentally flawed, or even detrimental, in brain-injured patients who have pulmonary abnormalities of such severity that they require a ventilatory strategy where higher CO2 tensions are being tolerated.