
Abstract
The authors examined the effect of selective endothelin (ET) receptor type A (ETA) antagonism on histological and functional recovery in cat at 24 hours after reversible middle cerebral artery occlusion (MCAO). A novel and specific ETA antagonist, Ro 61-1790 [5-methylpyridine-2-sulfonic acid-6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(2-1H-tetrazol-5-yl-pyridin-4-yl)-pyrimidin-4-ylamide sodium salt (1:2)] (Roche, Basel, Switzerland), was used at doses that produced steady-state plasma concentrations and abolished ET-induced pial arteriolar vasoconstriction. In a cranial window preparation, 8 nmol/L ET constricted pial arterioles by 33 ± 18% (mean ± SD), but this response was ablated by intravenous Ro 61-1790 treatment (10-mg/kg bolus, 4-mg/kg/h infusion). In additional animal cohorts, halothane-anesthetized cats were treated with 90 minutes of MCAO and 24 hours of reperfusion. Animals received Ro 61-1790 infusion beginning at the onset of reperfusion and continuing for 6 or 24 hours (n = 41). Control cats were treated with 0.9% saline by intravenous infusion throughout reperfusion. There was no difference in injury volume or neurologic evaluation score in saline-treated cats (n = 11; caudate 24 ± 28%, cortical injury 7.5 ± 5% of ipsilateral structure; score 52 ± 8) versus the results in cats treated with Ro 61-1790 for either 24 hours (n = 6; caudate 22 ± 23%, cortex 6 ± 5%, injury volume of ipsilateral structure; score 55 ± 3) or 6 hours (n = 11; caudate 33 ± 30%, cortex 12 ± 14%, injury volume of ipsilateral structure; score 50 ± 10). Mortality was greatest in the 24-hour drug treatment group. These data suggest that blockade of ETA receptor activity is not beneficial to tissue or functional outcomes from experimental stroke in cat.
Keywords
The endothelin (ET) family of isopeptides (ET-1, ET-2, and ET-3) has been implicated in numerous cerebrovascular pathologies, in part due to their potent vasoconstrictor capacity (Foley et al., 1994; Suzuki et al., 1992; Mima et al., 1989; Robinson and McCulloch, 1990; Gotoh et al., 1991). Topical or intraparenchymal application of exogenous ET-1 is associated with sufficient reduction in cerebral blood flow to cause irreversible ischemic brain damage (Robinson et al., 1990; Fuxe et al., 1992; Macrae et al., 1993; Sharkey et al., 1993). All three isoforms of ET are present in brain (Sakurai et al., 1992), and effects are mediated via two receptor subtypes, endothelin type A (ETA) and endothelin type B (ETB). The predominant effect of ETA receptor activity is vasoconstriction, whereas ETB receptors appear to mediate vasodilation in the cerebral circulation (Masaki et al., 1994). The effect of ET-1 on cerebral vessels has been best studied, but results vary with concentration in vivo, presumably reflecting proportionate ETA and ETB receptor activation. Blockade of ETA receptor prevents ET-1-induced vasoconstriction in isolated pial vessels but has little effect on pial arteriolar diameter under physiological conditions (Patel et al., 1996a). Endogenous ETs have been implicated in the pathophysiology of cerebral ischemia (Suzuki et al., 1992; Ziv et al., 1992; Patel and McCulloch, 1996; Patel et al., 1996a, b ), hypothetically by initiating or amplifying postischemic vasoconstriction and microvascular dysfunction. However, combined ETA/ETB antagonists do not improve hypoperfusion after global cerebral ischemia (Patel and McCulloch, 1996). In contrast, selective blockade of ETA improves cerebral perfusion as measured by laser-Doppler flowmetry after focal ischemia, enhancing short-term tissue survival (Patel et al., 1996b).
In this study, we examined the effect of selective ETA antagonism on histological and functional recovery from middle cerebral artery occlusion (MCAO) in the cat. We employed a novel agent, Ro 61-1790 [5-methylpyridine-2-sulfonic acid-6-(2-hydroxyethoxy)-5-(2-methoxyphenoxy)-2-(2-1H-tetrazol-5-yl-pyridin-4-yl)-pyrimidin-4-ylamide sodium salt (1:2)] at concentrations confirmed in our preparation to have good tissue penetration. We show that although the agent readily inhibits ET-1-induced pial vasoconstriction, there is no benefit in experimental stroke. Further, prolonged ETA antagonism can increase stroke mortality.
MATERIALS AND METHODS
The study was conducted in accordance with National Institutes of Health guidelines for the use of experimental animals, and the protocols were approved by the Johns Hopkins University Animal Care and Use Committee. Initial experiments were conducted to determine the pharmacokinetics of Ro 61-1790 in the cat and allowed selection of the optimal dose for a test of drug efficacy in focal cerebral ischemia. In additional experiments, the effect of the drug on pial vessel response to ET was measured to demonstrate in vivo activity of the drug within the cerebral circulation.
Pharmacokinetics of Ro 61-1790 in cat plasma
Male cats (2.0 to 4.5 kg; n = 3) received a 10-mg/kg intravenous drug bolus over 30 seconds, with subsequent blood sampling (0.8 mL) at 5, 15, 30, 45, 60, 90, 120, and 180 minutes after injection. Ro 61-1790 (Roche, Basel, Switzerland) was quantified in plasma using high-performance thin layer chromatography after protein precipitation. Analysis was carried out on high-performance thin layer chromatography silica 60F254 (20 × 10 cm) plates using an eluent consisting of ethylacetate/methanol/water/diethylamine (70:10:20:15, by volume). Postchromatographic fluorescence enhancement was done by immersion into Triton X-100 in chloroform and n-hexane, before in situ fluorescence (λ emission >400 nm) by means of a thin layer chromatographic scanner. Quantification was based on external standards using peak height. The quantification limit of the high-performance thin layer chromatography assay for Ro 61-1790 was 0.1 μg/mL of plasma.
Measurement of pial arteriolar caliber
Male cats (2.0 to 4.5 kg; n = 3) were anesthetized with pentobarbital sodium (35 mg/kg intraperitoneally followed by 5-mg/kg/h infusion) and then mechanically ventilated with supplemental oxygen. Skeletal muscle paralysis was produced with pancuronium bromide (0.1 mg/kg). Femoral artery and venous catheters were placed for blood pressure monitoring, arterial blood gas determination, and infusion of drugs. A parietal cranial window was constructed as previously described (Hudak et al., 1989; Clavier et al., 1994) for intravital microscopy and determination of pial vessel diameter. Each agonist was superfused in artificial cerebrospinal fluid and then “washed out” with fresh artificial cerebrospinal fluid followed by new baseline diameter measurements as previously described (Clavier et al., 1994). Temperature was controlled in response to measurements via thermistors in the cranial window. Window, brain, and rectal temperatures were maintained at 38 ± 0.5°C.
After baseline diameter measurements, the cranial window was suffused with 10−5 mol/L acetylcholine to document baseline endothelium-dependent vasoreactivity. After a 15-minute artificial cerebrospinal fluid “washout” infusion, a new baseline measurement was obtained; then ET-1 (8 nmol/L) was suffused. Post-ET-1 diameters were recorded at 5, 10, and 15 minutes. After artificial cerebrospinal fluid superfusion to remove ET-1, systemic Ro 61-1790 was administered intravenously (10 mg/kg, then 4-mg/kg/h continuous infusion). Arteriolar diameters were recorded at 15 and 30 minutes of drug infusion to document any response to the systemic agent; then 8 nmol/L ET-1 was again superfused. Final pial diameters were recorded at 5, 10, and 15 minutes after ET-1 treatment. Nine pial vessels were studied: three per animal ranging from 30 to 100 μm in diameter.
Middle cerebral artery occlusion study
Ro 61-1790 was stored at −80°C as a stock solution of 50 mg/mL in sterile water until the day of each experiment. Stock solution was then diluted into vehicle containing 0.9% NaCl. The final concentration was adjusted so that the infusion rate was at 4 mL/kg/h. The infusion was changed every 6 hours to ensure stability within the vehicle at room temperature.
Focal cerebral ischemia was produced by 90 minutes of reversible MCAO with 24 hours of reperfusion. Surgical methods were as previously described (Harukuni et al., 1998; Takahashi et al., 1995; Bethel et al., 1997). In brief, male cats (2.0 to 4.5 kg) were anesthetized with halothane in oxygen, orally intubated, and mechanically ventilated. All experiments were performed under conditions of normoxia, normocarbia, and normothermia. The left MCA was exposed by transorbital approach using microsurgical techniques and then reversibly occluded near its origin by a microvascular clip. Somatosensory evoked potentials were measured to document a decrease in amplitude of the major cortical deflection to < 20% of preischemic value. After 90 minutes of MCAO, the microvascular clip was removed for reperfusion, and cats were allowed to emerge from anesthesia and allowed free access to food and water until the end of the experiment. Each animal received either 0.9% saline (control group) or Ro 61-1790 in a randomized and blinded fashion at the onset of reperfusion. In Ro 61-1790-treated cats, the first group received a 10-mg/kg intravenous bolus followed by an infusion of 4 mg/kg/h for 24 hours. The second drug-treated group received 10 mg/kg Ro 61-1790 followed by the 4-mg/kg/h infusion for 6 hours. After 6 hours of infusion, the infusion was changed from Ro 61-1790 to 0.9% saline at the same rate. Neurologic outcome scores (Baskin et al., 1994; Harukuni et al., 1998) were recorded at 24 hours of reperfusion by an observer who was blinded to treatment group. Neurologic score was based on level of consciousness (maximum score of 4 for normal), gait (maximum score of 10 for normal gait), motor function of the forepaw (maximum score of 7 for normal function for each forepaw), motor function of the hindpaw (maximum score of 6 for normal function for each hindpaw), circling (maximum score of 5 for no circling), sensory function of the forepaw (maximum score of 4 for normal reaction for each forepaw), and sensory functio of the hindpaw (maximum score of 4 for normal reaction for each hindpaw). The maximum overall neurologic assessment score possible was 61.
After completion of the neurologic evaluation, each animal was reanesthetized with halothane and cardiac arrest was induced with intravenous potassium chloride. The brain was removed, sectioned into 12 3-mm-thick coronal sections, and stained with 2, 3, 5-triphenyltetrazolium chloride (Sigma Chemical Co., St. Louis, MO, U.S.A.) for evaluation of infarction volume (Bederson et al., 1986; Cole et al., 1990), as previously described (Matsumiya et al., 1990; Takeshima et al., 1992).
Statistical analysis
Values are expressed as means ± SD. Statistical comparison to assess changes in measured physiologic variables within groups was performed by repeated measures analysis of variance. Comparison of physiologic variables and injury volume among groups was achieved with one-way analysis of variance. Post hoc analysis was performed with the Newman-Keuls test. Data for the pial arteriolar diameter were assessed by using analysis of variance followed by Student's t test with Bonferroni correction for multiple comparisons. Statistical differences were considered significant at P < 0.05.
RESULTS
Mean plasma clearance for Ro 61-1790 (n = 3) was 4.5 ± 1.6 mL/kg/min. Therefore, a maintenance infusion of Ro 61-1790 at 4 mg/kg/h was chosen to achieve a steady-state plasma level of 15 μg/mL. We tested this same infusion in the cranial window experiments to confirm that ET-1-induced vasoconstriction was eliminated by systemic Ro 61-1790. Baseline physiological variables including mean arterial blood pressure (101 ± 8 mm Hg), pH (7.43 ± 0), Pa
Middle cerebral artery occlusion experiments
There were no physiologically significant differences between treatment groups in inhaled halothane concentrations (1.9 to 2.2%), body temperature, mean arterial blood pressure, arterial blood gases, or blood glucose concentration during baseline and ischemia. However, mean arterial blood pressure was significantly higher in all groups at 24 hours of reperfusion as compared with baseline (Table 1). Overall mortality (failure to survive 24 hours of reperfusion) was higher in cats that received 24 hours of RO 61-1790 infusion (5 of 11) than in those that received a 6-hour infusion (3 of 15) and saline (4 of 15). Because mortality was unacceptably high in the 24-hour infusion group, this treatment group was truncated early in the study. Autopsy of these early mortalities indicated gross cerebral edema in each animal, particularly severe within the hemisphere ipsilateral to MCAO.
Physiologic variables at baseline, middle cerebral artery occlusion (MCAO), and reperfusion
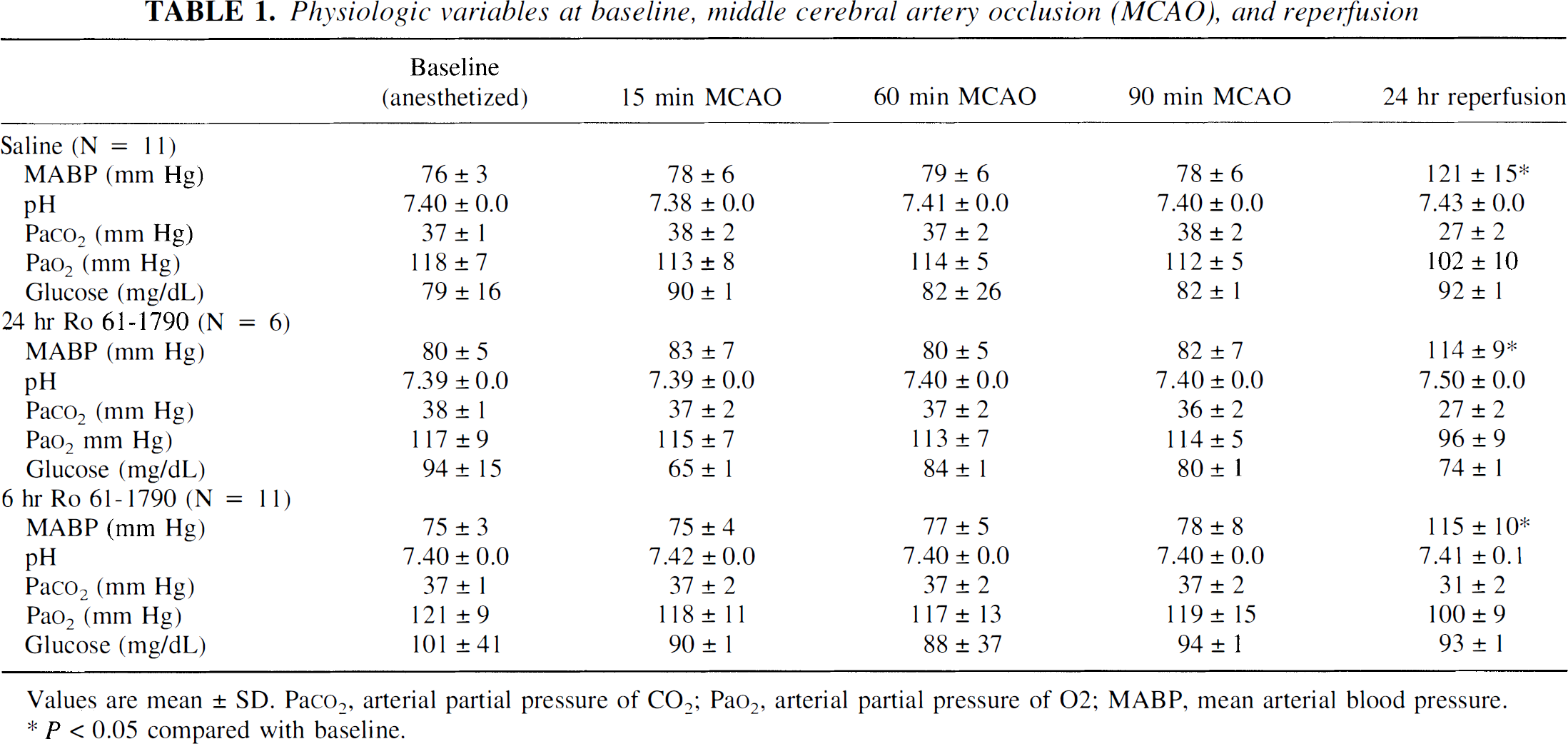
Values are mean ± SD. Paco2, arterial partial pressure of CO2; Pao2, arterial partial pressure of O2; MABP, mean arterial blood pressure.
P < 0.05 compared with baseline.
Somatosensory evoked potential amplitude was similarly ablated in all groups during MCAO and during the first 15 minutes of reperfusion. There was no difference in injury volume (Fig. 1) in saline-treated cats (n = 11; caudate 24 ± 28%, cortical injury 7.5 ± 5% of ipsilateral structure) as compared with the 6-hour drug infusion group (n = 11; caudate 33 ± 30%, cortex 12 ± 14%). Similarly, there was no difference in neurological score between groups (52 ± 8 in saline; 50 ± 10 in 6-hour drug treated). In the 24-hour Ro 61-1790 infusion group that was discontinued due to high mortality, histological injury was not different in the survivors as compared with the saline or 6-hour treatment groups (n = 6; caudate 22 ± 23%, cortex 6 ± 5%), and neurological score was 55 ± 3.
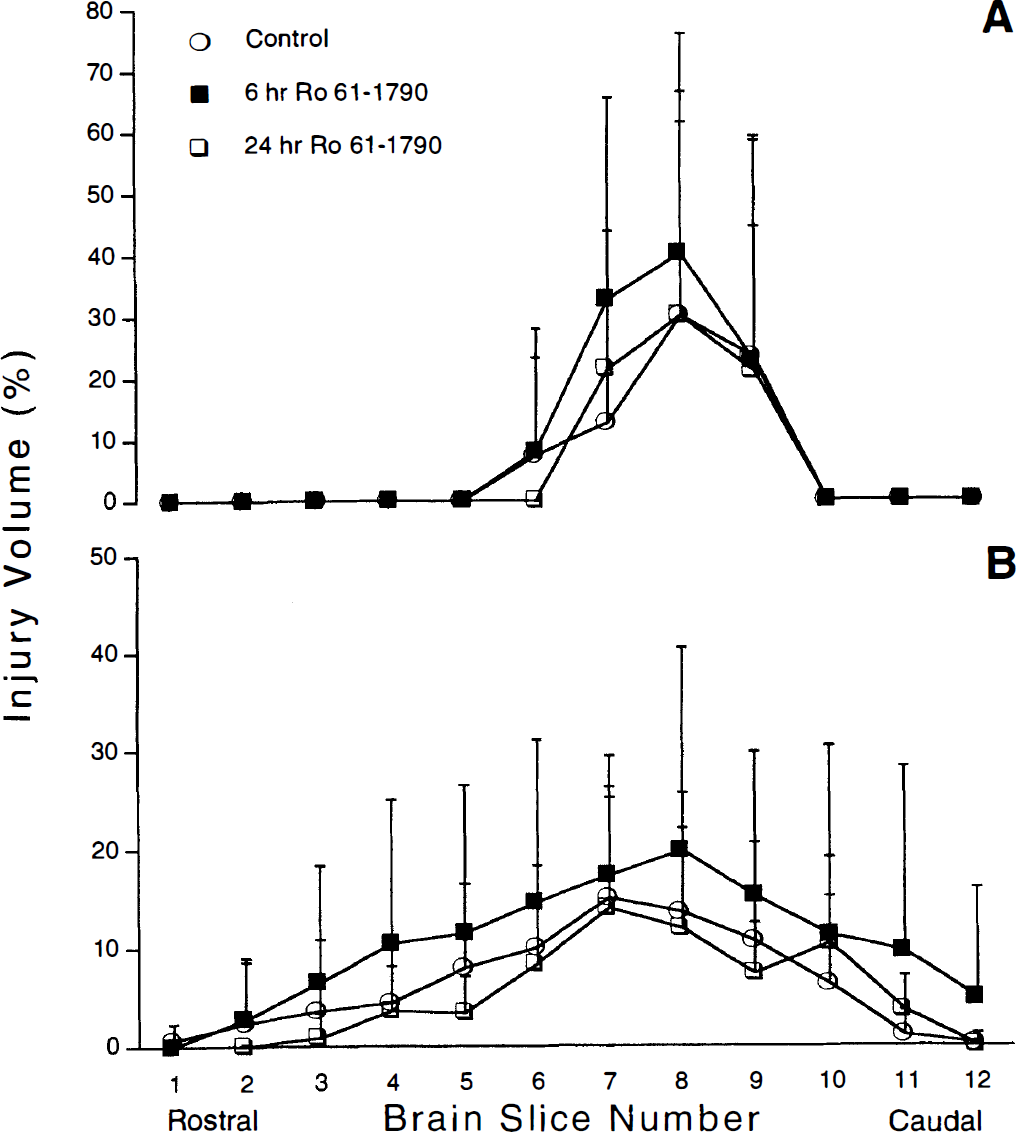
Infarction volume (% of ipsilateral structure) of caudoputamen (
In a previous study using 90 minutes of MCAO in the cat, we reported a significant reduction of infarction volume by the α-receptor ligand 4-phenyl-1-(4-phenylbutyl)piperidine (Takahashi et al., 1995). A statistical difference was apparent when infarction volume was reduced by 51% in the caudate-putamen (n = 10/treatment group). In the present study, treatment with Ro 61-1790 numerically increased the damage observed in caudate—putamen and in cortex; however, this result failed to reach statistical significance by analysis of variance or by t test. Using the goal of a 50% treatment effect, we examined post hoc the statistical power of our design and sample size of 11 animals/group. For the caudate—putamen, the power of our hypothesis testing was 0.73, which was more than adequate to establish a lack of difference between drug-treated and control cats. Because of the small size of cortical lesions in the control cohort (7.5 ± 5% of ipsilateral cortex), the ability to detect a 50% reduction of damage was conducted at a lower statistical power (0.38). To observe a significant difference in cortical infarction volume and reject the null hypothesis (power 0.75), we calculated the minimum required treatment effect to be 36% of the ipsilateral cortex, assuming an unchanged pooled standard deviation. In fact, the cortical infarction volume in the Ro 61-1790-treated cohort (6-hour treatment, n = 11) was 12 ± 14%.
DISCUSSION
The main finding of this study is that postischemic administration of the specific ETA antagonist Ro 61-1790 does not alter histological injury or neurological outcome when assessed at 24 hours after reversible MCAO in cat. Further, prolonged infusion of the antagonist resulted in high mortality, potentially due to enhanced cerebral injury. Ro 61-1790 as administered in the study was biologically active, as indicated by the agent's ability to attenuate the vasoconstrictive effects of local ET-1 on nonischemic pial arterioles studied in situ.
Ro 61-1790 is a trifunctionalized heteroarylsulfonamide pyrimidine with high water solubility for parenteral use (Roux et al., 1997). It is a competitive ETA antagonist with an affinity for the ETA receptor in the subnanomolar range with 1, 000-fold selectivity for the ETA versus ETB receptor (Roux et al., 1997). The compound has previously been demonstrated to prevent cerebral vasospasm in the canine model of subarachnoid hemorrhage in a dose-dependent manner (Roux et al., 1997). However, efficacy of this class of drugs in experimental stroke when administered after the onset of reperfusion has not been previously tested. Because previous reports suggest that ETA receptor antagonists must penetrate the blood-brain barrier for efficacy (Edvinsson et al., 1993; Patel et al., 1996b), the pharmacokinetics of the compound were examined in cat plasma. Based on these data, we employed an intravenous drug regimen of 10 mg/kg as a single bolus followed by a continuous infusion of 4 mg/kg/h. This same dose attenuated exogenous ET-1-induced vasoconstriction of nonischemic arterioles, demonstrating that bioactive amounts of Ro 61-1790 gain access to the adventitial surface of pial arteriolar vessels. Therefore, it seems unlikely that limited drug availability or access to target tissue would explain the compound's lack of efficacy in experimental stroke.
Endothelin, originally isolated from cultured aortic endothelial cells (Yanagisawa et al., 1988), is involved in a wide variety of physiological and pathophysiological events in brain. Numerous studies have provided evidence for a deleterious role for the ET family in focal (Suzuki et al., 1992; Gang-Zhi et al., 1993; Barone et al., 1994) and global (Ohlstein et al., 1994) cerebral ischemia and cardiac arrest (Takasu et al., 1997). The ability of exogenous ET-1 to overwhelm homeostatic mechanisms that maintain cerebral blood flow, even in the normal brain, has attracted interest in its role in influencing postischemic cerebral blood flow (Robinson et al., 1990; Macrae et al., 1993; Sharkey et al., 1993). Previous studies have reported no significant effect of ET receptor antagonists on cerebral arterial or pial arteriolar caliber in nonpathologic conditions in vivo (Foley et al., 1994; McAuley et al., 1996; Patel et al., 1994), but topical application of ET-1 antagonists increases the caliber of cortical arterioles in vivo in the ischemic penumbra during a focal cerebral ischemic challenge (Patel et al., 1996b). Therefore, a therapeutic potential for ET antagonists has been suggested (Ziv et al., 1992; Rubyani and Polokoff, 1994; Patel et al., 1996b), particularly if these agents reduce vasoconstrictive pathology in collateral vessels supplying the tissue at risk.
Several low molecular weight ET-1 antagonists with good blood—brain barrier penetration (for example, bosentan, PD155080, and PD156707) have been studied in focal cerebral ischemia (Patel et al., 1994, 1996b). Postischemic treatment with the potent and selective ETA antagonist PD156707 reduced infarction volume at 6 hours of permanent MCAO (Patel et al., 1996b). One speculation is that reduced tissue injury is the result of enhanced peri ischemic cerebral perfusion due to antagonism of the ETA receptor-mediated vasoconstriction and perhaps the unmasking of the ETB receptor-mediated vasodilation (Kobari et al., 1994; Patel et al., 1996b). Tatlisumak et al. (1998) have shown with diffusion perfusion imaging during focal ischemia that ET antagonists can protect brain independently of vascular mechanisms. A role for ETs as neuromodulators has been suggested (Berrino et al., 1994), but direct effects of ET receptor antagonists on neuronal function are unclear. In contrast, the present results indicate that blocking ET action via the ETA receptor subtype is not efficacious when tissue injury or neurological function is assessed at 24 hours after MCAO. We measured drug clearance from the central systemic circulation and adjusted our drug administration rate to ensure steady-state drug concentration throughout the study infusion period. We do not know the precise concentration gradient in the arterial tree or the drug concentration in the capillary. Ro 61-1790 concentration used in our study blocked ET-induced constriction in pial arterioles down to 30 μmm in diameter. Thus, it seems that effective concentrations were presented to vascular endothelium to reduce intraischemic or postischemic ET-induced vasoconstriction in large and small resistance vessels. We did not measure cerebrospinal fluid drug concentration or brain penetration and so cannot comment on drug effects that potentially rely on parenchymal accumulation.
Differences in the present results and previous studies may reflect differences in ischemic models or our use of a longer window of observation that extends beyond the initial period of acute cerebral edema and brain swelling. Prolonged blockade of ETA receptors increased mortality to an unacceptable level, so the present study was unable to evaluate the possibility that ischemic damage was exacerbated rather than improved. However, this possibility cannot be excluded. Enhanced mortality from stroke could result if prolonged ETA antagonist treatment amplified ETB receptor-mediated vasodilation, leading to exacerbation of cerebral edema. Alternatively, long durations of treatment during reperfusion could exert nonspecific blockade of vasoconstrictor responses, again leading to enhanced hyperemia and edema.
As expected, we observed a brisk and significant vasoconstriction in response to ET-1 superfusion over pial arterioles, which was lost in the presence of Ro 61-1790. These findings are in agreement with previous reports that local ET-1 application produces strong vasoconstriction of both pial and basilar arteries in several species (Mima et al., 1989; Robinson and McCulloch, 1990; Gotoh et al., 1991); however, ET-1 can act as a dilator at low doses in pial arteries (Armstead et al., 1989; Faraci, 1989). Studies examining cerebrovascular effects of intravenous ET antagonists on basal ceerebral blood flow have yielded mixed results. For example, pretreatment with the nonselective ET antagonist bosentan did not alter cerebral blood flow after transient global ischemia in rats (Patel and McCulloch, 1996). Further, no effect was observed in canine basilar artery caliber as measured angiographically (Gotoh et al., 1991). However, others have demonstrated a dose-dependent reduction in laser-Doppler flowmetry (Willette et al., 1990). Our study clearly demonstrates that intravenous administration of Ro 61-1790 increases pial vessel diameter. It can be hypothesized that these differences may be due to (a) a differential effect of ETA antagonists on different vessels or (b) differences in methodology, species, and animal models.
In summary, intravenous administration of the potent and selective ETA antagonist Ro 61-1790 at a dose that attenuates vasoconstrictive pial arteriolar responses to ET-1 does not improve brain injury or neurological outcome when given to cats at 90 minutes after MCAO and at the onset of reperfusion. The interaction between ET receptor subtypes in the cerebral circulation under physiological and ischemic conditions requires further investigation.