
Abstract
To examine a possible protective effect of exogenous glial cell line-derived neurotrophic factor (GDNF) gene expression against ischemic brain injury, a replication-defective adenoviral vector containing GDNF gene (Ad-GDNF) was directly injected into the cerebral cortex at 1 day before 90 minutes of transient middle cerebral artery occlusion (MCAO) in rats. 2,3,5-Triphenyltetrazolium chloride staining showed that infarct volume of the Ad-GDNF-injected group at 24 hours after the transient MCAO was significantly smaller than that of vehicle- or Ad-LacZ-treated group. Enzyme-linked immunosorbent assay (ELISA) for immunoreactive GDNF demonstrated that GDNF gene products in the Ad-GDNF-injected group were higher than those of vehicle-treated group at 24 hours after transient MCAO. Immunoreactive GDNF staining was obviously detected in the cortex around the needle track just before or 24 hours after MCAO in the Ad-GDNF group, whereas no or slight GDNF staining was detected in the vehicle group. The numbers of TUNEL, immunoreactive caspase-3, and cytochrome c-positive neurons induced in the ipsilateral cerebral cortex at 24 hours after transient MCAO were markedly reduced by the Ad-GDNF group. These results suggest that the successful exogenous GDNF gene transfer ameliorates ischemic brain injury after transient MCAO in association with the reduction of apoptotic signals.
Keywords
Glial cell line-derived neurotrophic factor (GDNF) has a potent neuroprotective effect on a variety of neuronal damage in vitro or in vivo (Lin et al., 1993; Henderson et al., 1994; Beck et al., 1995; Li et al., 1995; Tomac et al., 1995). Our previous papers and another report have also demonstrated the amelioration of ischemic brain injury by GDNF application after middle cerebral artery occlusion (MCAO) model in rodents (Abe et al., 1997a; Wang et al., 1997; Kitagawa et al., 1998a). Therefore, GDNF could be one of the most potent candidates for acute cerebrovascular injury as well as neurodegenerative disorders such as Parkinson's disease (Lin et al., 1993; Tomac et al., 1995). However, in regard to clinical application of this protein, administration could remain difficult. GDNF cannot be effectively delivered to the brain parenchymal lesion after the vascular injection because of the blood brain barrier. Furthermore, intracerebroventricular or intraparenchymal injection could not be frequently applicable in the clinical situation.
Gene delivery systems using virus vectors have been reported in many fields including not only genetic diseases but also in some acquired diseases such as cancer or cardiovascular disease (Nabel, 1995; Verma and Somia, 1997). Some genes have also been successfully transferred into the brain and have shown protective effects against ischemic brain injury (Betz et al., 1995; Linnik et al., 1995; Lawrence et al., 1997; Yang G-Y et al., 1997; Yang et al., 1998; Yenari et al., 1998). Adenovirus-mediated gene expression of an interleukin-1 receptor antagonist reduced brain ischemic injury after MCAO in rodents (Betz et al., 1995; Yang G-Y et al., 1997; Yang et al., 1998). Herpes simplex virus -mediated HSP72 gene was neuroprotective in a rat model of stroke and epilepsy (Yenari et al., 1998). Therefore, gene therapy for cerebrovascular disease could become one potentially better therapy in the near future (Heistad and Faraci, 1996).
Recently, our group demonstrated successful adenovirus-mediated LacZ gene transfer into normal or ischemic rodent brain (Abe et al., 1991b,c; Kitagawa et al., 1998b, c ). Replication-defective adenovirus-mediated gene transfer has some advantages for gene therapy for stroke in comparison with those mediated by the other vectors such as retrovirus, herpes simplex virus, etc. Adenovirus vectors could efficiently infect the terminated cells as well as proliferative ones. Adenovirus vectors are less toxic than the others, although inflammatory reactions (Byrnes et al., 1995; Newman et al., 1995) or transient cytotoxicity has been reported (Kitagawa et al., 1998b, c ). Moreover, the transient nature of gene expression, considered a disadvantage for other types of disorders, could turn out to be positive for acute brain diseases (Abe et al., 1997b).
The mechanism of the neuroprotective effect of GDNF is not clearly understood. Some apoptotic signals may be important in the development of ischemic neuronal cell death. Recently, it has been reported that caspases-3 and cytochrome c may play a pivotal role in apoptotic cell death after mitochondrial apoptotic signal pathways regulated by bcl-2 family genes (Kluck et al., 1997; Yang J et al., 1997). Our previous reports showed an induction of caspases after ischemic injury in the rat brain and an attenuation of ischemic brain injury by GDNF associated with the suppression of caspases induction (Kitagawa et al., 1998a). Thus, we prepared an adenovirus vector containing the GDNF gene (Ad-GDNF), and a possible protective effect of the Ad-GDNF transfer into rat brain was examined after transient MCAO in association with modifications of apoptotic signals.
MATERIALS AND METHODS
Adenoviral vector
The recombinant adenoviral vector used in this study was based on type 5 adenovirus that was essentially the same as in previous reports (Bajocchi et al., 1993; Setoguchi et al., 1994; Abe et al 1997b). In brief, the E3 region of the adenovirus genome was deleted, and a cassette containing cDNA for rat GDNF or Escherichia coli lacZ gene (used as a control vector) driven by cytomegalovirus promoter and SV40 polyadenylation signal was inserted into the E1 region of the adenovirus genome by homologous recombination. With this promoter, the rat GDNF or E. coli lacZ gene of the episomally located adenovirus vector may be transcribed as mRNA in the nucleus, and the mRNA translated into the protein in the cytoplasm. The adenoviral vector, designated as Ad-GDNF or Ad-LacZ, was propagated in 293 cells (CRL1573; American Type Collection) and purified and titered as previously described (Bajocchi et al., 1993; Setoguchi et al., 1994; Abe et al., 1997b).
Western blotting for glial cell line-derived neurotrophic factor gene expression
The 293 cells were cultured with Eagle's Essential Medium containing 10% heat-inactivated fetal bovine serum, 2 mmol/L glutamine, 50 U/mL penicillin, and 50 μg/mL streptomycin with or without Ad-GDNF (108 plaque-forming units [pfu]/ mL) at 37°C for 48 hours. After incubation, media were collected, centrifuged at 2,000g, and the supernatants were used for Western blotting. Culture supernatants (12.5 μL) or standard GDNF (25 ng) (recombinant human GDNF; Promega, Madison, WI, U.S.A.) were dissolved in Laemmli's sample buffer (Laemmli, 1970), separated on 8 to 16% sodium dodecyl sulfate (polyacrylamide gel electrophoresis) and electrophoret-ically transferred to polyvinylidene difluoride membranes for 2 hours. Briefly, the polyvinylidene difluoride membranes were incubated for 1 hour at room temperature with 150 mmol/L NaCl, 50 mmol/L Tris/HCl, pH 7.5, (Tris buffered saline; TBS) containing 0.04% NP-40 and 3% bovine serum albumin (fraction V), followed by overnight incubation at 4°C with anti-GDNF polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, U.S.A.). The primary antibody was diluted in TBS containing 1% bovine serum albumin (1:100). The polyvinylidene difluoride membranes were washed three times for 15 minutes each at room temperature in TBS and then incubated for 60 minutes with alkaline phosphatase-conjugated goat antirabbit IgG (H+L). The blots were stained with 5-bromo-4-chloro-3-indoyl phosphate-p-toluidine salt and p-nitroblue tetrazolium chloride as described in the BIO-RAD instruction manual.
In vivo adenovirus-mediated glial cell line-derived neurotrophic factor gene transfer
Male adult Wistar rats (body weight: 250 to 280 g) were used in this experiment. Adenovirus vector injection was performed according to our previous report with a slight modification (Abe et al., 1997b). Briefly, the animals were anesthetized with an intraperitoneal injection of pentobarbital (10 mg/250 g rat) and were immobilized using a stereotactic frame (SR-5N, Na-rishige, Tokyo, Japan). A burr hole with a diameter of 2 mm was carefully made in the skull by an electric dental drill to avoid traumatic brain injury. The location of the burr hole was 3 mm posterior and 5 mm lateral to the right of bregma, which is located at the upper part of the MCA. Dura mater was preserved at this time. The animals were allowed to recover at ambient atmosphere.
On the next day, at about 24 hours after the drilling, the rats were anesthetized by inhalation of a nitrous oxide-oxygen-halothane (68%-30%-2%) mixture, and their heads were fixed in the stereotactic frame. Ad-GDNF, Ad-LacZ (108 pfu in 10 μL of vector vehicle consisting of 10 mmol/L Tris-HCl, pH 7.4, 1 mmol/L MgCl2, and 10% glycerol), or vehicle solution was administered to the ipsilateral cortex via the burr hole through the dura mater at a depth of 4 mm from the surface of the skull bone at an angle of 24° laterally using a 10-μL sterile Hamilton syringe (the Gastight Highperformance Syringe, Hamilton, Reno, NE, U.S.A.) and a 26-gauge needle (0.47 mm outer diameter). The injections (10 μL) were slowly completed in 10 minutes. During the injection, the needle was gradually withdrawn at a rate of 0.4 mm/min.
Transient middle cerebral artery occlusion
At approximately 24 hours after the virus vector injection, the rats were again anesthetized by inhalation of a nitrous oxide-oxygen-halothane (69%-30%-1%) mixture during the surgical procedure. The right MCA was occluded by the insertion of a nylon thread through the common carotid artery as described in our previous reports (Hayashi et al., 1998a; Kitagawa et al., 1998a). Body temperature was maintained at 37 ± 0.3°C during the surgical preparation for MCA occlusion using a heat pad (BWT-100, Bio Research Center Co., Ltd, Nagoya, Japan) and heating lamp. The blood flow was restored by removal of the nylon thread after 90 minutes of transient ischemia. Blood samples (90 μL) were collected before MCAO (−1.5 h) or at 0, 8, or 24 hours after 90 minutes of reperfusion from ventral tail artery for measurement of Pao2, Paco2, and pH (pH/Blood Gas Analyzer, CIBA CORNING 238, CHIRON, Tokyo Japan). Body temperature was also monitored before MCAO or at 0, 8, or 24 hours after reperfusion with a rectal probe (RET-2, Physitemp Instruments Inc., Clifton, AZ, U.S.A.). The animals were allowed to recover at ambient temperature (21 to 24°C) until the time of sampling. The experimental protocol and procedures were approved by the Animal Committee of the Okayama University Medical School.
Estimation of brain injury after transient middle cerebral artery occlusion
To examine an effect of Ad-GDNF on infarct size after transient MCAO, the rat forebrains were removed and divided into 6 coronal (2 mm each) sections at 24 hours of reperfusion with vehicle (n = 9), Ad-LacZ (n = 6), or Ad-GDNF (n = 9) treatment. The coronal sections (designated S-1 to S-6 from frontal pole to midbrain) were stained with saline containing 2% 2,3,5-triphenyltetrazolium chloride at 37°C for 30 minutes, after which sections were fixed in 10% neutralized formalin, according to a technique reported previously (Bederson et al., 1986). The five infarct areas between each adjoining slice were measured by Scion Image software, version 1.62a, and then the infarct areas on each slice were summed and multiplied by slice thickness to give the infarct volume. In this experiment, regional cerebral blood flow (rCBF) of right frontoparietal cortex region was measured before or immediately after occlusion (−1.5 hours) or reperfusion (0 hours) respectively, and at 8 or 24 hours after the reperfusion through the burr hole using a laser blood flowmeter (Flo-Cl, Omegawave, Tokyo, Japan).
Enzyme-linked immunosorbent assay for glial cell line-derived neurotrophic factor
For measurement of GDNF protein content, the S-4 coronal section, which contained the needle injection site, was homogenized with buffer (0.01 mol/L Tris-HCl buffer, pH 7.5, containing 0.1 mol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), and 1 mmol/L phenylmethylsulfonyl fluoride) using a Polytron. The homogenate was centrifuged at 10,000g for 20 minutes at 4°C, and then the supernatant was collected as a sample for GDNF enzyme-linked immunosorbent assay (ELISA) assay. A specific GDNF sandwich immunoassay with a chicken polyclonal antibody for human GDNF was performed in the 96-well immunoassay plate (Nunc MaxiSorp Nalge Nunc Int., Rochester, NY, U.S.A.), which was precoated with the anti-GDNF monoclonal antibody, using the kit (GDNF Emax Immunoassay Systems, Promega). After overnight incubation at 4°C, reaction of a second antibody-HRP conjugate (kit component) for chicken IgY was performed, followed by color development with TMB solution (kit component). After the stop reaction with 1 mol/L phosphoric acid, OD450 was measured by a 96 well plate reader, and GDNF protein content of each sample was calculated. Using this system, immunoreactive GDNF in the samples can be quantitated in the range of 16 to 1,000 pg/mL.
Histologic study
For the histologic staining of DNA fragmentation, GDNF, caspase-3, and cytochrome c, the rat forebrains were removed and quickly frozen in a powdered dry ice at 1 day after vehicle (n = 3) or Ad-GDNF (n = 3) injection, or at 24 hours after transient MCAO (2 days after injection) of both vehicle- (n = 3) and Ad-GDNF- (n = 3) treated groups. Sham control samples (n = 2) were also collected in the same way without the drug injection and MCAO. Coronal sections at the caudate and dorsal hippocampal levels were cut on a cryostat at −18°C to a 10-μm thickness and collected on glass slides.
For detection of DNA fragmentation, terminal deoxynucleotidyl dUTP nick-end labeling (TUNEL) was performed with a kit (TACS TdT in Situ Apoptosis Detection Kit #80-4625-00, Genzyme, Cambridge, MA, U.S.A.). In brief, after the “fixation with 10% neutralized formalin,” a set of brain sections was washed 3 times with cold phosphate-buffered saline (PBS, pH 7.4) and made permeable with CytoPore (kit component) for 20 minutes. After blocking endogenous peroxidase with 2% H2O2 for 5 minutes, double-strand breaks in genomic DNA were detected with the mixture of terminal deoxynucleotidyl transferase (TdT) dNTP Mix, TdT enzyme, and TdT Mn2+ (kit components) in 1× TdT labeling buffer for 1 hour at 37°C. The reaction was stopped with 1× STOP buffer, followed by incubation for 20 minutes with streptavidin-horseradish peroxidase, and the staining was developed with 2,3′-diaminobenzidine tetrahydrochloride (0.5 mg/mL in 50 mmol/1 Tris-HCl buffer, pH 7.4).
Immunostaining for GDNF, caspase-3, and cytochrome c was performed by avidin-biotin-peroxidase method using a kit (PK-6101 for GDNF or cytochrome c, or PK-6105 for caspase-3, Vector Laboratories, Burlingame, CA, U.S.A.). The fresh-frozen sections were fixed for 10 minutes in ice-cold acetone and air-dried. Then the sections were rinsed 3 times in PBS (pH 7.4). After blocking with 10% normal serum (kit components) for 2 hours, the slides were incubated for 16 hours at 4°C with a first antibody: a rabbit polyclonal antibody against GDNF (D-20), cytochrome c (H-104), and a goat polyclonal antibody against caspase-3 (CPP32 p20, L-18) (Santa Cruz Biotechnology Inc., catalogue #sc-328, sc-7159, or sc-1225, respectively), diluted in PBS (1/200) containing 10% normal serum and 0.3% Triton X-100. Some sections were treated simultaneously without the first antibody. The specificity of these antibodies was also confirmed by Western blot analysis using those antibodies that showed a 20-kDa, 12-kDa, or 20-kDa single band of GDNF (Fig. 1, lane 3), cytochrome c (data not shown), or caspase-3 p20 subunit (Hayashi et al., 1998b), respectively. Endogenous peroxidase was blocked for 20 minutes with PBS containing 0.3% H2O2 and 10% methanol. The sections were then washed and incubated for 2 hours with the biotinylated second antibody (1:200), in the buffer, followed by incubation for 30 minutes with avidin-biotin-horseradish peroxidase complex. Staining was developed with 2,3′-diaminobenzidine tetrahydrochloride (0.5 mg/mL, in 50 mmol/L Tris-HCL buffer, pH 7.4), and lightly counterstained with Mayer hematoxylin.
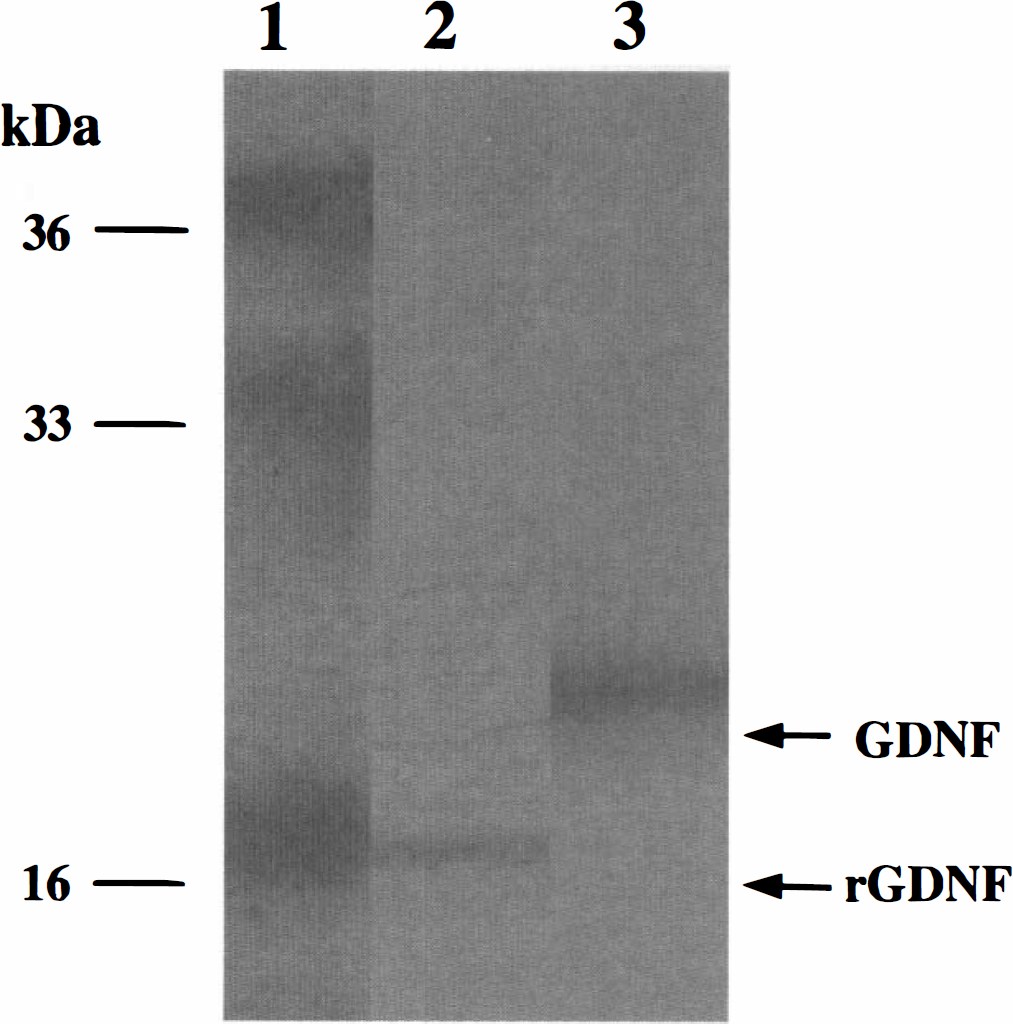
Western blot analysis of GDNF. Lane 1, molecular marker; lane 2, 16-kDa recombinant human GDNF (rhGDNF, 25 ng) as a standard; lane 3, the supernatant (12.5 μL) at 48 hours after coculture of 293 cells with Ad-GDNF. Rat GDNF was detected as a single band of 20-kDa glycoprotein.
The sections were examined by light microscope, and the stained cells in 0.25 mm2 of three random MCA areas were counted, summed, and categorized into 4 grades as follows: no staining, small (1 to 10), moderate (10 to 100), or large (100 to 500) number of stained cells as (−), (±), (+), and (2+), respectively. In addition, the number of TUNEL, caspase-3, or cytochrome c positive cells in the cerebral cortex of the Ad-GDNF treated samples (n = 6) was statistically compared with that of the vehicle group.
Statistical analyses
Statistical analysis was performed using Student's t-test for GDNF ELISA assay, one-way analysis of variance (ANOVA) for infarct volume, two-way analysis of variance followed by Bonferroni post hoc test for infarct area, analysis of variance repeated measure for physiologic and rCBF data, and Mann-Whitney test for the number of TUNEL, caspase-3, or cytochrome c positive cells, respectively.
RESULTS
Adenoviral glial cell line-derived neurotrophic factor gene expression in vitro
Rat GDNF protein content in the supernatant from the culture medium of 293 cells, which was infected by Ad-GDNF, was 71.5 ng/mL measured by GDNF ELISA assay using a kit, whereas GDNF protein was not detected in the control medium (data not shown). Western blotting demonstrated that the supernatant showed a clear 20-kDa band of rat GDNF (Fig. 1, lane 3), while standard recombinant human GDNF (rhGDNF) showed a 16-kDa single band (Fig. 1, lane 2).
Effect of adenoviral vector containing glial cell line-derived neurotrophic factor on infarction after middle cerebral artery occlusion
There were no significant differences in blood gases, pH, and rectal temperature among vehicle-, Ad-LacZ-, and Ad-GDNF-treated groups before MCAO, or at 0, 8, or 24 hours after the reperfusion (Table 1). Rectal temperature in both groups increased at 0 hour of the reperfusion and then declined to the basal level by 8 hours after the reperfusion.
Time course of physiologic parameters in MCAO rats
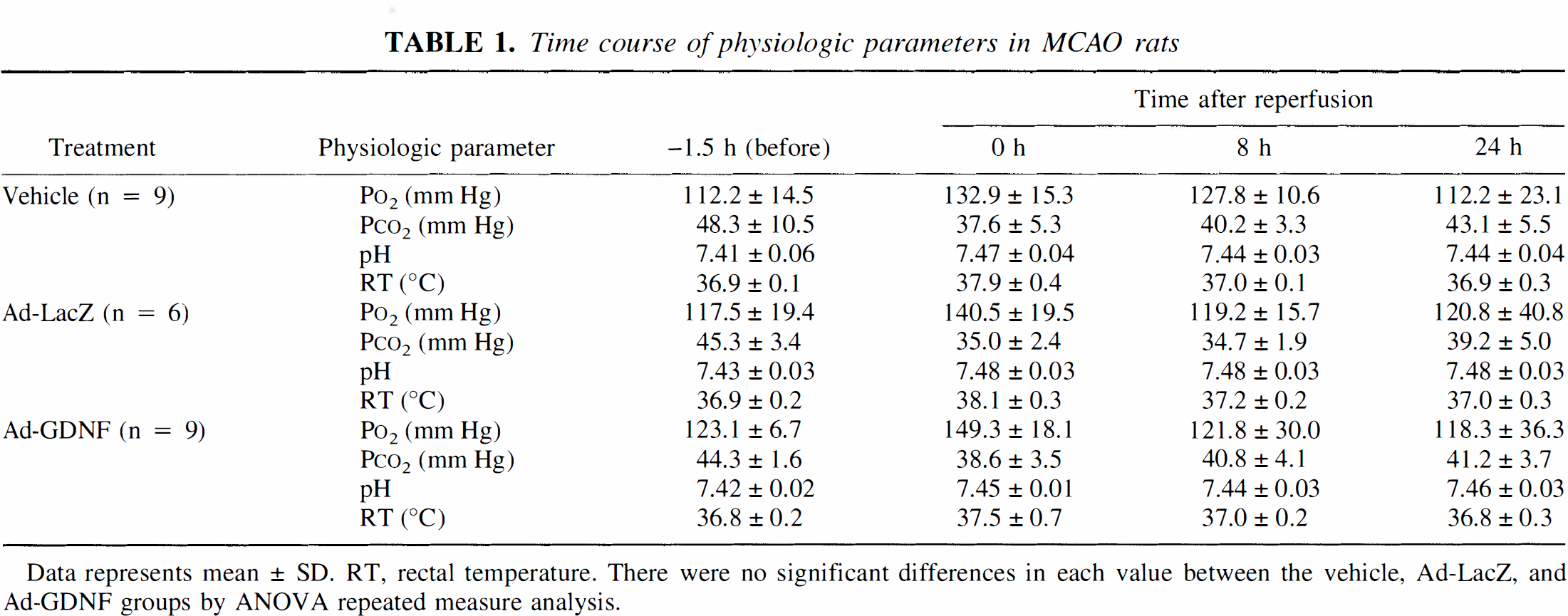
Data represents mean ± SD. RT, rectal temperature. There were no significant differences in each value between the vehicle, Ad-LacZ, and Ad-GDNF groups by ANOVA repeated measure analysis.
Regional CBF of both the vehicle-, Ad-LacZ-, and Ad-GDNF-treated groups was reduced to approximately 40% of the control immediately after MCAO and recovered to the baseline after reperfusion by withdrawing the nylon thread (Fig. 2). Although a slight increase of rCBF in the case of the Ad-GDNF-treated group compared to that of the vehicle- or the Ad-LacZ-treated group was observed, there was no significant difference between these three groups (Fig. 2). Although infarction in the brain sections of the sham control group could not be observed, infarct volumes of the vehicle-, Ad-LacZ-, and Ad-GDNF-treated groups at 24 hours after 90 minutes of transient MCAO were 209.2 ± 35.8 mm3 (mean ± SD, n = 9), 213.3 ± 59.1 mm3 (n = 6), and 97.6 ± 71.5 mm3 (n = 9; P < 0.001 versus the vehicle- and Ad-LacZ-treated groups), respectively (Fig. 3A). Infarct areas of two or three coronal sections (4, 6, and 8 mm caudal from frontal pole) from the Ad-GDNF-treated group were also significantly smaller than those of the vehicle or the Ad-LacZ groups (Fig. 3B), respectively.
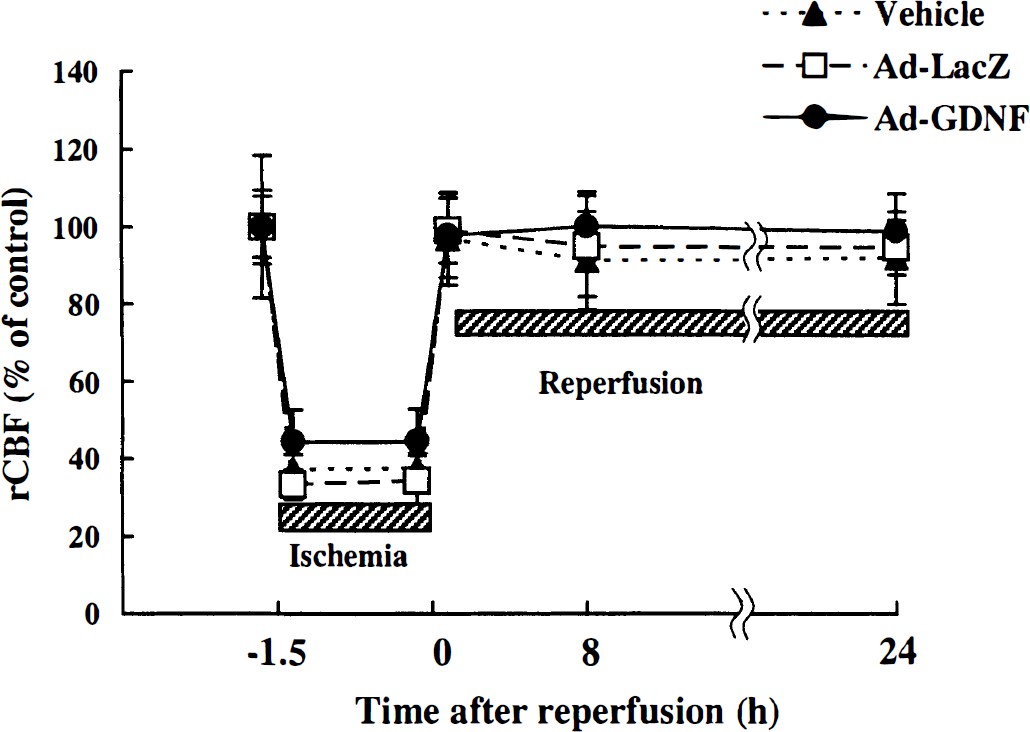
Temporal profile of regional CBF after 90 minutes of transient MCAO. Regional CBF reduced to about 40% of basal line immediately after MCAO occlusion (−1.5 hours) and restored to baseline after reperfusion (0 hours) in both vehicle- (n = 9, —▴—), Ad-LacZ- (n = 6, —•—), and Ad-GDNF-treated (n = 9, —•—) groups. There was no significant difference in CBF between these three groups. Data are expressed as mean ± SD.
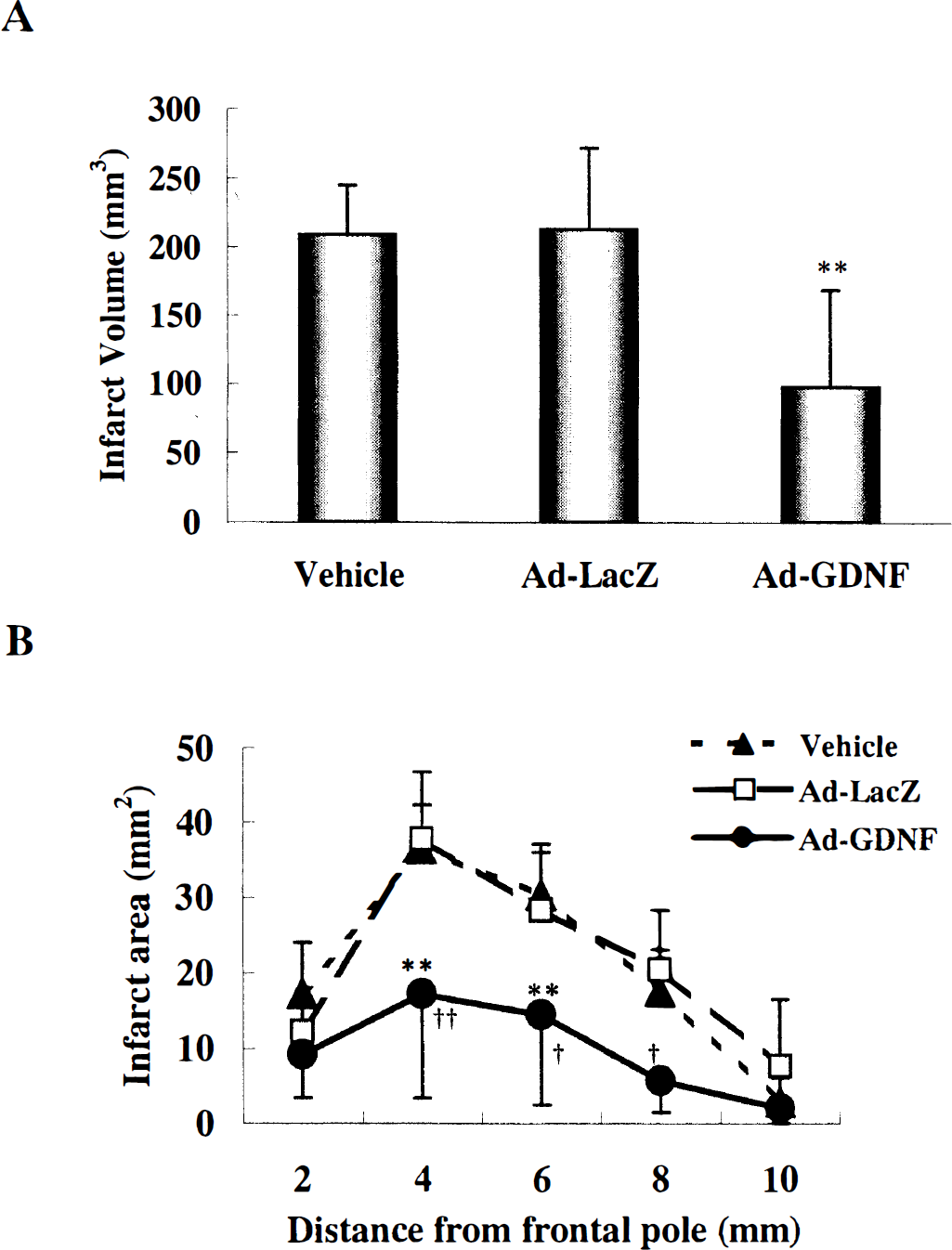
Effect of Ad-GDNF on infarct volume
Detection of immunoreactive glial cell line-derived neurotrophic factor
ELISA assay for GDNF revealed that rat GDNF contents at 24 hours after MCAO in an S-4 slice of the Ad-GDNF-treated group (222.6 ± 49.5 pg/100 mg tissue, n = 9) were about 10 times higher (P < 0.01) than those of the vehicle-treated group (22.9 ± 34.6, n = 9) (Fig. 4). Immunoreactive GDNF was not detected in any region including around the needle track (Fig. 5a, arrows) before MCAO (24 hours after vehicle injection) in the vehicle-treated group, whereas GDNF-positive cells were detected in the cortex (Fig. 5b, arrowheads) near the needle track (Fig. 5b, arrows) before MCAO in the Ad-GDNF-treated group. At 24 hours after the reperfusion, GDNF-positive cells (Fig. 5d, arrowheads) were also detected in the cortex near the needle track in the Ad-GDNF-treated group, but not or only slightly in the vehicle group (Fig. 5c). There were no staining cells in the sample treated without primary antibody as a negative control (data not shown).
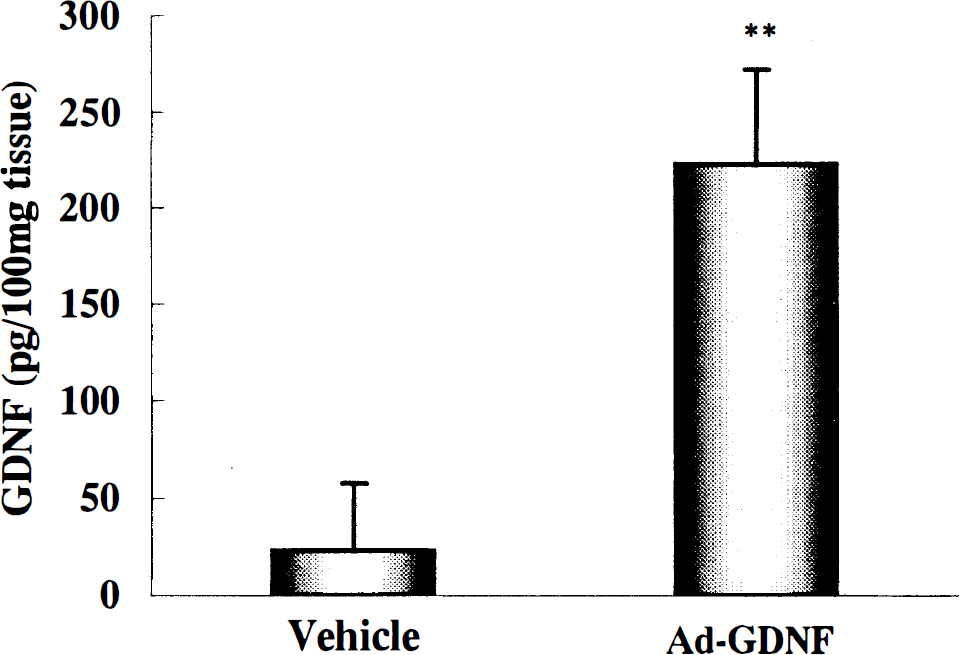
ELISA assay for GDNF content in S-4 coronal slice at 24 hours after 90 minutes of transient MCAO. Immunoreactive GDNF content in Ad-GDNF-treated group (n = 9, **P < 0.01 versus vehicle group) was significantly higher than that in the vehicle-treated group (n = 9). Data represent mean ± SD.
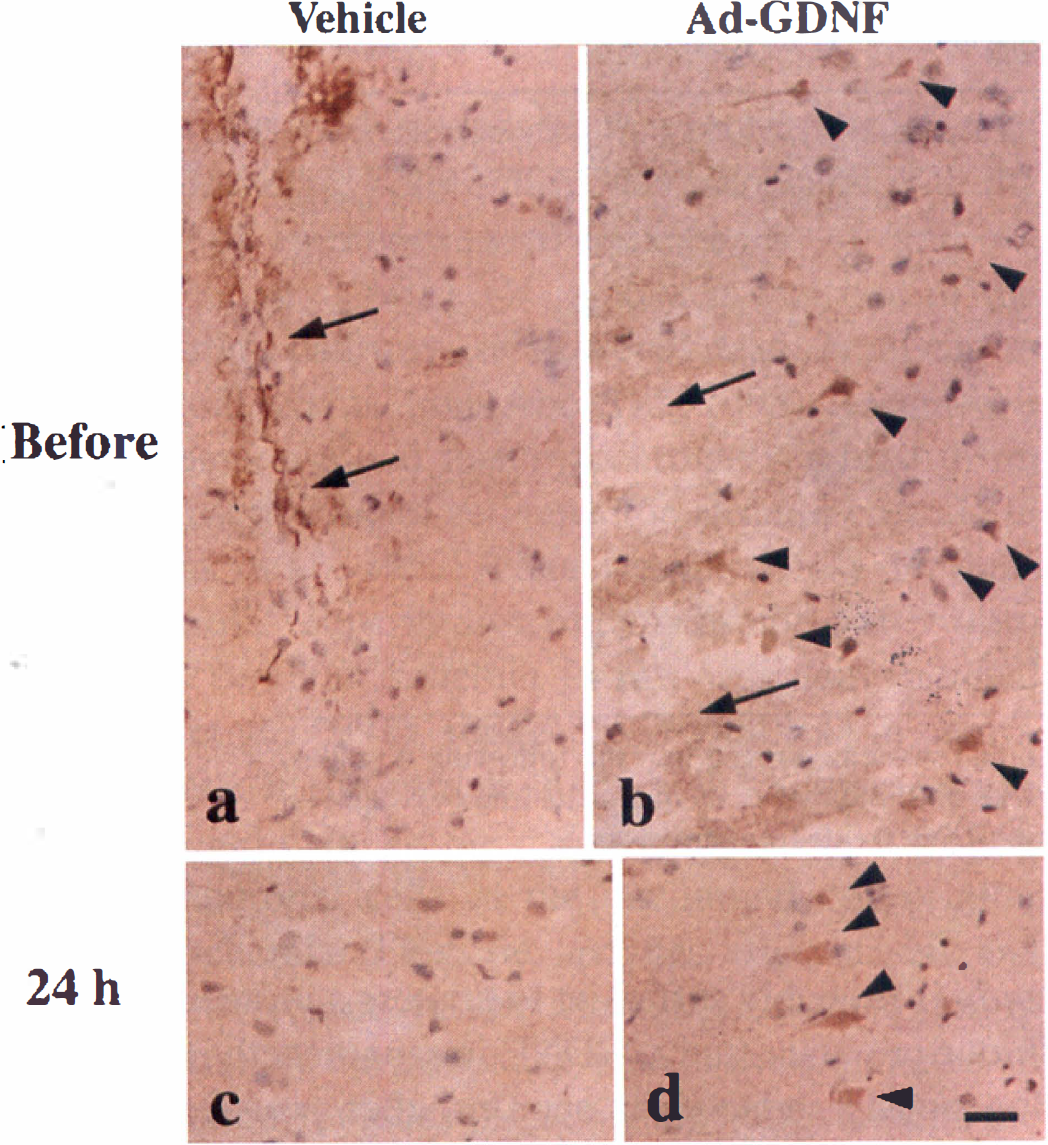
Representative staining for immunoreactive GDNF in the cortex near the needle track at 24 hours after Ad-GDNF injection just before transient MCAO
Effect of adenoviral vector containing glial cell line-derived neurotrophic factor on the changes of TUNEL, caspase-3, or cytochrome c stainings
There were no TUNEL or caspase-3-positive cells in the sham control brain (Figs. 6a and 6d), whereas cytochrome c-stained cells were slightly detected in layers IV to V of the ipsilateral (Fig. 6g, arrowheads), or contralateral frontoparietal somatosensory cortex. TUNEL -positive cells were markedly increased in ipsilateral cortex (Fig. 6b, Table 2) and caudate (Table 2) in the vehicle-treated group at 24 hours after transient MCAO. In the Ad-GDNF-treated group, the number of TUNEL-positive cells was obviously smaller, especially in the cortex (Fig. 6c, arrowhead), than that in the vehicle group (Table 2). Caspase-3- or cytochrome c-positive neurons were also markedly increased in the vehicle group at 24 hours after transient MCAO in both ipsilateral cortex (Figs. 6e and 6h, arrowheads) and caudate, whereas those positive cells were reduced in the Ad-GDNF-treated group (Figs. 6f and 6i, arrowheads, Table 2). Nonparametric statistical analysis (Mann-Whitney test) also showed that the number of positive cells in the cortex of the Ad-GDNF-treated group was significantly smaller than that in the vehicle group (TUNEL, or caspase-3, P < 0.01; cytochrome c, P < 0.05). In the ipsilateral hippocampus and in all parts of the contralateral side, TUNEL staining or caspase-3 immunoreactivity was not found in this experiment. Moreover, changes in cytochrome c immunoreactivity were not detected in any parts except for the ipsilateral ischemic area. No staining cells were detected in the sample treated without each primary antibody as negative control (data not shown). There was no leukocyte infiltration except that around the needle track in one vehicle treated-rat, and traumatic injury was observed only around the needle track (data not shown).
Changes of TUNEL, caspase-3, and cytochrome c immunoreactivities after transient MCAO in rats

Staining was performed at 24 hours after transient MCAO, and was categorized into the following 4 grades: no staining, or a small (2–10), moderate (10–100), or large (100–500) number of stained cells into (−), (±), (+), and (2+), respectively. Sham control, n = 2; vehicle and Ad-GDNF treated groups, n = 3.
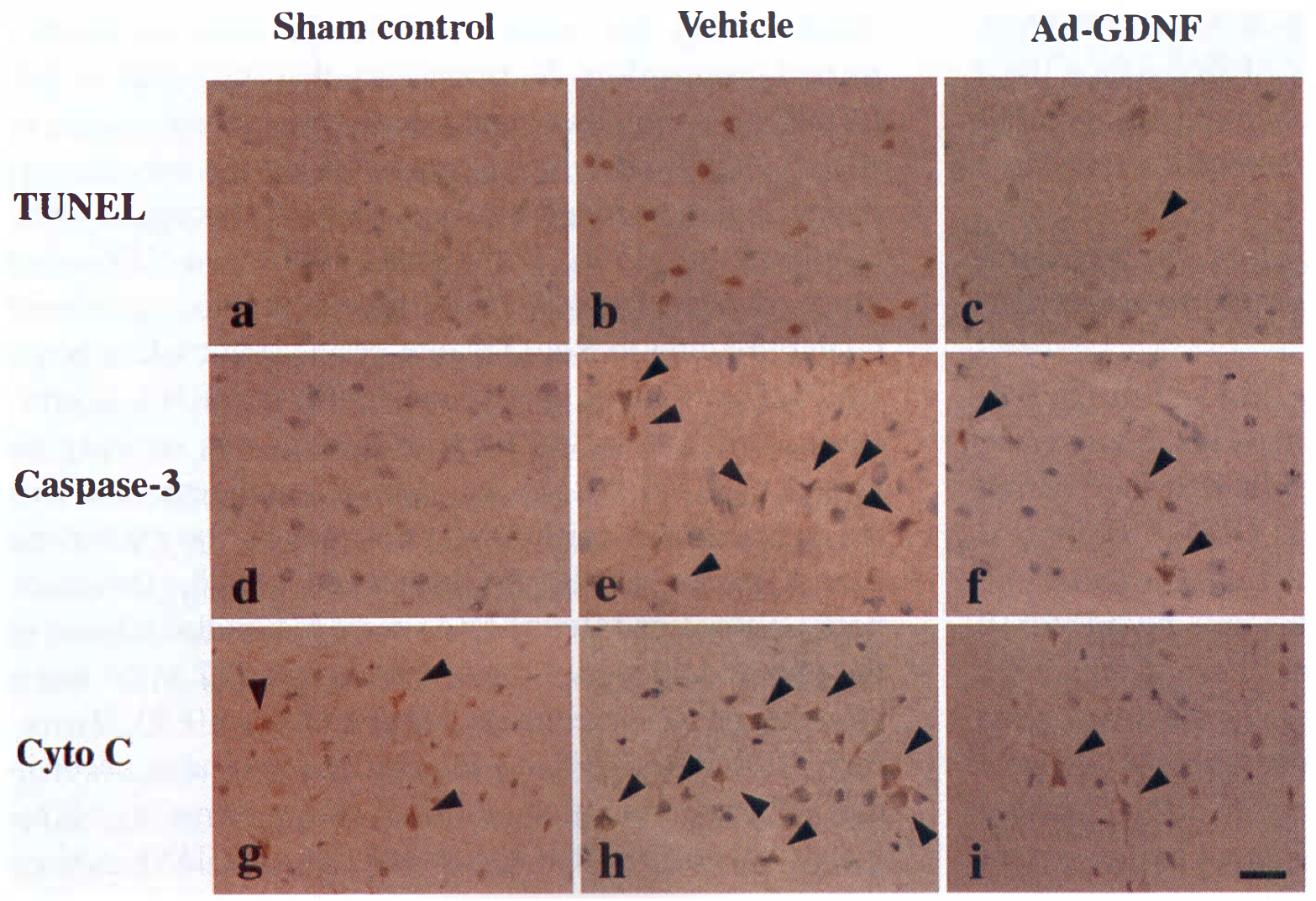
Representative staining for TUNEL
DISCUSSION
We demonstrated that Ad-GDNF used in this study had the ability to infect 293 cells and produce 20-kDa GDNF (Fig. 1, lane 3) in vitro. The culture supernatant from 293 cells infected by Ad-GDNF induced the neurite growth and the survival of cultured dopaminergic neurons in primary neuronal culture (data not shown). Therefore, Ad-GDNF could produce biologically active GDNF protein. Adenovirus or adeno-associated virus-mediated GDNF gene transfer prevents dopaminergic neuron degeneration (Choi-Lundberg et al., 1997; Kojima et al., 1997; Mamdel et al., 1997; Fan et al., 1998) and improves behavioral impairment in the rat model of Parkinson's disease (Bilang-Bleuel et al., 1997; Lapchak et al., 1997). Neuroprotective activity of adenovirus-mediated GDNF gene transfer against axotomy-induced motoneuron death was also reported (Baumgartner and Shine, 1997). However, it has not been reported if adenovirus-mediated GDNF gene transfer could protect more severe types of neuronal death such as with ischemia or stroke. In this experiment, we first demonstrated that adenovirus-mediated exogenous GDNF gene was successfully transferred into the cortical neurons (Figs. 5b and 5d, arrowheads) and attenuated ischemic brain injury after the transient MCAO model (Fig. 3).
Several reports have shown that adenovirus vector itself induces proteins such as inflammatory cytokines (Cartmell et al., 1999), adhesion molecules (Newman et al., 1995), or stress proteins (Kitagawa et al., 1998b, c ). There is a possibility that injection of the vector itself could influence or protect ischemic brain injury after transient MCAO. However, Ad-LacZ injection (used as a control vector) did not affect the infarction that occurred after transient MCAO (Fig. 3). Therefore, the neuroprotective effect of Ad-GDNF may be due to exogenous GDNF gene product.
GDNF gene expression was only detected in the cortical neurons around the injection site (Figs. 5b and 5d), and GDNF contents around needle route were 300 pg/100 mg tissue (Fig. 4). GDNF binds GDNF receptor α (GFRα-1) with a Kd value of 3 pmol/L and mediated the survival response of cultured neurons at 1 pg/mL to 10 ng/mL in in vitro study (Treanor et al., 1996; Klein et al., 1997). Moreover, previous gene transfer studies against neurodegeneration showed that similar levels of the gene product produced evident neuroprotective effect (Mandel et al., 1997). Therefore, the GDNF content in the present study is probably sufficient to possess a protective effect. In our previous study, GDNF immunoreactivity was endogenously induced in neurons after transient MCAO (Abe and Hayashi, 1997). However, neither GDNF mRNA nor protein immunoreactivity was detected in the normal rat brain, and the induction of GDNF protein was only temporally seen (1 to 3 h) in the early phase of transient MCAO (Abe and Hayashi, 1997). Therefore, the immunoreactive GDNF detected before or 24 hours after 90 minutes of transient MCAO (Figs. 5b and 5d) in the present study may predominantly be of adenovirus-mediated origin.
The protective mechanism of GDNF has not been fully understood, although previous work has demonstrated that GDNF diminished ischemia-induced nitric oxide release (Wang et al., 1997) or reduced caspase immunoreactive neurons (Kitagawa et al., 1998a). In the present study, there was no significant difference in rCBF among the vehicle-, Ad-LacZ-, and Ad-GDNF-treated groups (Fig. 2), suggesting that the effect of Ad-GDNF was not associated with the improvement of rCBF. GDNF signaling is mediated by the receptor tyrosine kinase encoded by the c-ret proto-oncogene (Ret) (Durbec et al., 1996; Trupp et al., 1996), and GFRα-1, a glycosyl-phosphatidylinositol-linked protein, assists in GDNF binding to Ret (Treanor et al., 1996; Klein et al., 1997). Distribution of Ret and GFRα-1 mRNA expressions in the rat central nervous system was recently reported. GFRα-1 mRNA was detected in the cerebral cortex, whereas Ret mRNA was not seen in the normal rat brain (Trupp et al., 1997; Glazner et al., 1998). However, both GFRα-1 and Ret mRNAs were markedly induced in the pyramidal layer of the cerebral cortex 12 to 24 hours after kainic acid treatment (Trupp et al., 1997). Therefore, GDNF/Ret/GFRα-1 interactions may also occur in the ischemic condition. Further study seems to be required to confirm that the interaction of GDNF and its receptors could be essential for its neuroprotective effect in ischemic brain injury.
Our previous study suggested that protective effect of GDNF on ischemic brain injury was associated with the inhibition of immunoreactive caspases −1 and −3 (Kitagawa et al., 1998a). Of interest is that one of the most important pathways of neuronal death is related to mitochondrial dysfunction (Abe et al., 1995). Recently, it has been demonstrated that cytochrome c release from mitochondria to cytosol activates the caspase cascade in vitro (Kluck et al., 1997; Yang J et al., 1997). Furthermore, cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats was demonstrated (Fujimura et al., 1998), suggesting a vital role in neuronal cell death. In the present study, both immunoreactive caspase-3 and cytochrome c were induced in the cytoplasm of neuronal cells in the penumbral cortex after transient ischemia (Figs. 6e and 6h, arrowheads) and obviously decreased in the Ad-GDNF-treated group (Figs. 6f and 6i, arrowheads) with correspondence to TUNEL-staining profiles (Figs. 6b and 6c). Although the TUNEL method is not specific for apoptotic neurons in the ischemic brain, the TUNEL-positive neurons in the ischemic penumbral region are mainly apoptotic cells, whereas those in the ischemic core are necrotic (Charriaut-Marlangue et al., 1996). Therefore, some of the TUNEL-positive neurons observed in the present study died by apoptotic process. Thus, the apoptotic pathway via cytochrome c and caspase-3 seems to be one of the targets of protection by GDNF. In this study, we have demonstrated the change of immunoreactive caspase-3 protein after transient MCAO, but the pro- or activated form of caspase-3 has not been examined. Further study of the change of caspase-3 activity would provide us the precise information about the protective mechanism of GDNF.
Gene therapeutic study for stroke has not yet been tried in the clinical situation. Although it has been demonstrated that adenovirus-mediated neuronal apoptosis inhibitory protein or an interleukin-1 receptor antagonist had a protective effect against ischemic brain injury, adenoviral vectors were injected 5 days before ischemia (Betz et al., 1995; Xu et al., 1997). Herpes simplex virus-mediated HSP72 gene transfer markedly improved striatal neuron survival in focal ischemia, but the vector was injected 8 hours before occlusion (Yenari et al., 1998). In this study, adenoviral vector was also injected 24 hours before MCAO, because it takes more than 8 hours to express the gene product using the adenoviral vector in the normal rat brain (Abe et al., 1997b). However, gene therapy should be applied after an occurrence of stroke in the clinical situation. Therefore, therapeutic study with these vectors should be examined when delivered after ischemia. Furthermore, each vector has some disadvantages such as toxicity or efficacy of gene expression.
Improvement of vectors or development of a safer vector may be necessary for more practical gene therapy. Finally, the route of administration may be of major concern for gene therapy. In the present study, we administrated the vector directly into the cerebral cortex. Of course, direct injection of the vector may not be practical in clinical application. Although intraarterial or venous administration could be more suitable for human ischemic diseases, there may be great difficulty in delivering the vector specifically to the ischemic area. When these problems are successfully resolved, gene therapy could have great potential for stroke therapy in the future.
Footnotes
Acknowledgments:
The authors thank Dr. Warita and Dr. Hayashi for their advice and technical support.