
Abstract
Functional alterations of barium-sensitive potassium inward rectifier (Kir) current, which is involved in the vasodilation of middle cerebral arteries (MCA) in rat brain, have been described during brain ischemiaireperfusion (I/R). The authors investigate the effects of I/R on Kir current recorded in isolated myocytes from MCA of control rats and from contralateral and ipsilateral MCA of ischemic rats by the whole-cell patch-clamp technique, and the relationship between its alteration and The severity of brain injury. The vascular smooth muscle cells exhibited similar morphologic features in all conditions, and the Kir was present in the three groups of myocytes, exhibiting a characteristic inward rectification and a normal external potassium dependence. The Kir density was significantly reduced in cell of MCA ipsilateral to occlusion with a maximum at −135 mV, whereas there was no difference between control and contralateral cells. This alteration in Kir density in occluded MCA was significantly correlated with severity of brain injury and brain edema. These results suggest that the alteration of Kir density in MCA myocytes after I/R and the consecutive impaired dilation of MCA may contribute to aggravation of the brain injury.
Keywords
The vascular tone in cerebral circulation is governed by different physiologic mechanisms that may be relevant to the endothelium. One of the mechanisms regulated by the interactions between the endothelium and the myocytes are the potassium (K+) channels present in the vascular smooth muscle cell membrane. Their activation or their inhibition checks the vasodilation or the vasoconstriction of the cerebral vessels (Faraci and Heistad, 1998). Four different types of K+ channels have been identified in arterial smooth muscle: (1) voltage-dependent K+ channels; (2) calcium-activated K+ channels; (3) ATP-sensitive K+ channels; and (4) inward rectifier K+ (Kir) channels (Nelson and Quayle, 1995).
The Kir current was first observed in vascular smooth muscle cells (VSMC) from rat resistance-sized cerebral arteries (Quayle et al., 1993), but more generally, the Kir channel is expressed in small arteries and in arterioles (Quayle et al., 1997). The Kir current is characterized by a strong inward rectification, a specific block by low external concentration of BaCl2, and an external K+ dependence of the conductance. The Kir channel plays an essential role in the vasodilation of small cerebral arteries in response to a rise of the external K+ concentration ([KC1]0). Thus, contrary to larger arteries, the small cerebral arteries hyperpolarize and dilate if extracellular K+ increases. During neuronal activity, the extracellular K+ concentration increases, producing a high, local K+ concentration near the blood vessels (Paulson and Newman, 1987). The activation of Kir channels increases and induces the repolarization of the membrane potential, inducing closure of calcium channels and then relaxation of the cerebral vessels. The Kir channels represent a physiologic intermediate between cerebral metabolic activity and CBF. Thus, K+ ions have been proposed to constitute an endothelium-derived hyperpolarizing factor (Edwards et al., 1998).
After focal cerebral ischemia/reperfusion (I/R) in rats, Marelli et al. (1998) demonstrated an attenuated vasodilation response of the occluded middle cerebral artery (MCA) to an increased external K+ concentration. Moreover, the application of BaCl2 has no significant effect on occluded MCA dilation, whereas it is significantly reduced on control MCA, suggesting an impaired function of Kir channels after I/R. Because of its potential importance in the control of CBF, it has been proposed that impairment of its function is likely to exacerbate the brain injury induced by I/R, but this hypothesis remains to be demonstrated. Our study highlights, by electrophysiologic patch-clamp studies, the effect of Kir current on rat MCA smooth muscle cells after I/R compared with normal conditions and studies its potential relationship to the severity of brain injury.
MATERIALS AND METHODS
Male Wistar rats weighing 280 to 300 g were used; all experiments were performed in strict accordance with guidelines of the National Institutes of Health and the French Department of Agriculture.
Focal ischemia/reperfusion model
Anesthesia was induced with chloral hydrate administered intraperitoneally at a dose of 300 mg • kg−1. A rectal probe was inserted, and core temperature was maintained with a heating pad and heating lamp at 37° ± 0.5°C. The caudal artery was exposed and cannulated with a 24G polyethylene catheter and connected to a blood pressure monitor. The MABP (mm Hg) was monitored throughout the experiment, and blood samples were taken before, during, and after ischemia to measure blood pH, Pao2 (mm Hg), and Paco2 (mm Hg).
The ostium of the right MCA was occluded intraluminally with a modified method previously described (Zea-Longa et al., 1989; Sydserff et al., 1995). The right carotid arteries were exposed through a midline cervical incision, and the common carotid and external carotid arteries were ligated with a silk suture. The pterygopalatine artery was exposed by developing a plane alongside the internal carotid artery and was ligated at its origin with fine silk. An aneurysm clip was placed across the internal carotid artery, and an arteriotomy was made in the common carotid artery stump, allowing the introduction of a 4–0 monofilament nylon suture with its tip rounded by flame heating. This was secured in place, and the aneurysm clip on the internal carotid artery was removed. The suture was gently advanced into the internal carotid artery and passed into the intracranial circulation to lodge in the narrower lumen of the proximal anterior cerebral artery (20 to 22 mm distal to the carotid bifurcation), thereby occluding the origin of the MCA. Mild resistance to this advancement indicated that the intraluminal occluder had entered the anterior cerebral artery. After 60 minutes, the suture was carefully removed, until its tip was blocked by ligature placed on common carotid artery, to permit reperfusion. The caudal arterial catheter was removed, and the artery was ligated to prevent bleeding. The animals were placed in a cage to recover from anesthesia at room temperature and were allowed to eat and drink freely.
Histology
Twenty-four hours after reperfusion, the rats were killed by an overdose of pentobarbital injected intraperitoneally, and brains were rapidly removed. The MCA were carefully dissected to the VSMC preparation, and thereafter brains were frozen and coronally sectioned into 50-μm thick slices on a cryostat at 12 levels separated by 1-mm intervals according to stereotactic sections maps (Paxinos and Watson, 1986). Sections were stained with cresyl fast violet. The unstained area of the brain sections was defined as infarcted. Cortical and subcortical infarcted areas and total hemispheric areas were calculated separately for each coronal slice by image analysis software (Color Image 1.32, National Institute of Mental Health, Bethesda, MD, U.S.A.) after digitization by a scanner process. Total, cortical, subcortical infarct volumes, and hemispheric volumes (in cubic millimeters) were calculated using numerical integration of the respective areas for all of the sections and the distance between them. A corrected total infarct volume was calculated to compensate for the effect of brain edema (Lin et al., 1993). The corrected volume was calculated using the following equation: corrected infarct volume = total infarct volume − (right hemisphere volume − left hemisphere volume). The edema volume was represented by the difference between right and left hemisphere volumes. The infarct volume quantification served as an index of brain injury level.
Cell preparation
All experiments were done on freshly dissociated cerebral VSMC obtained from MCA contralateral (I/R contralateral) and ipsilateral (I/R ipsilateral) to occlusion in ischemic rats and from MCA of control animals. The MCA myocytes were obtained by an enzymatic procedure described by Holland et al. (1996). Briefly, after dissection, the MCA were placed in an ice-cold buffer containing (in mmol/L) the following: 136 NaCl, 5.6 KCl, 4.17 NaHCO3, 2.6 CaCl2, 10 HEPES, 1 MgCl2, 0.44 NaH2PO4, 0.42 Na2HPO4, and 10 glucose, pH 7.4. They were transferred to the same buffer, except for the lower calcium (0.1 mmol/L) concentration, for 10 minutes. The artery then was exposed to two successive low-calcium buffer baths at 37°C containing (1) papain (1.5 mg/mL), dithiothreitol (1 mg/ mL), and bovine serum albumin (1 mg/mL) for 30 minutes; and (2) collagenase F (1.5 mg/mL), hyaluronidase type I-S (1 mg/ mL), and bovine serum albumin (1 mg/mL) for 10 minutes. Finally, the enzymatic bath was replaced by fresh low-calcium buffer containing 1 mg/mL bovine serum albumin. The single cells were obtained by gentle trituration with a wide-bore pipette. Cells were used within 7 hours.
Data recordings
Cells were placed in Petri dishes mounted on the stage of a phase-contrast inverted microscope. The whole-cell patch-clamp technique (Hamill et al., 1981) was used to evaluate Kir current by using a RK 400 patch-clamp amplifier (Bio-Logic, Claix, France) connected to a microcomputer equipped with pClamp 5.5 software (Axon Instruments, Inc., Foster City, CA, U.S.A.). The currents were low-pass filtered at 3 kHz and digitized at 10 kHz using a 12-bit analog-to-digital converter (Lab-master, TL-1, Scientific Solutions, Inc., Solon, OH, U.S.A.). The membrane capacitance of the cells was measured by integrating the area of capacitive transient elicited by a 5-mV depolarizing pulse from a holding potential of −50 mV. The pipette capacitance was not substrated from membrane cell capacitance. The pipettes were pulled from thin borosilicate glass capillaries (Clark Electromedical Instruments, Reading, U.K.) and had a resistance of 2.7 to 3.3 Mω; when they are filled with the pipette solution containing (in mmol/L): 130 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, and 5 creatine phosphate, pH 7.2. The bath solution contained (in mmol/L) 134/80 NaCl, 6/60 KCl, 1 MgCl2, 0.1 CaCl2, and 10 HEPES, pH 7.4. In some experiments, the [KCl]0 was raised to 60 mmol/L and the NaCl concentration was decreased to 80 mmol/L, so that NaCl + KCl = 140 mmol/L. Control and test solutions were applied to the exterior of the cell by placing the cell at the opening of 300-μm inner diameter catheters fixed on the motorized rotating head of a RSC 200 (Rapid Solution Changer, Bio-Logic, Claix, France). All chemicals were obtained from Sigma-Aldrich (Saint Quentin Fallavier, France). All experiments were done at room temperature (19° to 22°C).
Data analysis
The standard voltage clamp protocol consisted of 185-millisecond voltage ramps from −140 to +50 mV, from a holding potential of −60 mV. In each experiment, currents in response to four voltage ramps were averaged to give the final traces displayed in this work. The current density was calculated and preferentially used in this study to compare the different cells to eliminate variation of the cell size. To compare Kir density between the three groups, we extracted the density values at 14 determined potential levels from the current trace elicited by the ramp potential. The alteration of Kir density after I/R was estimated by the difference between the mean Kir control density (at 6 or 60 mmol/L KCl) and the Kir density value obtained for each ipsilateral cell.
Statistical analysis
Mean values ± SD were calculated. The whole Kir density values between the three experimental cell groups were compared by repeated-measures analysis of variance and a post hoc protected least significant difference Fischer test. The Kir density values at each different potential level (from −135 to −5 mV, 10-mV increment) then were compared between the three groups by one-way analysis of variance followed by a post hoc protected least significant difference Fischer test. To determine the relationship between infarct volumes (total infarct volume, subcortical infarct volume, cortical infarct volume, edema volume, and total infarct volume corrected from the edema volume) and alteration of current density, linear regression was used. For all statistical analyses, significance was assumed if P < 0.05.
RESULTS
The patch-clamp experiments have been performed on myocytes originated from MCA of 12 control and 10 ischemic rats. Cells from control, contralateral, and ipsilateral MCA exhibited the same morphologic aspect (Fig. 1), with lengths typically ranging from 60 to 80 μm and diameters from 5 to 10 μm. The capacitance values were not different among the different groups of cells: 19.53 ± 3.00 pF (n = 16) for control cells, 19.65 ± 1.79 pF (n = 8) for I/R contralateral cells, and 19.38 ± 2.41 pF (n = 19) for I/R ipsilateral cells.
Microphotographs of freshly enzymatically dissociated vascular smooth muscle cells obtained from control middle cerebral artery (MCA)
Characterization of potassium inward rectifier current
The transmembrane currents were measured on isolated VSMC by using the whole-cell configuration of the patch-clamp technique. Four successive potential ramps were applied from a holding potential of −60 mV (Fig. 2A) to obtain the typical averaged current trace illustrated in Fig. 2B (control trace). In this example, external [KC1]0 is 60 mmol/L, giving a K+ equilibrium potential (EK+) of −19.5 mV according to the Nernst equation. For potentials negative to EK+, an inward current is observed, which met the voltage axis close to EK+ and flowed in the outward direction for potentials positive to EK+. When a solution containing 0.5 mmol/L BaCl2 (this concentration being 10 times the half-block constant of the Kir current at 0 mV [Quayle et al., 1993], is perfused on the cell, the inward current is significantly reduced, whereas the outward one is affected only slightly. For potentials positive to EK+, the current is mainly carried through voltage-dependent K+ channels, since it is blocked by extracellular application of 4-aminopyridine (data not shown). To accurately evaluate the Kir component on the different population of cells (i.e., control, I/R contralateral, I/R ipsilateral), a BaCl2 solution was systematically applied in each experiment to completely inhibit the Kir current. Then, the residual barium-insensitive current was subtracted from the total one to unmask the Kir component (lower trace); its characteristic inward rectification is illustrated in Fig. 2C. The same kind of experiment also was realized in a 6 mmol/L external KCl concentration. A negative shift of the reversal potential then was observed, with a current that reversed at −73.53 ±3.22 mV (n = 4), close to the predicted K+ equilibrium (−77.7 mV), as well as a decrease of the Kir current density from −2.31 ± 0.72 pA/pF (n = 8) in 60 mmol/L [KC1]0 to −1.30 ± 0.46 pA/pF (n = 8) in 6 mmol/L [KC1]0 at −135 mV in control cells. In both [KC1]0, for the potentials positive to EK+, outward currents were small, in the order of few pA.
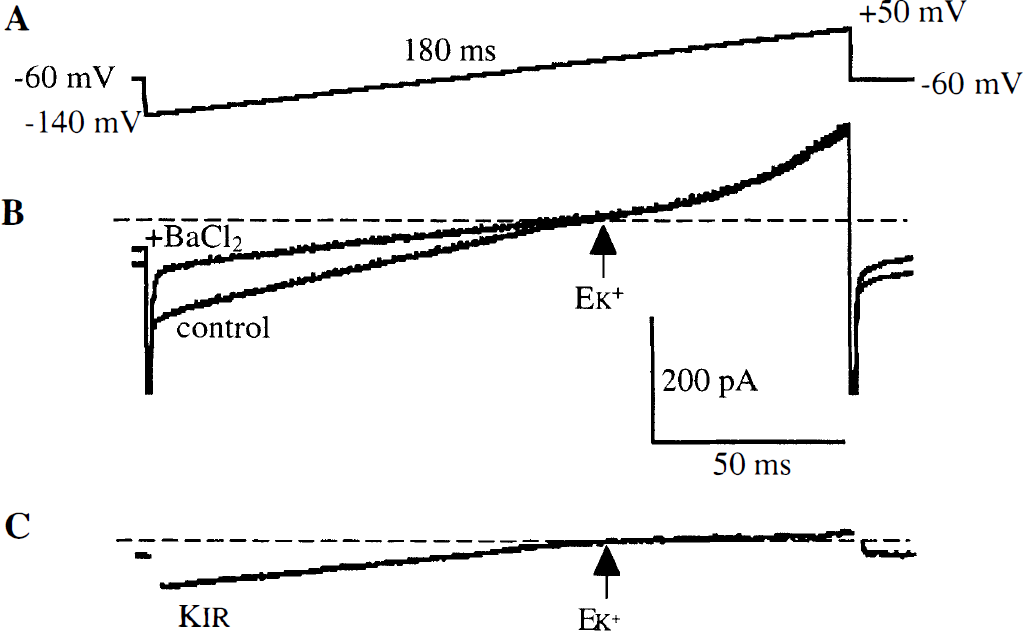
Measurement of potassium inward rectifier (Kir) amplitude in MCA.
Effect of ischemia on potassium inward rectifier current density
After application of the same protocol on cells originated from the MCA of ischemic rats, a Kir component showing inward rectification could be recorded in I/R contralateral and I/R ipsilateral cells. Examples of Kir current in control and I/R ipsilateral cells in 6 mmol/L KCl are shown in Figs. 3A and 3B. The mean Kir normalized for cell capacitance recorded in 6 mmol/L or 60 mmol/L [KC1]0 in the three experimental groups of cells are represented in Figs. 3C and 3D. Although the cell capacitance was similar in the three groups of myocytes, there was a significant (P < 0.005) reduction of the Kir density in I/R ipsilateral cells compared with control and I/R contralateral cells, regardless of the potential level in 6 as well as in 60 mmol/L [KC1]0.
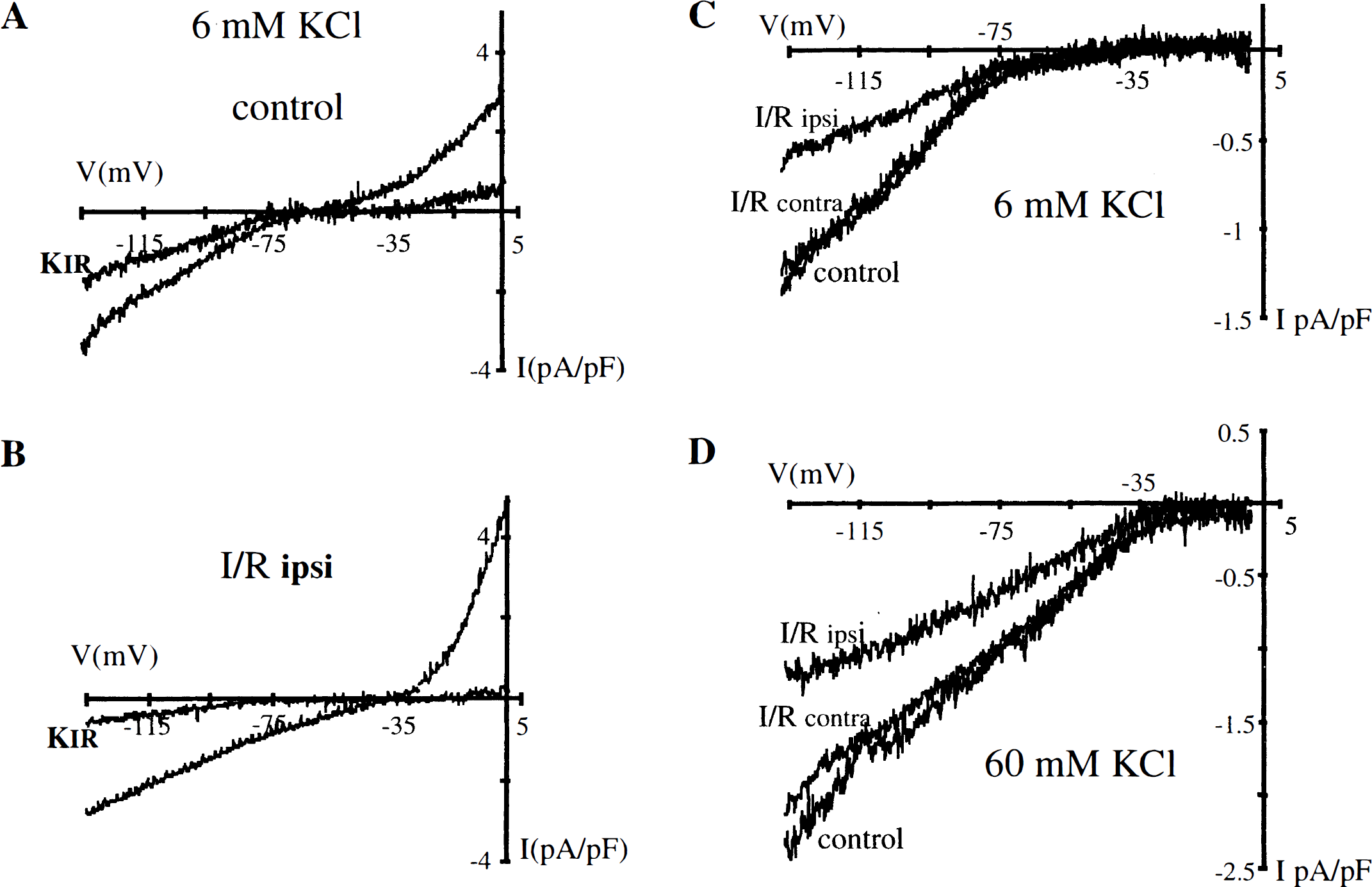
The potassium inward rectifier (Kir) density (pA/pF) in control, ischemia/reperfusion (I/R) contralateral, and I/R ipsilateral cells.
The means of Kir densities obtained at each of 14 potential levels (from −135 to −5 mV) have been plotted for the three groups of cells in Fig. 4, for 6 mmol/L [KC1]0 in Fig. 4a and for 60 mmol/L [KC1]0 in Fig. 4B. In both conditions, Kir density in I/R ipsilateral cells was significantly (P < 0.005 in 6 mmol/L [KC1]0, P < 0.05 in 60 mmol/L [KC1]0) different from that in control cells for potentials negative to EK+. In 6 mmol/L [KC1]0, this significant difference also was present between I/R ipsilateral and I/R contralateral cells. For potentials positive to EK+, the difference between the three cell populations was not statistically significant, although a decrease of Kir density was observed at each potential level for I/R ipsilateral cells compared with control and I/R contralateral cells. In this range of potentials, Kir outward amplitudes were small (5 pA or less). Whole-cell currents of so small an amplitude are difficult to discern from substraction of two current traces elicited by voltage ramps. Accurate measurements were more difficult to realize because of a low signal-to-noise ratio, which distorts the traces and smoothes the differences. The maximal Kir density alteration was observed at −135 mV; the mean Kir density was −1.30 ± 0.46 pA/pF (n = 8) in control versus −0.57 ± 0.38 pA/pF (n = 12) in I/R ipsilateral cells in 6 mmol/L [KC1]0 (P < 0.005) and −2.32 ± 0.72 pA/pF (n = 8) in control versus −1.31 ± 0.47 pA/pF (n = 7) in I/R ipsilateral cells in 60 mmol/L [KC1]0 (P < 0.05). In I/R contralateral cells, at −135 mV, Kir density was −1.19 ± 0.21 pA/pF (n = 4) and −2.09 ± 0.81 pA/pF (n = 4) at 6 and 60 mmol/L [KC1]0, respectively.
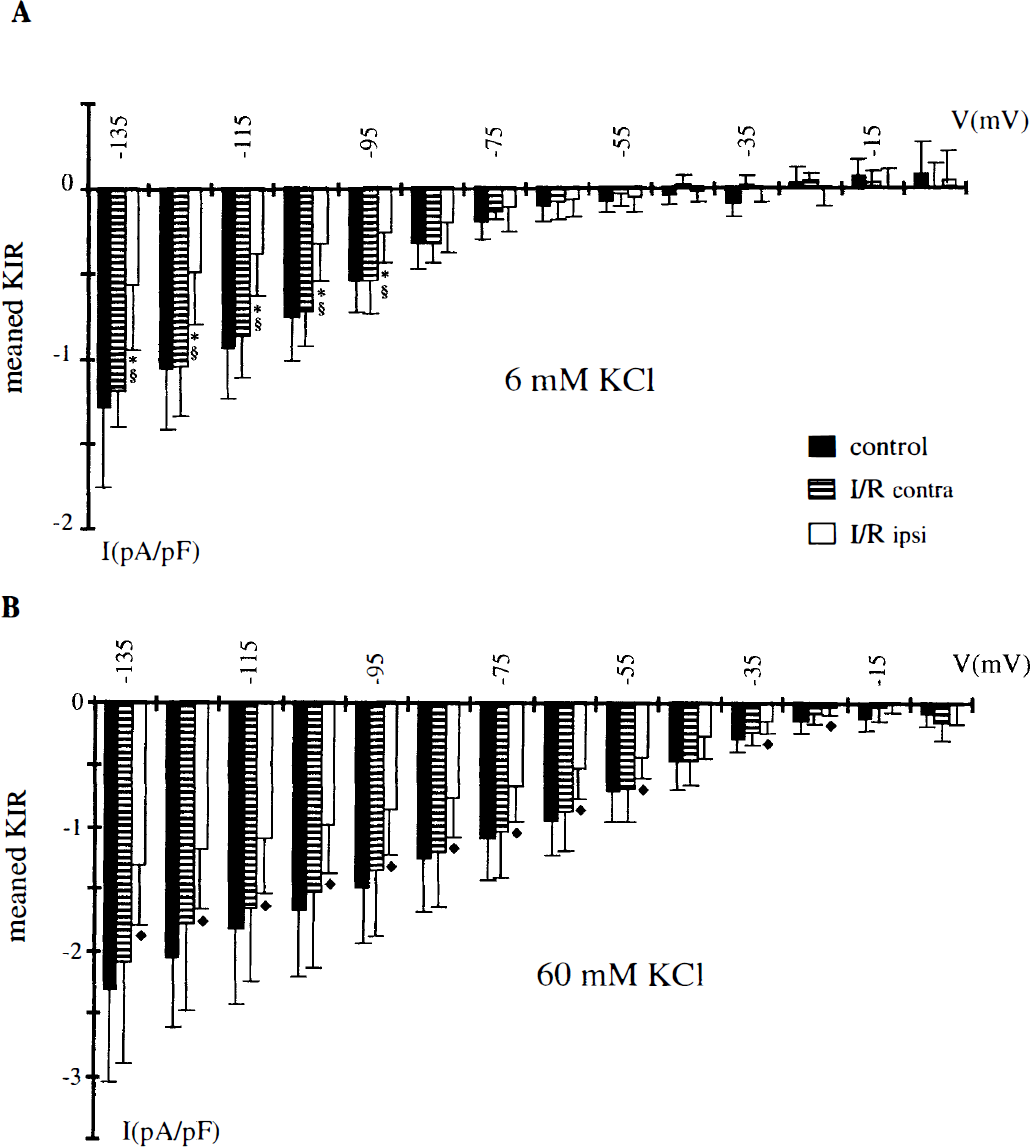
Summary of the averaged potassium inward rectifier (Kir) densities obtained in 6 mmol/L
Relationship between the alteration of potassium inward rectifier current density and infarct volume
The decrease of Kir density in I/R ipsilateral cells at −135 mV was an index of Kir impairment for correlation with brain injury. During ischemia, the main recorded physiologic parameters remained stable. One-hour MCA occlusion induced cortical and subcortical infarcts ipsilaterally to intraluminal occlusion in the 10 animals used in this study, whereas there were no contralateral lesions. The volumes of infarct are summarized in Table 1. There is a significant positive correlation between alteration of Kir density in I/R ipsilateral cells (expressed as the difference between the mean Kir control density and each Kir I/R ipsilateral density in 6 or 60 mmol/L [KC1]0) and the volumes of total infarct (r = 0.85, P < 0.001), cortical infarct (r = 0.82, P < 0.005), subcortical infarct (r = 0.88, P < 0.0005), brain edema (r = 0.87, P < 0.0005), and total infarct corrected for edema (r = 0.71, P < 0.05, Fig. 5).
Quantification of infarct and edema volumes

Values are mean ± SD. Volumes are expressed in mm3.
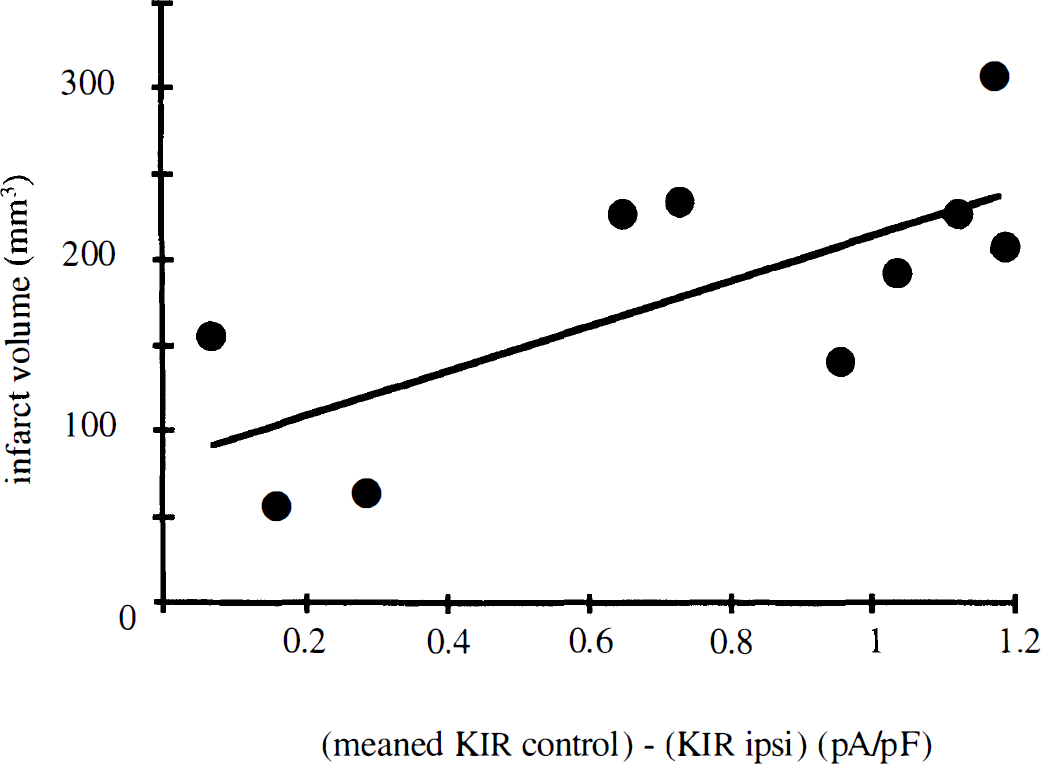
Relationship between the total infarct volume corrected from edema volume and the alteration of potassium inward rectifier (Kir) densities (at −135 mV) in I/R ipsilateral myocytes. Infarct volumes (mm3) of each brain ischemic rat (n = 10) are plotted versus the difference between the mean control Kir density and Kir density measured in I/R ipsilateral myocytes. Results obtained in 6 or 60 mmol/L [KCI]0 according to the cell. Solid line represents first-order linear regression (Y = 131.52X + 82.64; correlation coefficient r = 0.71; P < 0.05).
DISCUSSION
In this study, we examined the effects of I/R on the current density of Kir on freshly isolated myocytes obtained from MCA of control rats and from ipsilateral and contralateral MCA of ischemic 24-hour reperfused rats. Several main conclusions can be drawn from our experiments: (1) Kir shows its characteristic inward rectification and external K+ sensitivity in each cell group; (2) Kir density is similar in control cells and in I/R contralateral cells; (3) Kir density is significantly reduced in I/R ipsilateral cells; and (4) Kir alteration is positively correlated to the brain injury. These results agree with an impaired function of Kir channels after I/R and suggest a relationship between this modified Kir channel activity and the development of the brain infarct.
Involvement of Kir in pathophysiologic mechanism of brain ischemia has been suspected by Marelli et al. (1998). Using vasomotion experiments, they demonstrated that vasodilation consecutive to an increase of [KC1]0 was attenuated on rat ischemic MCA, with a lack of effect of BaCl2 compared with normal MCA, suggesting that Kir stimulation was significantly impaired after I/R. We found, with a direct electrophysiologic approach, that the density of Kir was decreased in VSMC of MCA after I/R, compared with contralateral or control MCA. The decrease in vasodilation response to change in [KC1]0 could be so explained by a decrease in Kir density induced by I/R. A link between this altered Kir channel function and the severity of cerebral ischemia has been previously suspected because of physiologic role of Kir in CBF regulation but has not been demonstrated directly.
In the last few years, the preponderant role of Kir channels in vasodilation of resistance-sized cerebral arteries has emerged from different investigations (McCarron and Halpern, 1990a; Quayle et al., 1993; McPherson and Keily, 1995; Knot et al., 1996; Johnson et al., 1998). Small cerebral arteries have been shown to be extremely sensitive to changes in [KC1]0, increasing in diameter when external [KC1]0 increases in response to neuronal metabolic activity (Paulson and Newman, 1987). These dilations are insensitive to blockers of the calcium-activated K+ channels (tetraethylammonium), voltage-dependent K+ channels (4-aminopyridine), ATP-sensitive K+ channels (glibenclamide), and to the removal of endothelium (Knot et al., 1996). Below 5 mmol/L KCl, an ouabain-sensitive transient dilation is observed, resulting from the Na/K-ATPase stimulation; above 5 mmol/L KCl, an ouabain- and barium-sensitive sustained dilation occurred, caused by an increase of outward Kir current (McCarron and Halpern, 1990a). In the normal physiologic situation, the membrane potential of the VSMC is positive to EK+, and Kir current flows through the channels in the outward direction, contributing to the maintain of a relatively hyperpolarized membrane potential. This induces a closure of calcium channels and a relaxation of the vessels.
We demonstrated here that there was a positive correlation between alteration of Kir current and brain injury. The alteration of Kir current could contribute to the extent of neuronal necrosis, as suggested by the correlation with the size of infarct volume after correction for edema. The alteration of Kir channels induces the loss of the K+-sensitive dilation property and prevents a normal vasodilation. Because there is a link between Kir function and the regulation of CBF to neuronal metabolism, Kir alteration could be responsible for the uncoupling of flow and metabolism after ischemia (Ginsberg, 1990). This alteration of CBF related to Kir impairment might explain that cerebral circulation may not be able to respond to the increase of brain metabolism, in particular, in the ischemic penumbra area, where the neuronal damages are not definitive and can be reversed by an early reperfusion. Indeed, reduction in cerebral vasodilation could prevent a reperfusion sufficient to minimize the neuronal necrosis. Moreover, Kir channel alteration might contribute to change in vascular permeability, as suggested by the positive correlation between Kir density decrease and importance of brain edema. More studies are needed to explore this hypothesis.
These results concerning Kir alteration in VSMC after I/R support the hypothesis of the involvement of cerebral vessel wall impairment in the physiopathologic mechanism of brain ischemia. Whereas functions of smooth muscle layers could be involved in I/R physiopathologic pathways, functional interrelationships between endothelium and VSMC also could be altered (Bari et al., 1996). Using vasomotion experiments, it has been shown that myogenic properties, as well as endothelial function of MCA, were impaired after I/R contributing to brain injury (Cipolla et al., 1997). Vascular abnormalities (alteration of vascular anastomoses, VSMC disorganization, endothelial dysfunction) also have been thought to explain the increased susceptibility to cerebral ischemia in spontaneously hypertensive rat strains, as proved the larger size of experimentally induced brain infarct than in the normotensive rat (Gratton et al., 1998). Because the K+-induced, barium-sensitive dilation has been reported to be absent in spontaneously hypertensive rat cerebral arteries, this alteration of Kir current could be one of the mechanisms explaining the increase of susceptibility to brain ischemia, underlining again the role of Kir channels in the physiopathologic mechanism and the severity of I/R (McCarron and Halpern, 1990b).
The known physiologic regulations of Kir channels consist in variations of external K+ concentration and variations of passive wall tension, which modify Kir channels activation, and in variations of intracellular magnesium and polyamine concentrations responsible of the inward rectification (McPherson and Keily, 1995; Johnson, 1996). The drastic cellular modifications that occurred after ischemia activity, like the ionic gradient alterations or the disturbed polyamines metabolism, might influence the electrophysiologic characteristics of Kir channel. For example, neuronal polyamine synthesis was increased during I/R, and polyamines participate in the setting of brain injury. Reperfusion injury also has been involved in development of MCA endothelial dysfunction during I/R, as proved by the lack of effect of ischemia alone on endothelium function, whereas a dysfunction developed after 24 hours of reperfusion (Cipolla et al., 1997). The release of free radicals is one of the first events occurring after reperfusion. Free radicals are synthesized from oxygen reactive species by different enzymatic systems in smooth muscle cells and in endothelial cells, and are strongly vasoactive. They are known to interfere with numerous cellular transduction signals and to react with many target molecules. Alteration of Kir channels also could be cause by free radical actions, since it was reported for the impaired function of ATP-sensitive K+ channels, which participate in the abolition of calcitonin gene-related peptide-induced vasodilation of MCA after I/R (Bari et al., 1996; Louis et al., 1996). At the ionic channel level, particularly the K+ transporting one, different in vitro experimental results show that free radicals applied to cerebral vessels are able to modulate the activity of the calcium-activated K+ channels by an increase of intracellular calcium concentration because of a blockade of calcium extrusion systems, and the ATP-sensitive K+ channels by a decrease of ATP concentration or oxidation of the channel proteins (Wei et al., 1996). An action of free radicals on Kir channel protein itself or on its regulation might be considered.
In conclusion, it is widely admitted that the faster the CBF is restored, the smaller the subsequent neuronal dysfunction (Pulsinelli, 1992). Hence, the K+ channels of the cerebral vascular bed constitutes promising pharmacologic targets for the treatment of the I/R pathologic mechanism. Nevertheless, it remains to be demonstrated whether the reduction of Kir density results from modification of the single-channel Kir characteristics or from a down-regulation of Kir channel protein expression. Further experiments also are necessary to achieve a better understanding of the involvement of Kir channel dysfunction in the cascade of pathophysiologic events occurring during I/R. Treatment that minimizes or reverses Kir channel dysfunction will be helpful in improving CBF, which will provide a better answer to the metabolic needs of neuronal ischemic cells. A successful outcome in the prevention of Kir alteration, together with the resulting neuroprotective effect, would confirm the involvement of Kir in the physiopathologic mechanism of I/R.
Footnotes
Acknowledgments:
The authors thank Dr. Bruno Bastide for helpful discussions, and Mrs. S. Duriez and N. Pruvost for secretarial assistance.