
Abstract
Brain reperfusion causes prompt, severe, and prolonged protein synthesis suppression and increased phosphorylation of eukaryotic initiation factor 2α [eIF2α(P)] in hippocampal CA1 and hilar neurons, The authors hypothesized that eIF2α(P) dephosphorylation would lead to recovery of protein synthesis. Here the effects of insulin, which activates phosphatases, were examined by immunostaining for eIF2α(P) and autoradiography of in vivo 35S amino acid incorporation. Rats resuscitated from a 10-minute cardiac arrest were given 0, 2, 10 or 20U/kg of intravenous insulin, underwent reperfusion for 90 minutes, and were perfusion fixed. Thirty minutes before perfusion fixation, control and resuscitated animals received 500 μCi/kg of 35S methionine/cysteine. Alternate 30-μm brain sections were autoradiographed or immunostained for eIF2α(P). Controls had abundant protein synthesis and no eIF2α(P) in hippocampal neurons. Untreated reperfused neurons in the CA1, hilus, and dentate gyrus had intense staining for eIF2α(P) and reduced protein synthesis; there was little improvement with treatment with 2 or 10 U/kg of insulin. However, with 20 U/kg of insulin, these neurons recovered protein synthesis and were free of eIF2α(P). These results show that the suppression of protein synthesis in the reperfused brain is reversible; they support a causal association between eIF2α(P) and inhibition of protein synthesis, and suggest a mechanism for the neuroprotective effects of insulin.
Reperfusion after global brain ischemia results in a prompt inhibition of translation initiation (Cooper et al., 1977; DeGracia et al., 1996) and suppression of protein synthesis that is especially severe and prolonged in the neurons of the CA1 and hilar regions of the hippocampus. It is generally accepted that this depression of protein synthesis results from inhibition of translation initiation (reviewed in Krause and Tiffany, 1993). Initiation, generally believed to be the rate limiting-step in translation, is coordinated by eukaryotic initiation factors (eIF) (Merrick and Hershey, 1996) that orchestrate the assembly of the ribosomal subunits, the mRNA to be translated, and the initiator methionyl-tRNA (Met-tRNAi). During translation initiation, Met-tRNAi is joined to the ribosome in a complex with eIF2 and GTP. At a later stage of initiation, this GTP is hydrolyzed, and before eIF2 can participate in another round of translation initiation, an exchange of GDP for GTP must be carried out by eIF2B (guanine nucleotide exchange factor) (Rowlands et al., 1988).
Under conditions of cellular stress (e.g., heat shock, viral infection, starvation), rates of global protein synthesis are down-regulated by phosphorylating Ser51 on the α subunit of eIF2 (Redpath and Proud, 1994; Pain, 1996). Phosphorylated eIF2α [eIF2α(P)] competitively inhibits the eIF2B-catalyzed GDP–GTP exchange reaction (Rowlands et al., 1988). Because the ratio of eIF2 to eIF2B is approximately 5:1 in the brain (Alcazar et al., 1995), translation initiation will be nearly completely inhibited when approximately 20% of eIF2 contains a phosphorylated α-subunit. Hu and Wieloch (1993) found that formation of the eIF2/GTP/Met-tRNAi complex was diminished after reperfusion, and Burda et al. (1994) report enhanced phosphorylation of eIF2α by 30 minutes of brain reperfusion. Previously, we have shown that after 10 minutes' reperfusion, there is an approximately 20-fold increase in eIF2α(P) (DeGracia et al., 1996) so that it represents about 23% of total brain eIF2α, and this reperfusion-induced eIF2α(P) is localized to vulnerable neurons between 1 and 4 hours of reperfusion (DeGracia et al., 1997). These observations suggest that eIF2α(P) is likely to be a major cause for the inhibition of protein synthesis seen in vulnerable neurons during postischemic reperfusion.
Insulin has previously been shown to affect several aspects of the translation initiation system other than eIF2α phosphorylation (Proud and Denton, 1997), and in general these insulin-induced modifications increase protein synthesis. In addition, one study suggests that in calf chondrocytes, insulin reduced eIF2α(P) (Towle et al., 1984). Furthermore, several studies show that administration of insulin during reperfusion substantially reduces both neuronal death and the final extent of neurologic disability (LeMay et al., 1988; Voll et al., 1989; Strong et al., 1990; Shigeno et al., 1991; Voll and Auer, 1991b) independent of hypoglycemia (Voll and Auer, 1991a). Thus, we hypothesize that reduction of the levels of eIF2α(P) would enhance protein synthesis in vulnerable hippocampal neurons during reperfusion, and that this might be accomplished by insulin administration. Therefore, we used immunostaining for eIF2α(P) and autoradiography of in vivo 35S amino acid incorporation to investigate the effect of insulin on this initiation factor and protein synthesis in hippocampal neurons during reperfusion.
MATERIALS AND METHODS
All animal experiments were approved by our institutional review board and were conducted following the Guide for the Care and Use of Laboratory Animals (NIH publication #86-23, revised 1985). Male Long Evans rats weighing 425 to 500 g were anesthetized with ketamine and xylazine; instrumented for vascular access; monitored for arterial pressure, electrocardiogram, and core temperature; and subjected to 10 minutes of global brain ischemia by cardiac arrest followed by resuscitation and 90 minutes of reperfusion with postarrest intensive care as previously described (DeGracia et al., 1996). Nonischemic controls (n = 3) were instrumented in the same way. Core temperature was maintained between 36.5° and 37.5°C. When the animals resumed spontaneous perfusion, they received 0, 2, 10, or 20 U/kg of intravenous recombinant human insulin (Novo Nordisk Pharmaceuticals, Princeton, New Jersey, n = 3 each group). Arterial blood glucose was measured every 30 minutes, and intravenous injection of 50% dextrose was used as needed to maintain blood glucose above 100 mg/dL. Thirty minutes before perfusion fixation, all animals were given 500 μCi/kg intravenously of 35S methionine/cysteine labeling reagent (ICN Radiochemicals, Costa Mesa, CA, U.S.A.). The animals were perfusion fixed with 50% methanol/10% glacial acetic acid, and 30-μm frontal sections were prepared as described (DeGracia et al., 1997). Alternate slices were used for autoradiography and immunohistochemical study.
Immunohistochemical study was performed using our antibody specific for α-subunit phosphorylated eIF2 (DeGracia et al., 1997), and hippocampal photomontages were obtained with a ×40 lens. Sections from the same anatomical level in all brains were used for autoradiography and were incubated overnight in 50% ethanol and 0.5 mol/L sodium salicylate, transferred to gelatin-coated slides, dried, lightly stained with chrome alum/gallocyanin for 30 minutes, and air dried. The slides then were dipped in a 1:1 dilution of NTB2 Kodak silver emulsion in distilled H2O and sealed in a light-tight box with a desiccant for 60 days. The slides were developed for 2 minutes at 15°C in a Kodak developer, (Eastman Kodak, Rochester, N.Y., U.S.A.) rinsed in distilled H2O, placed in a Kodak fixer for 5 minutes, and then rinsed in distilled water. The slides then were serially dehydrated in ethanol (70% to 100%) and washed for 2 minutes twice in xylene; then coverslips were applied. Photomontages of the CAl, dorsal and ventral blades of the dentate gyrus (DGd and DGv, respectively), and hilus from each animal were obtained using a × 100 oil-immersion lens. To evaluate labeled amino acid incorporation, the “colony count” mode (default settings) of the Intelligent Quantifier program (BioImage, Ann Arbor, MI, U.S.A.) was used to count the autoradiographic silver grains in 120 × 96 μm areas of the CA1, DGd, DGv, and hilus on 1700× photomontages. Representative sections are shown in the figures.
RESULTS
Ten minutes of ischemia followed by 90 minutes of reperfusion produced the expected inhibition of protein synthesis, measured by 35S amino acid autoradiography, in all areas of the hippocampus examined (Fig. 1). Treatment at reperfusion with 2 U/kg of insulin did not induce improvement of protein synthesis by 90 minutes' reperfusion in any of these areas. There was a trend toward improved protein synthesis in the DGd and DGv after treatment with 10 U/kg of insulin. In contrast, treatment at reperfusion with 20 U/kg of insulin induced near-normal protein synthesis at 90 minutes' reperfusion in all areas examined (Fig. 1).
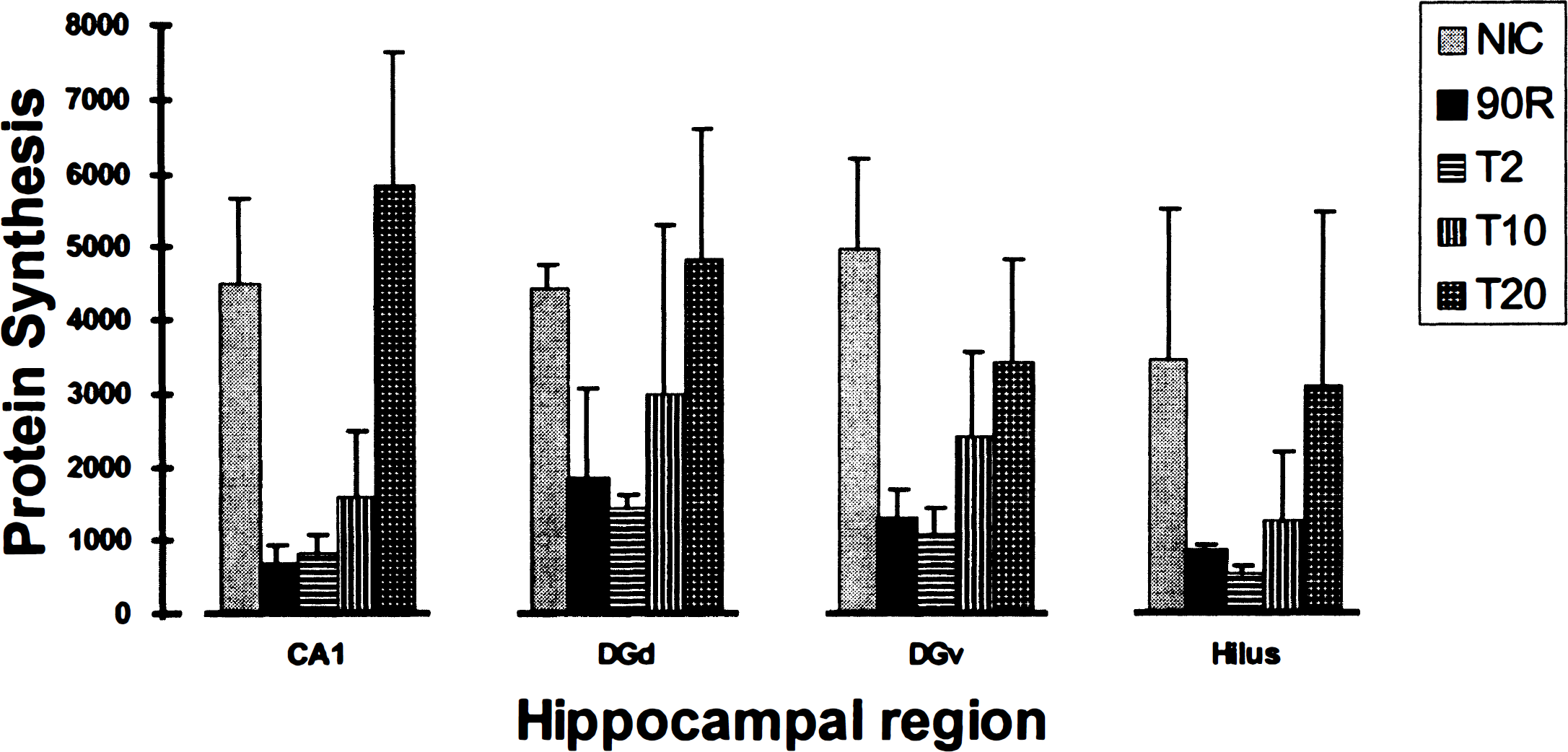
Protein synthesis. Comparison of protein synthesis in the hippocampal CA1, dorsal blade of the dentate gyrus (DGd), ventral blade of the dentate gyrus (DGv), and hilus from nonischemic controls (NIC) and rats reperfused 90 minutes after a 10-minute cardiac arrest and treated with 0 (90R), 2 (T2), 10 (T10), or 20 (T20) U/kg of intravenous insulin at resuscitation. Data are shown as mean ± SD. Protein synthesis was evaluated by counting silver grains in 11,520 μm2 areas from autoradiographs of brain sections obtained after in vivo incorporation of 35S-labeled amino acids during a 30-minute interval before perfusion fixation (see text).
Low-power micrographs showed that immunostaining for eIF2α(P) was negligible in nonischemic sections (Fig. 2, NIC). This contrasted with intense eIF2α(P) immunostaining in the CA1, DGd, DGv, and hilus of the untreated 90-minute reperfused group (Fig. 2, 90R). Treatment at reperfusion with 2 and 10 U/kg of intravenous insulin had little effect on eIF2α(P) immunostaining (not shown). However, 20 U/kg of intravenous insulin induced by 90 minutes' reperfusion essentially complete elimination of eIF2α(P) (Fig. 2, T20).
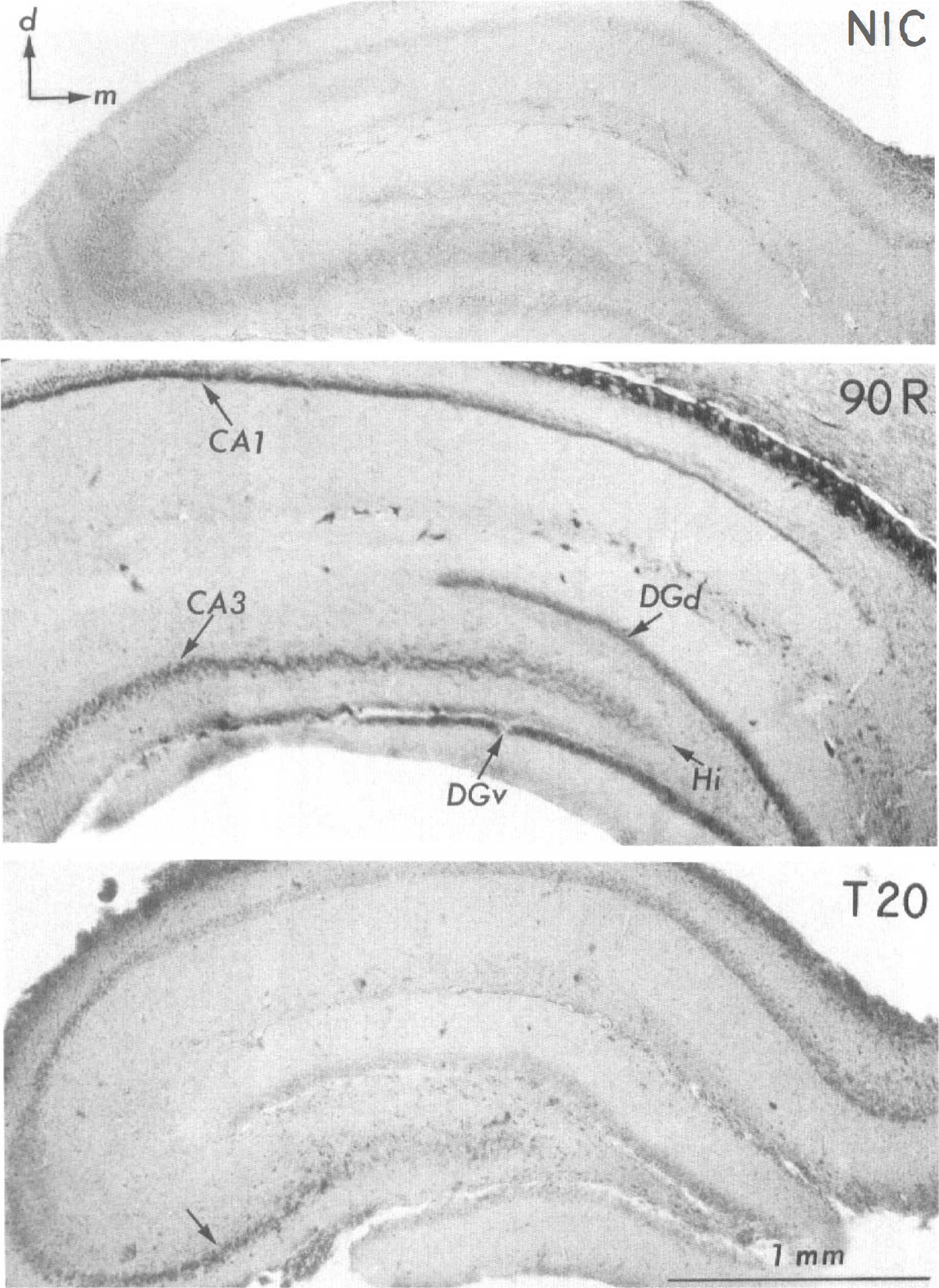
Phosphorylated eukaryotic initiation factor 2α [eIF2α(P)). Immunostaining for eIF2α(P) in low power (45×) photomicrographs of coronal sections through the hippocampal formation from the nonischemic control (NIC), 90-minute reperfused (90R), and the 20-U/kg insulin-treated 90-minute reperfused (T20) groups. The NIC section has no immunostaining, but the 90R section has deep staining of cell body layers, including the CA1 and CA3 sectors, hilar region (Hi), and the dorsal (DGd) and ventral (DGv) blades of the dentate gyrus. In the T20 section, little or no eIF2α(P) immunostaining is seen in the CA1 or dentate gyrus but persists in a portion of the CA3 sector (arrow). Orthogonal arrows in the NIC section indicate dorsal (d) and medial (m) directions for the three sections. Scale bar, 1 mm.
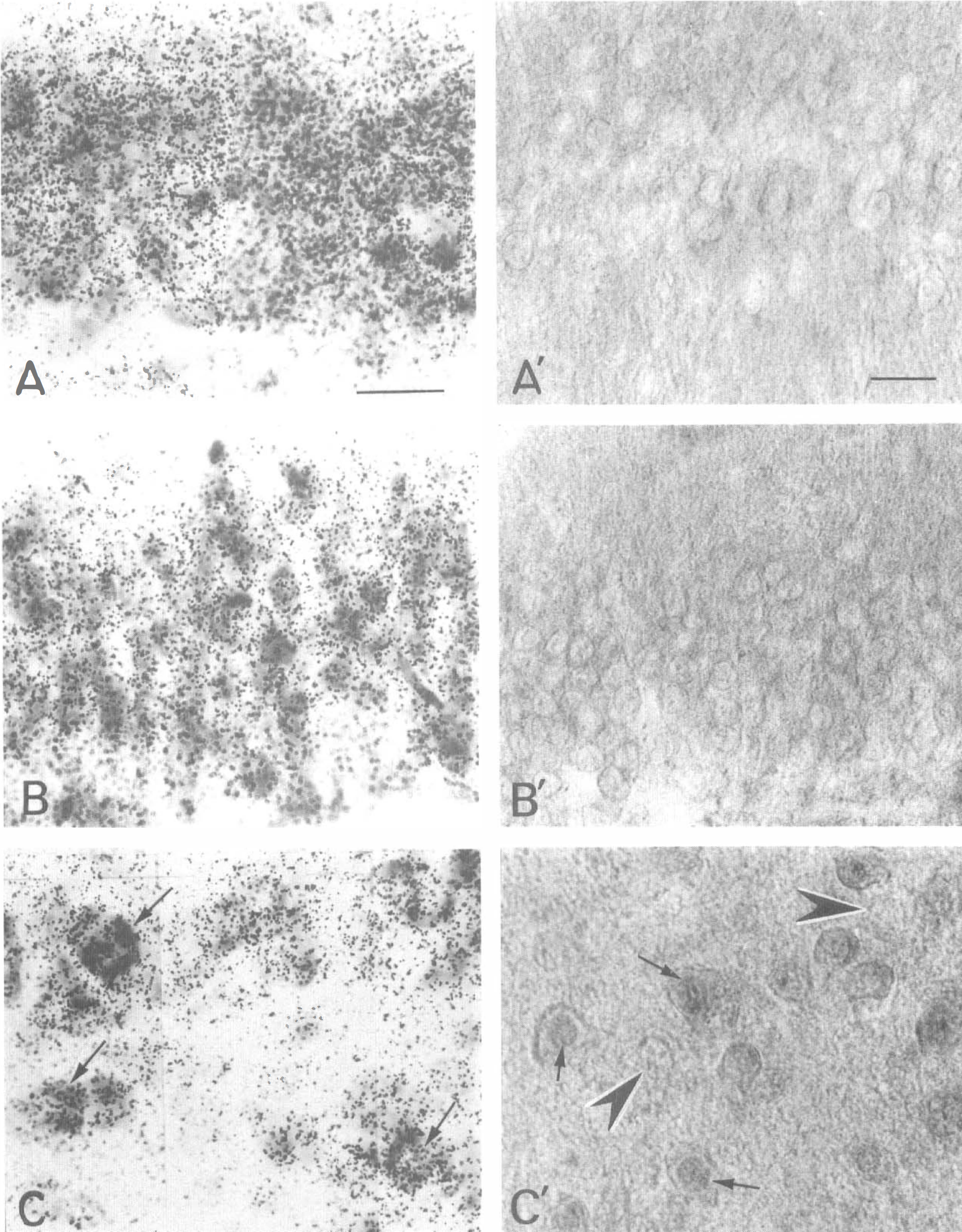
At higher magnification, nonischemic sections showed in the CA1, dentate gyrus (DG), and hilar regions active protein synthesis, primarily in cell bodies (Figs. 3A through 3C). Immunostaining for eIF2α(P) was negligible (Figs. 3A′ through 3C′). This contrasted with a prominent reduction in labeled amino acid incorporation (Fig. 4A through C) and intense eIF2α(P) immunostaining (Fig. 4A′ through C′) in the CA1, DG, and hilus of the untreated 90-minute reperfused group.
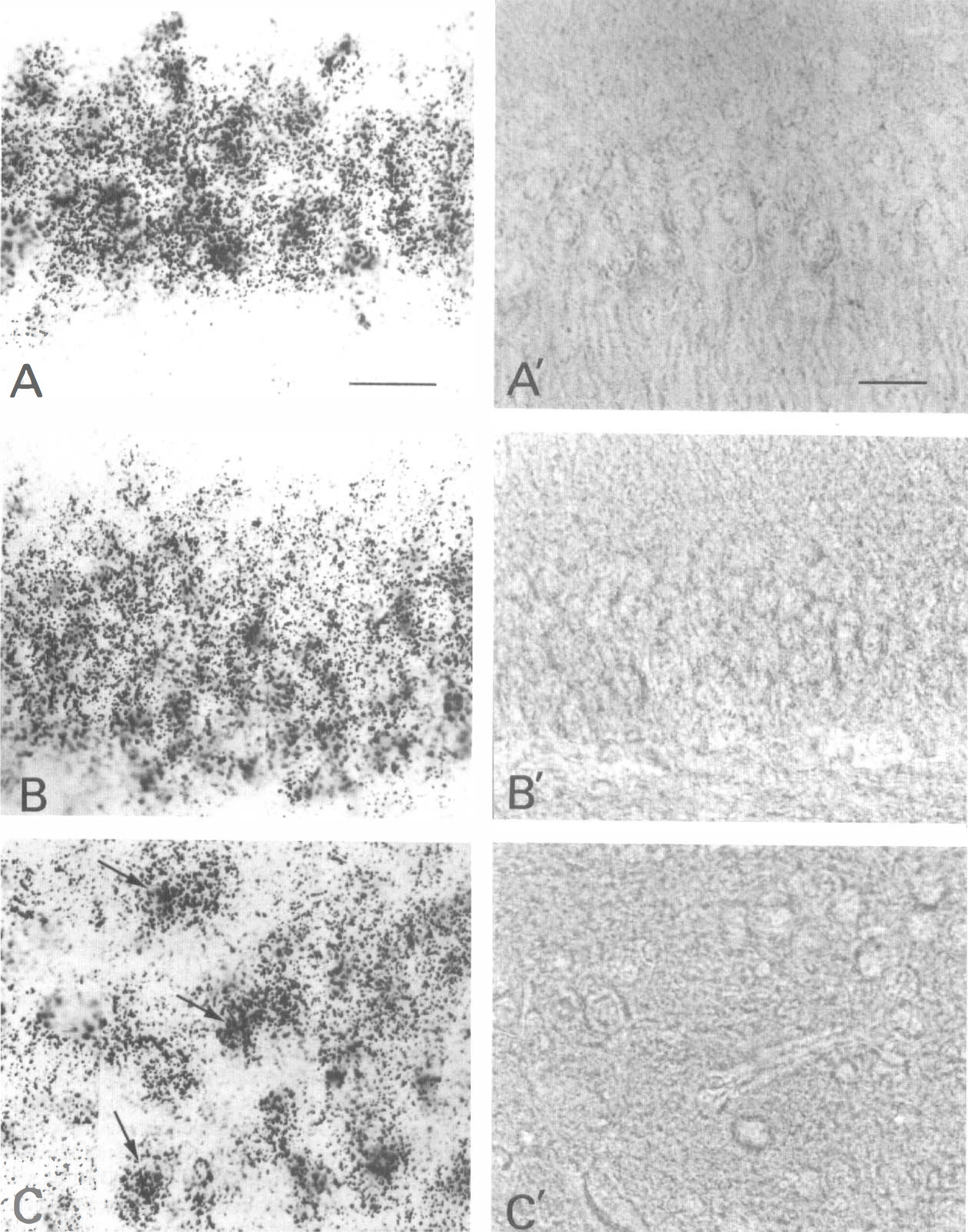
Protein synthesis and eIF2α(P) in a non-ischemic control. Autoradiography of 35S-labeled protein synthesis (
Protein synthesis and eIF2α(P) in an untreated 90 minute reperfused. Autoradiography of 35S-labeled protein synthesis (
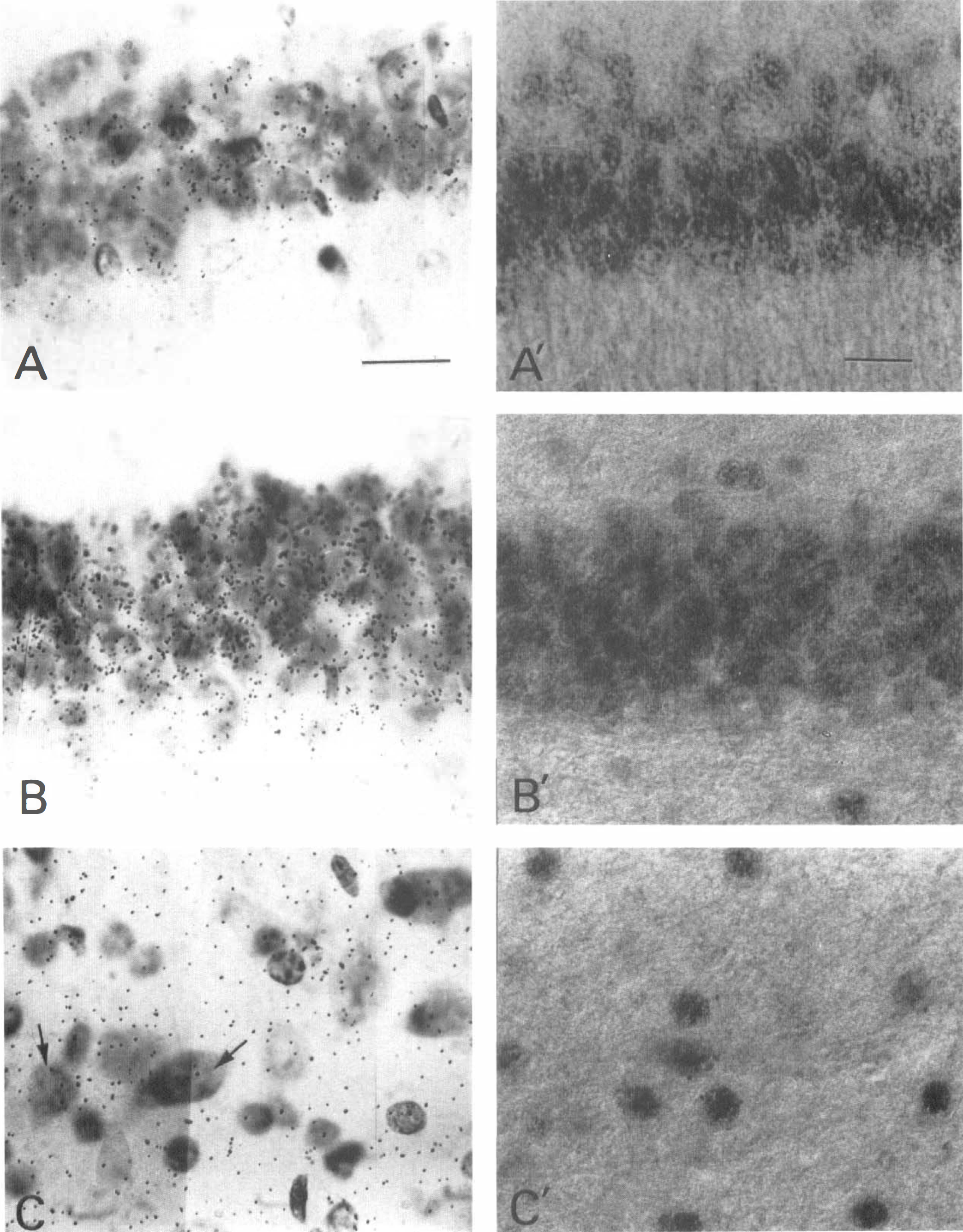
Treatment at resuscitation with 20 U/kg of intravenous insulin induced, by 90 minutes' reperfusion in the CA1 and DG, essentially complete recovery of protein synthesis (Figs. 5A and 5B) and clearing of eIF2α(P) (Figs. 5A′ and 5B′). This insulin dose did not prevent the initial phosphorylation of eIF2α in these cells during the first 5 minutes of reperfusion (Fig. 6). Taken together, these results demonstrate that insulin exerts its effect by promoting dephosphorylation rather than by inhibiting phosphorylation. Although 20 U/kg of intravenous insulin also improved protein synthesis in the hilus (Fig. 1), reduced eIF2α(P) immunostaining (compare Figs. 4C′ and 5C′) persists in some hilar cells (Fig. 5C′, small arrows) but not in others (Fig. 5C′, large arrowheads). Hilar cells that continue to display eIF2α(P) show staining in the nucleus and have small cell bodies (Fig. 5C′, small arrows). These cells may represent either atrophic neuronal elements or glia. Larger hilar cells do not display eIF2α(P) immunostaining (Fig. 5C′, large arrowheads) and correspond to hilar neurons that have recovered protein synthesis (Fig. 5C, arrows).
Protein synthesis and eIF2α(P) in 20 U/kg insulin-treated 90 minute reperfused. Autoradiography of 35S-labeled protein synthesis
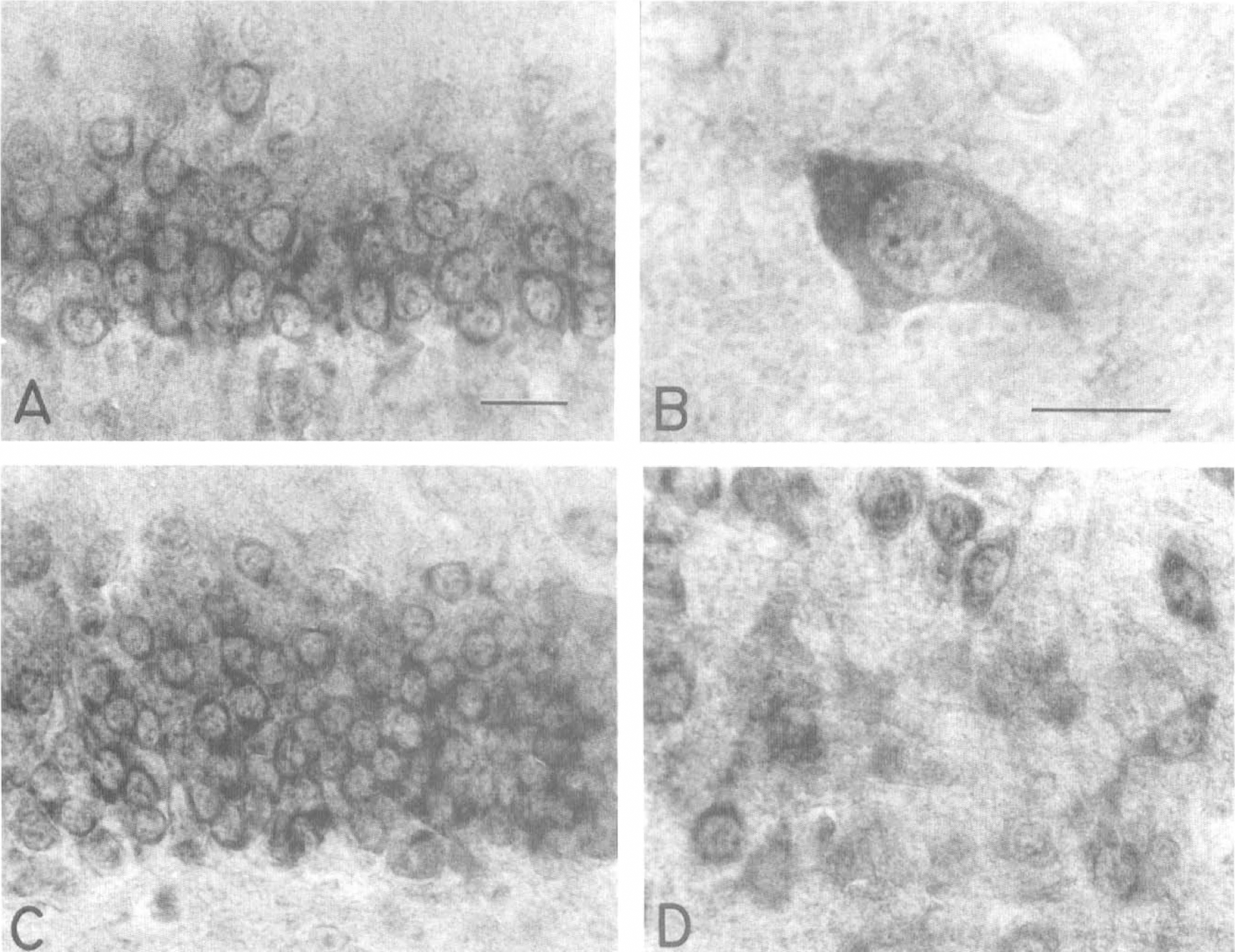
Insulin-treated 5 minute reperfused. After a 10 minute cardiac arrest, treatment at reperfusion with 20 U/kg insulin does not prevent phosphorylation of eIF2α in hippocampal neurons during the first 5 minutes reperfusion. Intense eIF2α(P) immunostaining is present in the cytoplasm of the neurons in the CA1 pyramidal layer
As an incidental finding, we noted that there was at 90 minutes' reperfusion a zone of CA3 neurons that exhibited persistent eIF2α(P) immunostaining (Fig. 2, T20, small arrow) and inhibited protein synthesis (not shown) that were not improved at any dose of insulin. As discussed later, this may be related to the paucity of insulin receptors on these cells.
DISCUSSION
The large reduction in protein synthesis in vulnerable neurons that accompanies postischemic brain reperfusion reflects alterations in the translation initiation system that occur during reperfusion rather than during ischemia itself (Cooper et al., 1977). Here we confirm the prominent depression of protein synthesis in various hippocampal neurons during reperfusion after global brain ischemia and demonstrate that this is accompanied by phosphorylation of eIF2α. Moreover, we found that high-dose insulin administered at return of circulation results, by 90 minutes' reperfusion, in dephosphorylation of eIF2α(P) and concomitant restoration of protein synthesis in CA1, DG, and large hilar neurons. These results show that the suppression of protein synthesis in the reperfused brain is reversible; they support a causal association between eIF2α(P) and inhibition of protein synthesis in vulnerable neurons and suggest a mechanism for the neuroprotective effects of insulin.
Insulin has an established neuroprotective effect in the setting of ischemia and reperfusion. Based on the idea that preischemic mild hyperglycemia had a detrimental impact on neurologic outcome, LeMay et al. (1988) intraperitoneally administered up to 2.4 U/kg of insulin 1 hour before 20 minutes' cerebral ischemia induced by vessel occlusion in rats, and observed a beneficial effect on neurologic deficit scores associated with a reduction in the preischemic elevation of blood glucose that was induced by surgical stress. Voll et ai. (1989) demonstrated a reduction in spatial learning deficits in rats treated with 2.0 U/kg of insulin given subcutaneously twice daily for 1 week beginning after 10.5 minutes' carotid occlusion during controlled hypotension, and they correlated this improved function with reduced loss of CA1 neurons after 12 weeks. Voll and Auer (1991a) went on to show that the neuroprotective effect of insulin (7 U/kg) did not derive from hypoglycemia, and Zhu and Auer (1994) demonstrated that after 10 minutes' forebrain ischemia in rats, continuous infusion of 85 U insulinlkg/day into brain ventricles reduced loss of hippocampal and cortical neurons more effectively than infusion of either 26 U insulin/kg/day or 185 μg/kg/day of insulin-like growth factor-1 (IGF-1). Other studies have shown that insulin improves neurologic deficit scores and cognitive function and prevents necrotizing brain damage after transient forebrain ischemia (Fukuoka et al., 1989; Strong et al., 1990; Voll and Auer, 1991b). This evidence has been interpreted to suggest that insulin has a neuron-sparing effect that is mediated by direct interaction with receptors in the central nervous system (Zhu and Auer, 1994).
Insulin is known to bind to only two receptors: the insulin receptor and the IGF-1 receptor (LeRoith et al., 1993). It does not bind to the insulin-like growth factor-2 receptor or the insulin receptor–related receptor. Insulin receptors are found in specific regions within the brain. Autoradiographic studies shown high-density [125I]insulin binding in the DG and CA1 regions (Hill et al., 1986; Werther et al., 1987; Kar et al., 1993). Immunohistochemical localization of the insulin receptor showed the densest immunoreactivity on the perikarya and processes of the CA1 and CA2 pyramidal cells, whereas the granule cell layer of the DG had moderate density, and much lower density was seen in CA3 and CA4 neurons. Glial cells did not stain (Unger et al., 1989).
There are several possible pathways by which insulin could reverse eIF2α phosphorylation in vulnerable neurons during early reperfusion. Insulin might induce down-regulation of an eIF2α kinase, up-regulation of an eIF2α phosphatase, or both. There are three known eIF2α kinases: (1) GCN2, found in yeast, Drosophila, and recently, in mammals (personal communication, Douglas Cavener, Vanderbilt University and Ronald Wek, Indiana University, Bloomington, IN), (2) the hemin-regulated inhibitor found primarily in reticulocytes but not conclusively identified in brain, and (3) the double-stranded RNA-activated kinase PKR. The PKR kinase is present in the brain, but eIF2α phosphorylation during early reperfusion is undiminished in mice bearing a homozygous knockout of PKR (DeGracia et al., 1999). Thus, the identity of the kinase responsible for phosphorylation of eIF2α during brain reperfusion is not known, and a role for insulin in regulating such an enzyme during cerebral reperfusion is speculative. However, there is precedent for insulin-induced inhibition of an eIF2α kinase; activation of Ras, an intermediate in insulin signaling, leads to activation of a 97-kd inhibitor of PKR (Bandyopadhyhay and Sen, 1992).
The reversal of eIF2α phosphorylation in vulnerable neurons by insulin during early reperfusion also might result from the activation of an eIF2α(P) phosphatase. Four classes of serine-threonine protein phosphatases (PP) have been described, based on their catalytic subunits and sensitivity to various inhibitors. PP1 appears to be the enzyme responsible for dephosphorylating eIF2α(P) in vivo (Redpath and Proud, 1990), and isoforms PP1α and PP1 γ are present in the brain and concentrated in the neocortex and hippocampus (Takizawa et al., 1994). PP1 is activated in response to insulin signaling (Srinivasan and Begum, 1994); thus, activation of PP1 could play a role in the insulin-induced reversal of eIF2α phosphorylation in vulnerable neurons during early reperfusion.
Up-regulation of the activity of eIF2B is another potential mechanism for the enhancement of protein synthesis by insulin (Webb and Proud, 1997). As noted earlier, the GDP in the eIF2/GDP complex formed at the end of initiation must be exchanged for a GTP before the next round of initiation, and eIF2B performs this function by increasing the off-rate of GDP (Price and Proud, 1994). Insulin has been shown to increase eIF2B's activity and protein synthesis in vivo (Welsh and Proud, 1992). However, this mechanism is unlikely to be effective in the reperfused brain, where high eIF2α(P) levels would continue to competitively inhibit eIF2B because the affinity of eIF2B for eIF2α(P) is 150-fold higher then for unphosphorylated eIF2α (Rowlands et al., 1988).
There an several possible explanations for the high dose of insulin required to achieve the dephosphorylation of eIF2α(P) and recovery of protein synthesis in the CA1, DG, and hilus. Insulin transport into the brain or the responsiveness of its receptor may be inhibited as a result of ischemia and reperfusion. The glucocorticoid dexamethasone inhibits insulin transport into the CNS (Baura et al., 1996), and glucocorticoid levels are elevated in humans after brain ischemia (Fassbender et al., 1994). Alternatively, there may be decreased responsiveness of the insulin receptor. Tumor necrosis factor α levels are elevated in the reperfused brain (Lavine et al., 1998), and tumor necrosis factor α induces insulin resistance by increasing serine/threonine phosphorylation of the insulin receptor and of the major insulin receptor substrates IRS-1 and IRS-2 (Paz et al., 1997).
The neuroprotective effects of high doses of insulin also might involve crosstalk with another intracellular signaling pathway or with another receptor. The mitogen-activated protein kinase system provides an example of crosstalk in intracellular signaling. Mitogen-activated protein kinase is efficiently activated by phosphoinositide 3-kinase but also can be stimulated by a protein kinase C family member when numerous receptors are activated (Duckworth and Cantley, 1997). It also is possible that the neuroprotective effects of high doses of insulin occur by activation of the IGF-1 receptor (Gluckman et al., 1993). Insulin and IGF-1 have significant sequence homology, although insulin has about a 100-fold lower affinity than IGF-1 for IGF-1 receptors (LeRoith et al., 1993). Autoradiographic studies have shown that [125I]IGF-1 binds in the DG (ventral more than dorsal) and the CA1, CA2, and CA3 areas of the hippocampus (Kar et al., 1993), and IGF-1 also has been shown to have a neuroprotective effect in transient forebrain ischemia (Zhu and Auer, 1994). However, in a direct comparison of intraventricular administration of IGF-1 and insulin, Zhu and Auer (1994) found that IGF-1 produced a significantly weaker neuroprotective effect than insulin. That study and our observation that high-dose insulin fails to reverse eIF2α phosphorylation in the CA3, which has a higher concentration of IGF-1 receptors than insulin receptors, argue against involvement of the IGF-1 receptor in the mechanism by which insulin induces dephosphorylation of eIF2α(P) during brain reperfusion.