
Abstract
The regional binding of N1′ -([11C]methyl)naltrindole (MeNTI), a selective δ-opioid antagonist, was studied in healthy human subjects with positron emission tomography (PET). After the bolus intravenous administration of high specific activity [11C]MeNTI, PET was performed over 90 minutes. Arterial plasma samples were obtained during the scanning period and assayed for the presence of radiolabeled metabolites. The data were analyzed with various kinetic (two-and three-compartment models, Patlak graphical analysis) and nonkinetic (apparent volume of distribution and activity at a late scanning time) approaches. This tracer showed irreversible binding characteristics during the scanning period used. The results of the analyses also were compared with the density and distribution of δ-opioid receptors in the human brain in vitro. Additionally, computer simulations were performed to assess the effects of changes in receptor binding and tracer transport changes on the perceived binding parameters obtained with the models. A constrained three-compartment kinetic model was demonstrated to be superior to other quantification models for the description of MeNTI kinetics and quantification of δ receptor binding in the human brain with 11C-labeled MeNTI.
Keywords
Changes in the function or number of opioid receptors have been proposed in several neuropsychiatric and neurode generative conditions, including Parkinson's, Huntington's, and Alzheimer's diseases (Gulya, 1990); substance abuse (Kosten, 1990); and seizure disorders (Mayberg et al., 1991). Three primary opioid receptor subtypes have been described pharmacologically and subsequently cloned: mu (μ), delta (δ), and kappa (κ) (Rei sine and Bell, 1993).
Considerable advances have been made in the development of radiotracers for the measurement of central opioid receptor densities in living human subjects using positron emission tomography (PET) (Mayberg and Frost, 1990). [11C]Carfentanil, a selective μ opioid agonist, has been validated for the quantification of μ opioid receptors in vivo (Frost et al., 1985, 1989). [11C]Carfentanil has been used in conjunction with [11C]diprenorphine, a nonselective opioid antagonist, for the estimation of non-μ receptor sites (Frost et al., 1990). Measurements of combined δ and κ densities have been performed through the use of the nonsubtype-selective antagonist [18F]cyclofoxy (Carson et al., 1993).
Although much progress has been made on the in vitro localization of δ-opioid sites in the mammalian brain (Mansour et al., 1993), a suitable tracer for selective in vivo imaging in humans has not been previously available. This is of interest for the investigation of the role of δ-opioid receptors in normal and pathologic brain function in human subjects. As examples, preclinical data suggest that δ-opioid receptors are involved in seizure kindling and threshold (Tortella et al., 1983), and in the reinforcing effects of psycho stimulants (Jones and Holtzman, 1992; Menkens et al., 1992).
Naltrindole, a selective, high-affinity δ-opioid receptor antagonist, is a non peptide compound that is centrally active after peripheral administration in the rat (Portoghese et al., 1990). This compound is 700-fold and 3000-fold more selective for δ than for μ and for κ sites, respectively. Yamamura et al. (1992) have characterized [3H]naltrindole binding in rat brain homogenates, demonstrating its high affinity for δ sites (Kd = 37 pmol/L). [3H]Naltrindole shows high binding densities in neocortical regions and basal ganglia, and moderate to low in hippocampus and thalamus (Drower et al., 1993) after the known distribution of δ-opioid receptors. Recently, a positron-emitting analog of naltrindole has been synthesized in our laboratories by methylation of the indole nitrogen with [11C]iodomethane (Lever et al., 1992, 1994). This methylated analog, N1′-([11C]methyl) naltrindole ([11C]MeNTI), retains the δ-opioid receptor affinity and specificity of naltrindole. Lever et al. (1992) have shown that [11C]MeNTI administered intravenously permeates the blood–brain barrier, has a distribution in mouse brain that correlates with δ binding in vitro, and that its binding in vivo is selectively blocked by δ but not μ or κ antagonists. The distribution of [11C]MeNTI in human brain has been demonstrated by PET (Madar et al., 1996). [11C]MeNTI also has been used to show increased δ-opioid receptors in patients with temporal lobe epilepsy (Madar et al., 1997).
Here we examine a variety of methodologic alternatives for the quantification of [11C]MeNTI binding to δ-opioid receptors in the human brain with PET. Both kinetic and nonkinetic approaches were applied to the data that were obtained through dynamic PET studies, after the bolus administration of high specific activity [11C]MeNTI to healthy human subjects.
METHODS
Subjects and PET acquisition
Eight young healthy subjects (mean age 31 ± 7 years), seven men and one woman, were recruited by advertisement and gave written informed consent to study participation. The subjects were screened by history and physical and neurologic examinations. They had no history of medical, neurologic, or psychiatric disorders; current use of psychoactive medications; or drug abuse.
An intravenous line was placed in an antecubital vein and an arterial catheter in the radial artery at the level of the wrist. Scanning was performed with a GE 4096 ± PET scanner (General Electric, Milwaukee, WI, U.S.A.), which acquires IS simultaneous slices with a separation between planes of 6.5 mm. Before data acquisition, each volunteer was fitted with a thermoplastic face mask that was used in conjunction with x-ray computed tomography to localize the planes for the PET study and to eliminate subject motion. The planes selected for imaging were standardized as follows: the glabellar-inion line was visually located on the computed tomography scan. An imaging plane that intersected the amygdalae, parallel to the glabellar-inion line, was selected as the fourth PET plane. The masks were worn during the PET study, allowing alignment of the PET scanner slices to the glabellar-inion line and the standardization of imaging planes across subjects.
After subject positioning in the scanner gantry, a 10-minute transmission scan (68RGe/68Ga source) was performed for attenuation correction immediately before the PET study. The [11C]MeNTI was synthesized as previously described (Lever et al., 1992), and approximately 20 mCi were administered intravenously as a bolus (mean specific activity 2000 Ci/mmol, range 1107 to 4030 at the time of administration). Twenty-five dynamic PET scans (6 × 30 seconds, 5 × 1 minute, S × 2 minutes, 9 × 8 minutes) were acquired over 90 minutes. Four of the subjects were scanned twice, before and after the oral administration of 100 mg naltrexone, approximately 1 hour before scanning.
The images were smoothed to a final in-plane resolution of 7.7 mm at full-width half-maximum. An image then was generated for placement of 4 × 4 pixel (8 × 8 mm) regions of interest by averaging serial images obtained from scans covering the entire scanning period to provide gross anatomical landmarks. Regions of interest were placed bilaterally on the parietal, cingulate, occipital, and frontal cortices, cerebellum, thalamus, caudate, and putamen. The average activity (nCi/cm3) in these regions then was quantified for each of the scanning intervals, producing activity data as a function of time.
Analysis of blood and plasma radioactivity
After radiotracer administration, approximately 20 arterial blood samples were collected in rapid succession for the first 3 minutes. An additional 20 samples were obtained during the remaining 87 minutes of the scan. The blood samples were centrifuged for S minutes at 2000 rpm. The plasma portion was withdrawn and counted for 1 minute in a gamma counter.
The proportion of metabolized [11C]MeNTI in plasma was determined from arterial blood samples obtained before injection and at 5, 10, 15, 30, 45, 60, 75, and 90 minutes after injection. The first four samples were 8 mL, and the remaining, 16 mL. These samples were centrifuged at 2000 rpm for 5 minutes, the plasma removed and passed through an activated C18 reverse-phase Sep-Pak (Waters Associates, Milford, MA, U.S.A.). The eluent was washed with 0.1 mmol/L ammonium formate and eluted with 100% methanol. The methanol fraction was diluted with an aqueous solution of 2% triethylamine and 3% acetic acid to a final solution of 40% methanol. This solution was passed through a C18 reverse-phase analytical HPLC column (Waters Associates). The HPLC mobile phase was composed of 1:1 acetonitrile and methanol (40%) and HPLC-grade water buffered with 2% triethylamine and 3% acetic acid (60%). The HPLC flow rate was set at 3 mL/min. Extraction efficiencies for the parent compound and radiolabeled metabolites were calculated and corrected for at each step of the Sep-Pak separation. The preinjection blood sample, with [11C]MeNTI added, was used as a control to determine whether metabolites were produced in vitro and the extraction efficiency of the parent drug. A sample of blood obtained 90 minutes after tracer administration, with high metabolite content, was used to calculate the metabolite extraction fraction. The total concentration of radioactivity in plasma then was corrected for the presence of radiolabeled metabolites by multiplying the plasma concentration by the fraction of unchanged drug using linear interpolation. Further details on these procedures can be found in prior publications (Frost et al., 1989; Price et al., 1993; Sadzot et al., 1991). Briefly, the HPLC system was equipped with two opposed 12.7-cm (5-inch) NaI (T1) detectors, a strip chart recorder, and a computer interface. Each radioactive peak was automatically integrated and the decay corrected back to the time of sample injection into the column. The percentage of total activity corresponding to unmetabolized [11C]MeNTI then was calculated for each time point.
The free fraction of [11C]MeNTI in plasma (f1) was determined as previously described for [11C]diprenorphine and [11C]flumazenol (Sadzot et al., 1991; Price et al., 1993). Before tracer administration, a blood sample was obtained from each of four subjects. Fresh tracer, in equal amounts, was added to 1-mL aliquots of plasma and 50 mmol/L Tris buffer (pH 7.4 at room temperature). Centrifree micropartition systems (Amicon, Bedford, MA, U.S.A.) were loaded with equal portions (≈ 0.4 mL) of these standard samples then centrifuged for 20 minutes at 1500 rpm in a fixed-angle (35°) rotor centrifuge. The value of f1 was calculated using the following relation:
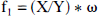
Theory and kinetic methods
In this work, six different approaches were used to analyze [11C]MeNTI kinetic data. These analyses involved the curve fitting of two- and three-compartment models to the data, graphical analyses, determination of the apparent volume of distribution during a late scan, and a single-scan estimate of specific binding obtained from a late scan.
General three-compartment model. The regional two- and three-compartment methods of analyses were based on a more complex four-compartment system, previously described in work from our laboratory (Frost et al., 1989) and other authors (Frey et al., 1985; Koeppe et al., 1991). Practically, the use of this model is precluded in most cases by its complexity and the increased variance associated with the calculation of six separate rate constants when only a single dose of tracer is administered. Therefore, in this work, a three-compartment model was investigated. This model consists of one vascular and two tissue compartments, differing from the four-compartment model in that the free and nonspecific ally bound tracer compartments are combined into a single compartment. Such a configuration assumes rates of exchange between free and nonspecifically bound tracer that are rapid relative to tracer transport across the blood–brain barrier and tracer–receptor association and dissociation. Following the scheme of Koeppe et al. (1991), the set of differential equations that describe the exchange between compartments are defined as follows:
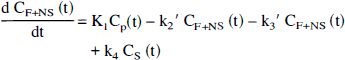
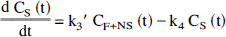
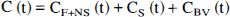
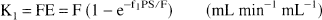
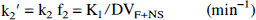
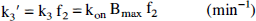

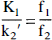
The ratio of K1/k2′ (or distribution volume of free and nonspecifically bound tracer, DYF+NS) was determined after the administration of naltrexone, a competitive antagonist, by modeling kinetic data from a region of negligible δ-opioid receptor concentration, the cerebellum. Calculation of f2 allows the determination of k3 = konBmax from the data. This approach to the estimation of DVF+NS is based on two primary assumptions. First, there is a negligible concentration of receptors in cerebellum after blockade with naltrexone. Second, DVF+NS is approximately the same across brain regions, and the tracer distribution in the cerebellum adequately reflects this volume.
Three-compartment, four-parameter estimation (model A). Since negligible dissociation of tracer from receptor was observed during the time period of the PET scan (see Results), the value of k4 was constrained to 0 to reduce variability in the parameter estimation that otherwise would result. Therefore, a three-compartment, four-parameter (K1, k2′, k3′, BV) kinetic model was applied to the [11C]MeNTI time–activity data.
Three-compartment, three-parameter estimation (model B). To reduce the variability in the estimation of k3′ for curve fits to specific-binding data, the distribution volume of free and nonspecifically bound tracer (DVF+NS) was specifically constrained. For each subject, the cerebellar DVF+NS was determined using a two-compartment model (three parameters–K1, k2′, and BV–were estimated), and this value was used as a constraint during curve fits to the specific binding data. Since the value of k4 was experimentally found to be ≈ 0, only K1, k3′, and BV were simultaneously determined for the remainder of the regions.
Two-compartment, three-parameter estimation (model C). A two-compartment, three-parameter kinetic model consisting of a vascular compartment and a single tissue compartment was applied to the specific binding data as a further means of model simplification. Three rate constants (K1, k2¶ime;, and BV) are calculated for this system. The definition of K1 remains the same as in models A and B, and k2¶ime; is defined as follows:
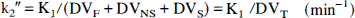
Optimal fits to the PET data for models A, B, and C were obtained using standard nonlinear least-squares curve fitting with Marquardt's method of minimization (Bevington, 1969). Time delay and BY parameters were included in the models. The time delay factor reflects the difference in time for the tracer to reach the radial artery and brain tissue. Similarly to Koeppe et al. (1991), this factor was determined for each study by fitting the first 10 minutes of plasma and PET data for whole brain slices at four different scanning planes (3, 5, 8, and 10) with a two-compartment model in which BV was assumed to be 3.5%. The average of the time delays for the four slices then was used to constrain this parameter in further model fits for each subject. The BY parameter, which accounted for contributions to brain activity measured by PET from the metabolized and unmetabolized drug in blood, was allowed to iterate with each of the model fits described here.
Graphical analysis: Patlak slope. The graphical analysis described by Gjedde (1982) and Patlak et al. (1983) has been used to measure the net accumulation of tracer in an irreversible compartment. The procedure entails graphing the activity in the region of interest divided by plasma activity (region of interest/plasma) versus normalized time ((∫Cp(t) dt)/Cp(T)). After an initial uptake period, this plot affords a linear portion with a slope equal to
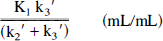
Normalized regional tracer concentration
Normalization to activity injected
The measure of regional tissue radioactivity concentration of activity at a late time after injection is the most simplified approach used here. The tissue concentrations are normalized to injected dose (mCi) to allow comparison between subjects. This method assumes that tracer equilibrium is rapid and that at late time points nearly all brain activity arises from specifically bound drug. Such an approach does not require blood sampling or mathematical analysis and is frequently used in the estimation of tracer activity in brain when blood sampling is not available or practical.
Computer simulations
Computer simulation studies were performed to investigate the errors associated with the parameter estimation by the various quantitative methods. First, estimates of the rate constants for this radiotracer were obtained using the most complex compartment model that we were able to use for all subjects (three-compartment estimation of K1, k2′, and k3′ and BV, with k4 = 0 (Eq. 1 through 3). The rate constants for all subjects then were averaged, as were their metabolite-corrected arterial input functions. The rate constants K1 and k3′ then were modified to examine how variations in either transport rate or receptor binding may influence the data estimated with a given model and to test the validity of assuming irreversible or reversible binding. Noise-free PET data then were simulated using the general three-compartment model equations. Each method of analysis then was applied to the simulated data to assess the accuracy of each approach. In the case of the three-compartment method, the rate constant k4 was allowed to iterate.
Statistics
Comparisons between the model fits were obtained using a modification of the Akaike information criterion (AIC) (Akaike, 1974), defined as AIC = -n In(SSQ) - 2p, where n is the number of data points, SSQ is sum of squared errors, and p is number of parameters. Larger values denote better fits.
Results are expressed as the mean ± 1 SD. When specifically indicated in the text, data variability is expressed as the percent coefficient of variation [%COY = (SD ÷ mean) × 100].
RESULTS
Blood and plasma radioactivity
The overall extraction efficiencies for the Sep-Pak C18 columns ranged from 70 ± 10% for the 10-minute plasma sample to 80 ± 4% for the 75-minute sample. No detectable metabolites were formed in vitro.
The HPLC chromatograms showed the parent compound peak (retention time ≈ 7.5 minutes) throughout the study and two primary metabolite peaks (retention time ≈ 3.5 minutes and ≈ 4.5 minutes) as early as 5 minutes after injection. Poorer resolution of parent and metabolite peaks were noted in the 90-minute HPLC chromatogram; this was attributed to low levels of radioactivity in plasma and the larger extent of parent compound metabolism. A representative plasma time–activity curve before and after metabolite correction is shown in Fig. 1 The average proportions of unmetabolized tracer in plasma decreased from 86 ± 4% at 5 minutes to 34 ± 7% at 90 minutes after injection. The free fraction of tracer in plasma (f1) was calculated for four of the subjects and averaged 0.074 ± 0.013.
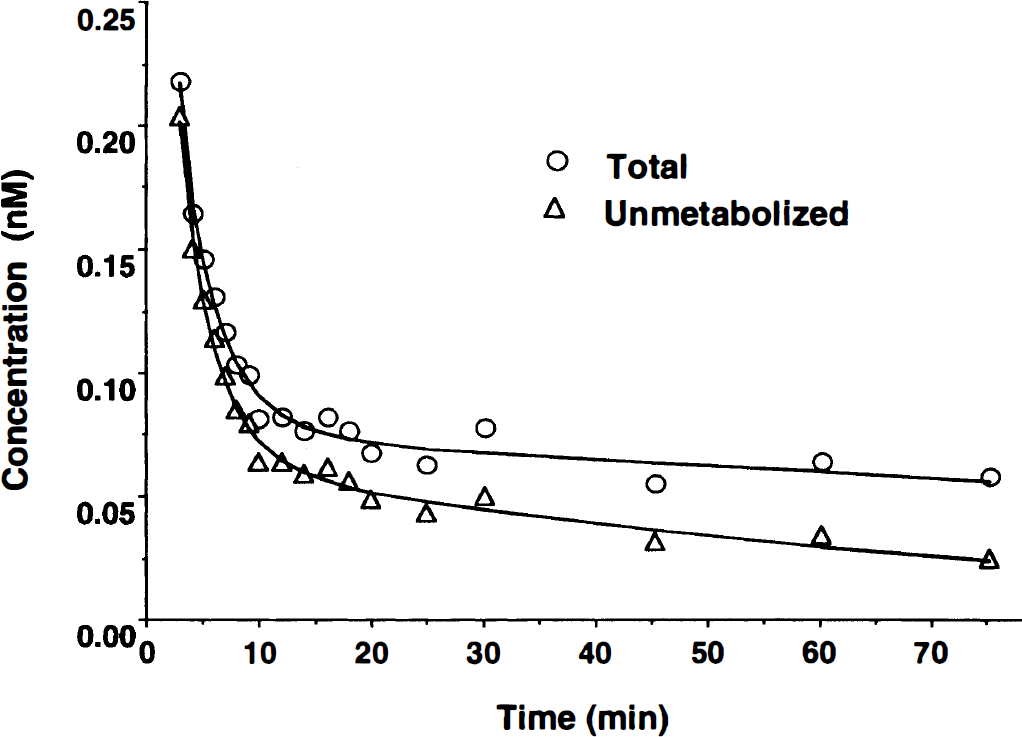
Examples of the arterial plasma concentrations that are typically obtained before (○, total) and after (Δ, unmetabolized) the correction of high specific activity N1′,-([11C]methyl) naltrindole ([11C]MeNTI) time–activity data for the presence of radiolabeled metabolites.
Regional tracer distribution and tracer kinetic analyses
Figure 2 (first row) presents four representative PET slices obtained from one of the subjects, averaged from 34 to 70 minutes after injection. Higher tracer concentrations at these times are observed in neocortex and basal ganglia, areas known to be rich in δ-opioid receptors. Lack of significant tracer activity in the cerebellum also is observed at these times. In the second row of Fig. 2, PET images from the same subject were processed similarly, approximately 1 hour after the oral administration of 100 mg of naltrexone. Significant displacement of tracer is observable in regions known to contain δ-opioid receptors (i.e., neocortex, basal ganglia, thalamus), with negligible changes in tracer activity in the cerebellum.
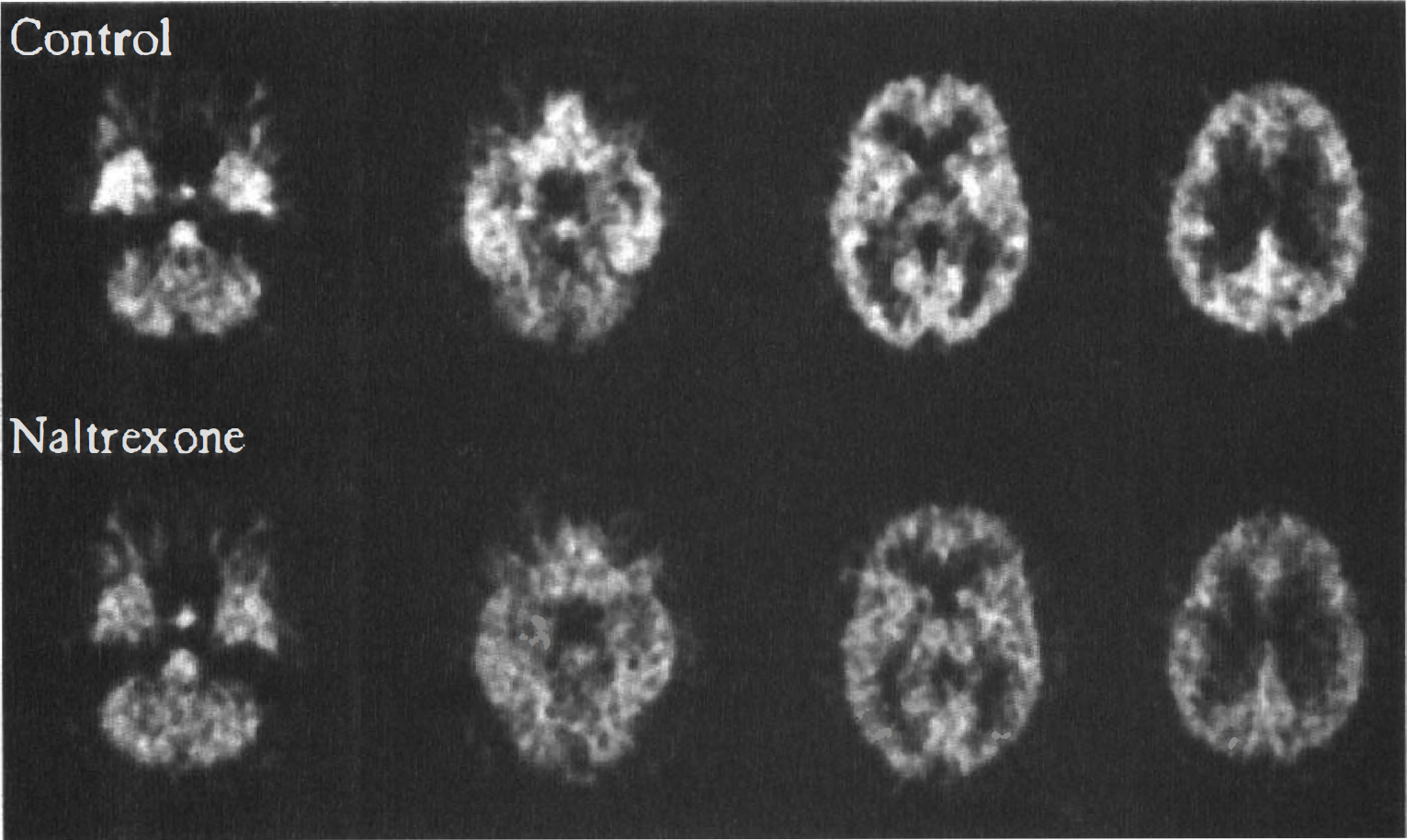
Images of [11C]MeNTI binding at four different levels, before (upper row) and after (lower row) the administration of 100 mg of naltrexone orally in a representative subject. The images are formed by averaging the data acquired 35 to 70 minutes after administration of the tracer and are expressed as a ratio to activity injected.
Typical time-activity curves of [11C]MeNTI in regions with high (putamen), intermediate (frontal cortex), and low receptor densities (thalamus, cerebellum) are presented in Fig. 3. A lower degree of binding and faster clearance is observed in the thalamus, as well as a greater retention of tracer in the putamen relative to frontal cortex. Average peak whole-brain tracer activity for the eight subjects was 0.56 ± 0.13 μCi/mL brain for doses ranging from 16 to 28 mCi. A low peak brain uptake of tracer coupled with low radioactivity concentrations in plasma and difficulty in resolving the 90-minute metabolite chromatogram resulted in poor data quality in the last two scan periods (74 to 90 minutes after injection), which were not used for the analyses presented.
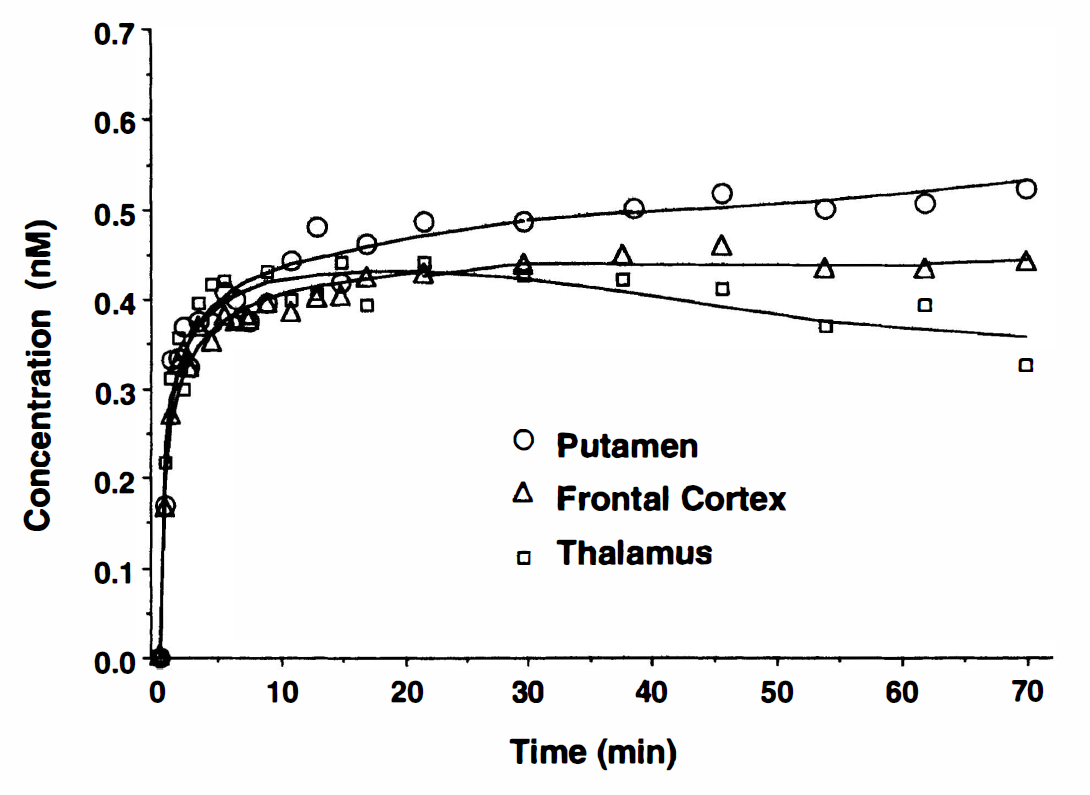
Examples of regional positron emission tomography data that were obtained after the high specific activity bolus injection of [11C]MeNTI. Data points correspond to decay-corrected regional concentrations of [11C]MeNTI. The lines represent the curve fits obtained with a three-compartment, two-parameter tracer kinetic model (model B). Regions with high (putamen, frontal cortex) and low receptor density (thalamus) are well differentiated.
As described earlier, the application of the general three-compartment model (non-zero k4) to the data revealed that the dissociation rate constant k4 was negligible and had high intersubject parameter variability and uncertainty in its estimation (data not shown). Therefore, k4 was subsequently fixed to 0 for the data presented. A summary of results for each of the applied analytical methods is presented in Table 1.
Comparison of methods for the quantification of [11C]MeNTI specific binding parameters in human brain
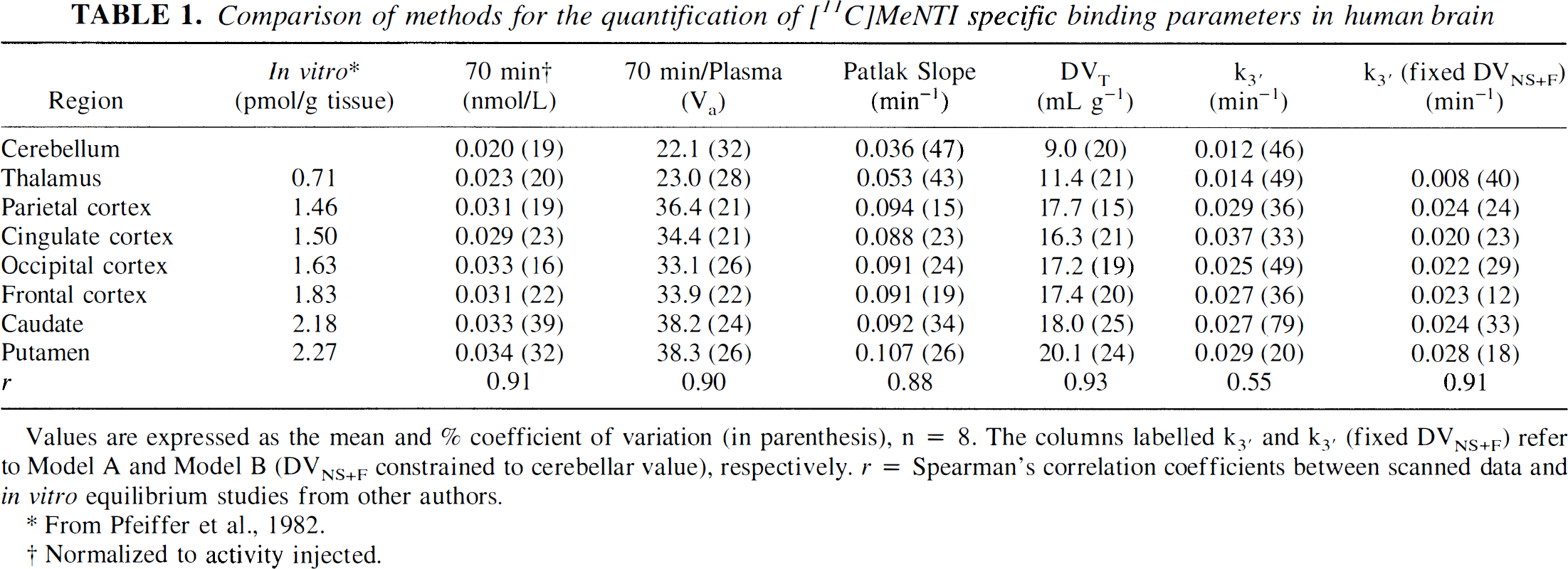
Values are expressed as the mean and% coefficient of variation (in parenthesis), n = 8. The columns labelled k3′ and k3′ (fixed DVNS+F) refer to Model A and Model B (DVNS+F constrained to cerebellar value), respectively. r = Spearman's correlation coefficients between scanned data and in vitro equilibrium studies from other authors.
From Pfeiffer et al., 1982.
Normalized to activity injected.
The variability of the specific binding parameter was largest for the most complex model (k3′ for model A), which corresponds to a three-compartment configuration with four estimated parameters (K1, k2′, k3′, and BV). The variability in k3′ was lower when model B was applied to the data (DVF+NS for specific binding curve fits was specifically constrained to cerebellar DVF+NS value). The DVF+NS calculated by model A averaged 8 ± 4 in areas of high receptor concentration (cortical regions and basal ganglia). Similarly, cerebellar DVF+NS values averaged 9 ± 2 for all subjects and were not significantly different from those of brain regions with high specific binding (paired two-tailed t test, P > 0.05), supporting the feasibility of the simplification introduced in model B. With this simpler approach, in which only K1, k3′, and BV were fitted to the data, intersubject variability was reduced, with %COV ranging from 12 to 40%. Similar performance was observed for the two-compartment model (calculation of DVT) and the graphical method (Patlak), although the uncertainty of the latter method was considerable in areas with low receptor concentrations (cerebellum and thalamus). The nonkinetic approaches, apparent volume of distribution at 70 minutes after injection (Va), and tracer concentration in a late scan (70 minutes after injection) normalized to injected dose and yielded variability estimates in the range of 16% to 39%.
The rank order of the specific receptor binding parameters estimated with each of these approaches were highly correlated with receptor concentrations obtained from equilibrium studies in vitro by other authors (Pfeiffer et al., 1982) (Table 1). On the other hand, for the nonkinetic analysis, Va and ratio of late scan activity to injected dose, the range of values obtained was severely restricted, compared with in vitro or kinetic analysis results. This restricted range of values for the various binding-related measures appeared to result from both an overestimation in areas of low receptor concentration and an underestimation in areas of high receptor content (confirmed in simulation studies; data not shown). Since both of the nonkinetic methods of analysis assume tracer equilibrium during the scanning period, a nonequilibrium condition even at late times after injection is the likely reason for these results.
The AIC values improved with the three-compartment configuration in all regions: from 81 in the two-compartment configuration in thalamus, to 83 using a three-compartment model with DVF+NS fixed to cerebellar values. Similarly, in frontal cortex it improved from 99 to 108, and in putamen, from 83 to 91. No improvements in the fits were observed when DVF+NS was not fixed from cerebellar values with exception of the thalamus. In this area, a small improvement was noted from the constrained (AIC = 83) to the unconstrained model (AIC = 85). Three-compartment model fits (k4 = 0) are shown in Fig. 3. Rate constants (K1 through k3) obtained with the three compartment model are presented in Table 2. Two compartment fits consistently underestimated the observed values during the later time points, confirming that the assumption of significant tracer-receptor dissociation during the study period is not valid for this radiotracer.
[11C]MeNTI regional parameter estimates from a three-compartment, three-parameter model configuration
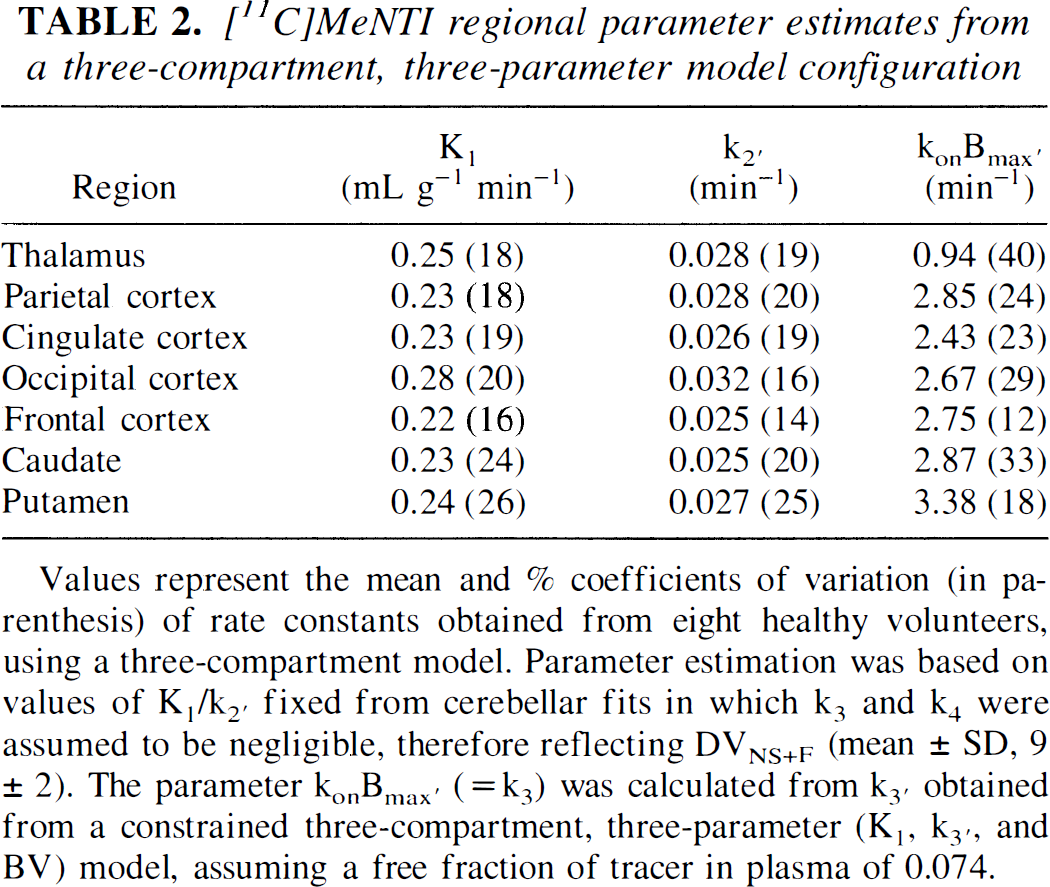
Values represent the mean and% coefficients of variation (in parenthesis) of rate constants obtained from eight healthy volunteers, using a three-compartment model. Parameter estimation was based on values of K1/k2′ fixed from cerebellar fits in which k3 and k4 were assumed to be negligible, therefore reflecting DVNS+F (mean ± SD, 9 ± 2). The parameter konBmax′ (= k3) was calculated from k3′ obtained from a constrained three-compartment, three-parameter (K1 k3′, and BV) model, assuming a free fraction of tracer in plasma of 0.074.
Computer simulations
The accuracy of various quantification models to describe and differentiate changes in tracer transport and receptor binding were tested in simulation experiments. In these, the rate of tracer transport (K1) and the receptor binding parameter (k3′) were varied over a wide range of values, and noiseless data were simulated based on the predetermined rate constants using a three-compartment model equation.
In Figs. 4A through C, the performance of the quantification methods, which appeared to be adequate based on the results of Table 1, is presented. We simulated a range of K1 changes from -100 to +200% of the original data (obtained from the frontal cortex of a representative subject). The putative receptor measure obtained from the graphical analysis (Fig. 4A) was demonstrated to be highly sensitive to modifications in the transport rate of this ligand, perhaps more so when reductions were simulated. Estimations in receptor binding directly obtained from the slope of the plot were likely to be biased by the contribution of K1 values to the equation defining the slope [slope = K1k3′/(k2′ + k3′)]. Two- or three-compartment models, which separate transport from binding, were not as biased in this regard (Figs. 4B and 4C). However, the higher the values of k3 simulated, the poorer the two-compartment fits, with systematic underestimations of the observed values by the end of the observation period (90 minutes). Longer observation periods may have compensated for the increases in k3 values, but those are impractical in actual experiments because of the short half-life of carbon-11.
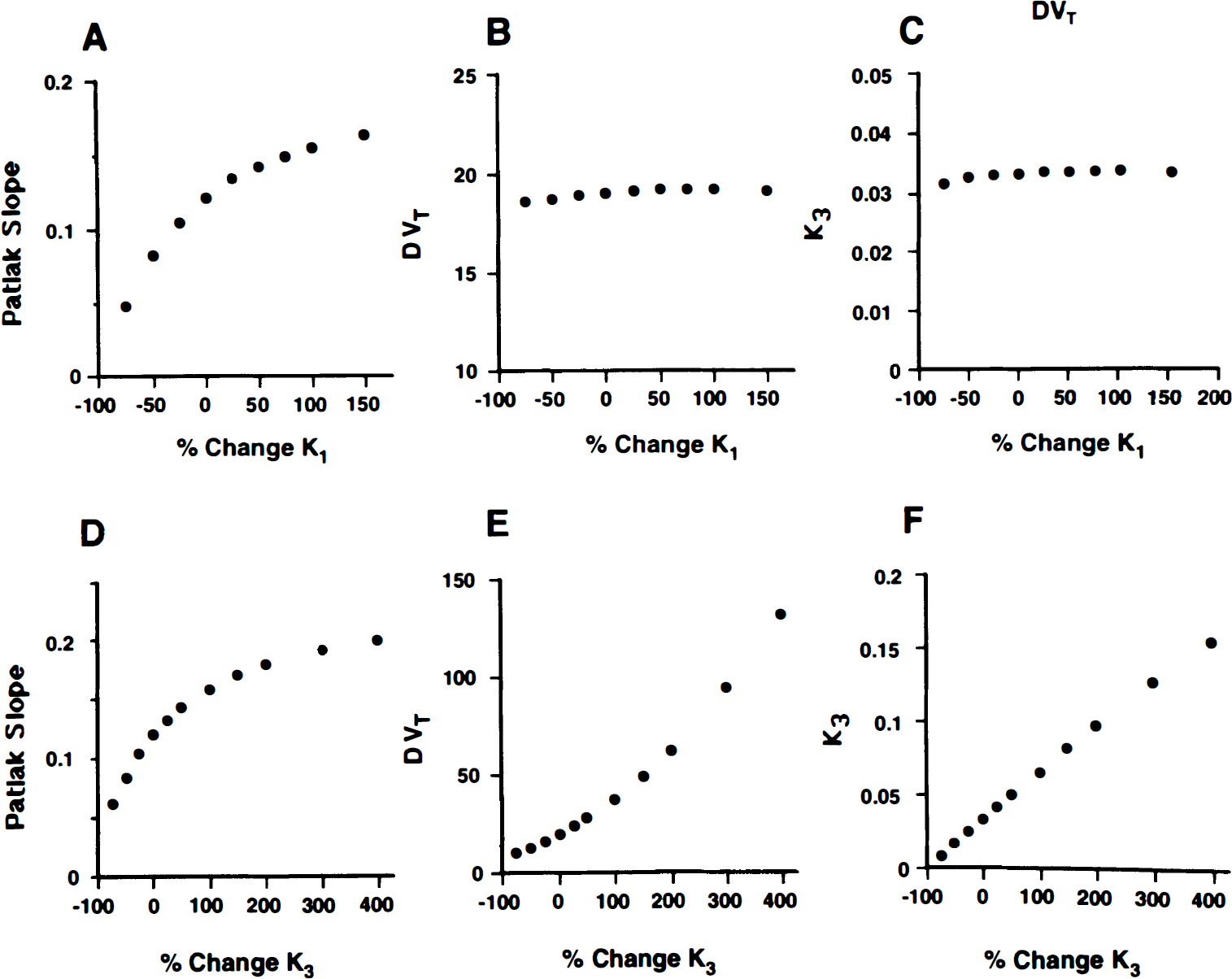
Effect of simulated changes in K1 on the receptor binding parameters obtained with a graphical method (slope of Patlak plots)
In Figs. 4D through 4F, the sensitivity of the various methods to simulated changes in k3′ was explored. In the case of the graphical analysis (Fig. 4D), the sensitivity of the Patlak slope to detect changes in receptor density was limited at high k3′ values. The relation between slope and simulated receptor binding became progressively nonlinear when increases in more than 50% of the original values in frontal cortex were simulated. Considerably more linear relations were observed for the two- and three-compartment configurations (Figs. 4E and 4F). However, because the data were originally obtained from a three-compartment model, it is possible that the results may have been biased toward better fits for this model.
A related issue is the condition of flow limitation described in detail by other authors in the study of tracers with slow ligand–receptor dissociation rates (Frey et al., 1992; Koeppe et al., 1994). Exploration of a possible flow-limitation condition also was performed using computer-simulated data. The calculated tissue response at a late time after injection (70 minutes) was plotted against the simulated ratio k3′/k2′ (Koeppe et al., 1994). As shown in Fig. 5, at ratios of k3′/k2′ above ≈ 1.5, the activity present in the late scan becomes progressively nonlinear with the simulated increase in k3′. The k3′/k2′ ratios observed experimentally in brain regions of healthy volunteers (Table 2) ranged from 0.3 to 1.1, below the nonlinear region of the graph. This should theoretically allow a good definition of binding densities in areas of high receptor concentration when tracer kinetic analyses are applied. Nonlinearity, with the resulting constriction of values, would be observed only when the actual receptor concentrations in the tissue of interest are higher than 50% above those observed in the basal ganglia (the highest binding region for this tracer), corresponding to k3′ values higher than 0.045, approximately.
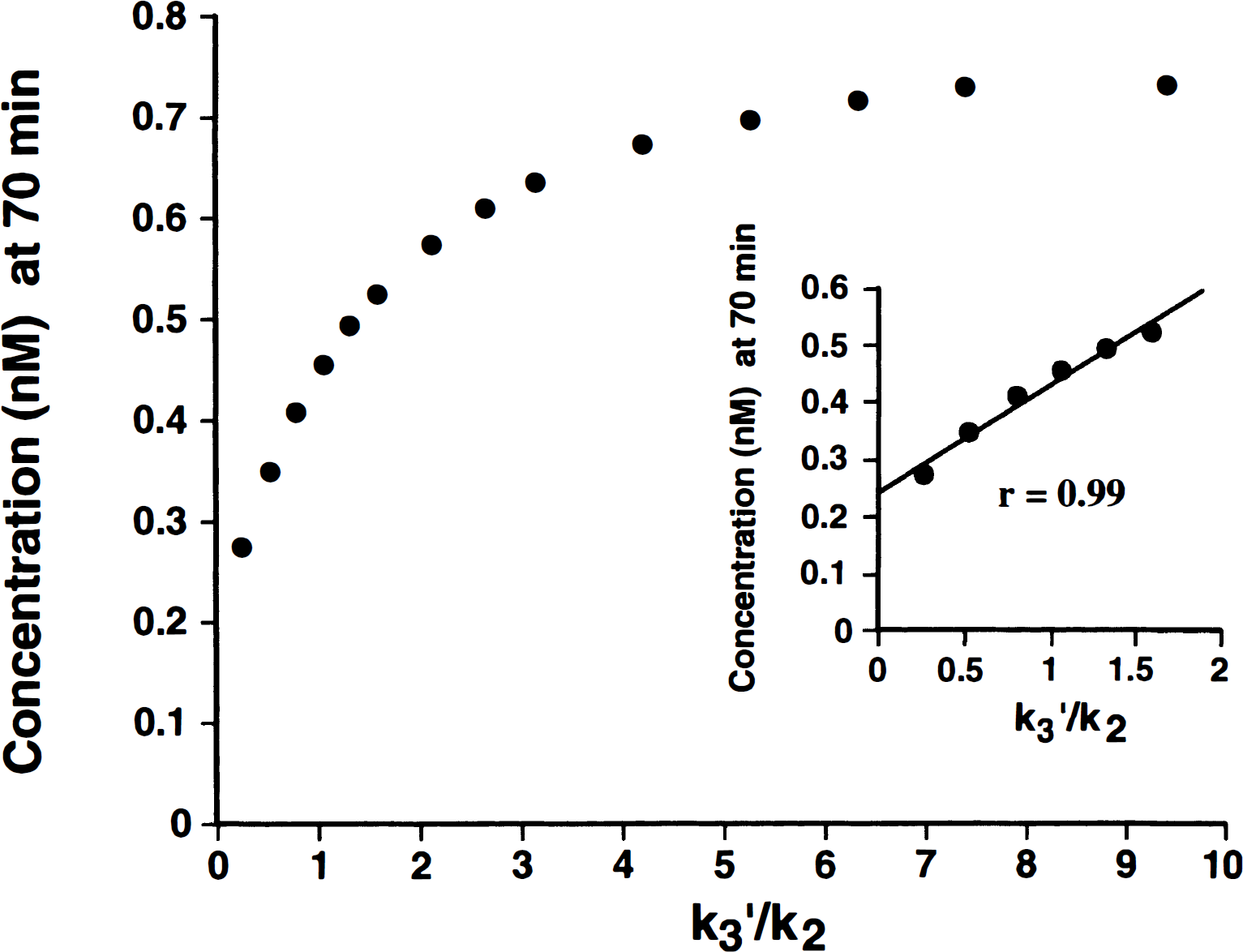
Effect of the ratio of ligand binding and clearance rates on the uptake of [11C]MeNTI at a late time after injection. Data represent the calculated concentration of tracer, based on noiseless computer simulations, when the ratios k3′/k2 were varied. Simulations were performed using an actual metabolite-corrected [11C]MeNTI input function from one of the subjects and typical values for K1, k2′, and k3′ for the frontal cortex. The inset shows the tissue response in the physiologic range observed in the subjects studied.
Figure 2 shows data obtained from a representative healthy subject before and after administration of 100 mg of naltrexone orally. Examination of the time–activity curves showed constant blockade over the scanning period with this long-acting opioid antagonist. K1 and K1/k2′ values were similar between blocked and unblocked studies for all regions examined, including the cerebellum (two-tailed, paired t tests, P > 0.05). For the four subjects in which two scans were performed, at baseline and after naltrexone administration, mean percent reductions in regional k3′ values after displacement were 68 ± 12% in frontal cortex, 72 ± 20% in occipital cortex, 62 ± 11% in parietal cortex, 70 ± 12% in putamen, 57 ± 15% in caudate nucleus, and 41 ± 21% in thalamus. The differences in mean percent blockade between regions probably result from uncertainties in the estimation of k3′. These data include one subject who, because of difficulties in radiotracer synthesis, was scanned approximately 3 hours after administration of 100 mg of naltrexone, in which the degree of [11C]MeNTI displacement was much lower than in other experiments. Without inclusion of that data set, k3′ values were reduced in the range of 40% to 95% across brain regions after naltrexone administration.
DISCUSSION
In this work, [11C]MeNTI and PET were used to obtain in vivo specific binding measures in healthy human subjects by applying a variety of analytic methods. The different methodologic approaches also were tested using computer simulation studies. These studies demonstrated that kinetic analyses (e.g., compartmental modeling or graphical analyses) yielded less biased binding-related measures compared with the nonkinetic methods. Specifically, this work demonstrated that a constrained three-compartment model was the analytic method of choice.
The nonkinetic binding measures that were evaluated corresponded to ratios of late scan data to either metabolite-corrected plasma or to the injected dose (mCi). These methods showed regional rank order of values that were highly correlated with previously reported in vitro binding data from human brain (Pfeiffer et al., 1982) and the autoradiographic distribution of δ-opioid receptors and δ receptor mRNA in experimental animals (Drower et al., 1993; Mansour et al., 1993). Tracer uptake was highest in basal ganglia and neocortical regions and low in thalamus and cerebellum. However, a significant degree of nonspecific binding is noted in the later region, which, at least based on the animal literature, should have negligible binding levels. Without further analysis, these measures appear to be adequate for the estimation of radiotracer-specific binding with the advantage of allowing short scanning periods (radiotracer binding estimated from a single scan performed late after radiotracer administration) and simpler methods of analysis.
These two nonkinetic quantification models rely on the assumption that tissue–plasma equilibrium has been achieved for all brain regions. In the absence of equilibrium, specific binding can be overestimated in regions with low receptor concentration (e.g., thalamus) and underestimated in regions with high receptor concentration (e.g., basal ganglia) because of regional differences in the time to reach equilibrium, which depends on the receptor concentration and radioligand dissociation rates of the region. The biases introduced by these methods are pronounced for radiotracers that have small dissociation rates relative to the rate of its clearance from plasma (Carson et al., 1993; Frost et al., 1989; Koeppe et al., 1994), and therefore can be considered irreversible. In addition, the range of the specific binding parameter values (for regions of high to low receptor density) is constricted for the single scan analyses and leads to difficulty in distinguishing regional specific binding differences.
An additional difficulty in attempting to quantify receptor densities from nonkinetic data is the possibility that the activity present in brain regions may be dependent on the degree of tracer transport from plasma to tissue. When the lack of equilibration during the scanning period is combined with high degrees of receptor binding relative to the rate of tracer transport from plasma to tissue, the observed concentration of tracer in the tissue will be limited in areas of high receptor density. This flow-limited condition has been observed for tracers that, similarly to [11C]MeNTI, present small rates of transport from tissue back to plasma (Frey et al., 1992; Koeppe et al., 1994). When k3, (Bmaxkon), is large relative to k2 in a specific region of interest, all available tracer in tissue is bound, and accumulation of the tracer in that region becomes proportional to the amount of tracer delivered (flow limited) (Koeppe et al., 1994). This may contribute to the biases observed with the nonkinetic methods of analysis (underestimation in areas of high receptor density) and may deteriorate the accuracy of kinetic approaches in the same regions by providing the model with flow limited data. In the case of [11C]MeNTI, simulation studies were performed in which the rate constant k3′ obtained from a representative subject was progressively increased with respect to k2′. This tracer demonstrated favorable ratios of k3′/k2′ across brain regions in scanned data (range of 0.3 to 1.1), whereas flow limitation effects were clearly observed only at k3′/k2′ ratios higher than 1.5 in the simulation studies. Under these circumstances, PET data obtained over 70 minutes after tracer administration appear to be adequate to obtain measures that are unbiased by flow limitation effects, provided that the methods used are otherwise valid in their assumptions.
For the kinetic methods (least-squares curve fitting and graphical analysis techniques), the parameter bias and variability depended on the complexity of the model configuration. The computer simulation studies defined when the specific binding parameters were independent of changes in radiotracer delivery, and described the sensitivity of the parameter to differences in receptor density. The variability in the receptor estimates was most pronounced with the more complex model tested, a three-compartment configuration in which K1, k3′, k2′, and BV contributions were fitted to the data. Coefficients of variation when individual rate constants were calculated ranged from 20% to 79% across brain regions, making it impractical for its routine use in the estimation of receptor densities in human subjects. A variation of this model assumed that an approximation to the combined nonspecific and free volumes of distribution could be obtained from a region with negligible δ-opioid receptor binding (i.e., cerebellum) (Mansour et al., 1987; Pfeiffer et al., 1982). This simplification also assumes that, in addition to equilibration between nonspecific and free compartments and plasma tracer concentration, the volume of distribution in those compartments is constant across brain regions and similar to that in the nonspecific binding region. This assumption is supported by the lack of significant differences between the DVF+NS values obtained in regions with low and high specific binding, and also by the lack of improvement in model fits across brain regions, when the DVF+NS parameters are allowed to vary in the estimation procedure, although fits for areas with low receptor concentration (e.g., thalamus) were minimally improved when the parameters were allowed to freely vary. Similar approaches have been applied successfully in other radiotracer PET binding studies (Frost et al., 1989; Koeppe et al., 1991; Price et al., 1993; Sadzot et al., 1991; Sawada et al., 1991).
The ability of the various models to accurately identify changes in apparent binding values from changes in tracer delivery is of importance in the application of the data analysis to patient data. Changes in regional CBF, which impact on the amount of tracer delivered to the tissue, are frequently found in various diseases (e.g., Alzheimer's disease, seizure disorders) (Bonte et al., 1993; Engel, 1988; Frackowiak et al., 1981) or physiologic states (i.e., during pharmacologic or cognitive challenges) (Holthoff et al., 1991; Zohar et al., 1989, Zubieta et al., 1998). If the specific binding process cannot be separated from radiotracer delivery effects, then differences in regional tracer uptake could be misinterpreted as differences in regional receptor concentration.
The kinetic methods that specifically describe radiotracer transport and specific binding (conventional two-and three-compartment models) were less sensitive to simulated changes in the plasma-to-tissue transport rate than those that did not (e.g., graphical analysis). However, the assumption of significant dissociation of the tracer from the specific binding compartment by the two-compartment model is not valid in this case. The fact that this tracer appears to have irreversible binding characteristics during the study period limits the number of approaches that can be used for its quantification. The graphical approach is easy to implement and uses a simple linear regression for the calculation of its binding parameter (Patlak et al., 1983). This method produced acceptable precision in its estimates, especially in areas of high receptor concentrations (%COV values from 19% to 34% in areas of high receptor density, 43% to 47% in low specific binding regions). Uncertainties in the estimation of the slope in low specific binding regions may result both from a high proportion of nonspecific binding and high K1 values for this radiotracer. The computer simulations of the [11C]MeNTI data demonstrated that the Patlak specific binding parameter (slope = K1k3′/k2′ + k3′) was highly sensitive to alterations in K1. Similar results have been reported for other radiotracers (Koeppe et al., 1994). Without the separate determination of K1 from the initial uptake phase of the plot, this model does not appear to be adequate for the unbiased calculation of receptor densities with [11C]MeNTI. Another graphical alternative, in which the blood plasma term of the Patlak plot is substituted by tissue measurements in an area devoid of specific binding, may be less sensitive to transport changes (Patlak and Blasberg, 1985). Although this method initially was tested for the analysis of scanned [11C]MeNTI data, it showed large degrees of intersubject variability in the binding estimates and was abandoned as a routine method of analysis.
The linearity of the receptor density measure with respect to simulated changes in k3′ also was tested for the various quantification models using computer simulations. Binding densities estimated from the slope of Patlak plots became nonlinear when increases larger than 50% in k3′ were simulated for the frontal cortex. This corresponded to values of k3′ larger than 0.051. As previously shown for other tracers with negligible dissociation from receptor sites (Koeppe et al., 1994), as k3′ increases, the term k3′/[k2′ + k3′] approaches unity. In this situation, the equation that defines the slope, K1 k3′/[k2′ + k3′], becomes proportional to K1 (flow dependent), with K1 then being a limiting factor for the accurate estimation of binding densities. In the case of two-and three-compartment models, these effects were less prominent and apparent only at large increases in the simulated k3′.
Since none of the available δ-opioid selective antagonists are currently approved for human administration at saturating doses, displacement of [11C]MeNTI binding by a competing ligand was tested after the administration of the nonselective antagonist naltrexone. This drug has lower affinity for δ receptors than for μ or κ sites, and the tracer displacement was high but not complete for most of the brain regions studied. Regarding the selectivity of [11C]MeNTI for δ-opioid sites, this has been demonstrated after in vivo administration in experimental animals (Lever et al., 1992). Moreover, the rank order of binding densities identified with [11C]MeNTI in the human brain is highly correlated with the known distribution of δ receptors and is clearly different from that reported for other tracers labeling μ (Frost et al., 1989), μ and κ (Carson et al., 1993; Kawai et al., 1990; Sawada et al., 1991), or all opioid sites (Frost et al., 1990; Sadzot et al., 1991).
In summary, a simplified three-compartment model, in which nonspecific and free tracer volumes of distribution were estimated from a brain region devoid of specific binding, proved to describe accurately the data obtained with PET and [11C]MeNTI. This method presents the advantages of acceptable precision in the estimation of binding parameters, excellent correlation with equilibrium in vitro data described by other authors, a linear relation between the model binding estimates and simulated binding densities, and independence from changes in tracer transport across the blood–brain barrier. These properties permit the application of the methodology presented here to the study of δ-opioid receptor involvement in both physiologic and pathologic brain function.