
Abstract
Two kynurenine hydroxylase inhibitors, (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61-8048), have been tested as neuroprotective agents on brain lesions induced by bilateral carotid occlusion in gerbils or by middle cerebral artery occlusion in rats. The percentage of lesioned pyramidal neurones found in the hippocampal CA1 region of gerbils subjected to bilateral carotid occlusion for 5 minutes decreased from 92 ± 10% in vehicle-treated animals to 7 ± 6% after mNBA (400 mg/kg intraperitoneally, three times at 1, 30, and 180 minutes after occlusion) or to 10±11% after Ro 61-8048 (40 mg/kg intraperitoneally, three times). A significant reduction in infarct volumes also was found when the kynurenine hydroxylase inhibitors were given to rats after permanent middle cerebral artery occlusion (from 207 ± 111 mm3 in vehicle-treated rats to 82 ± 18 and to 62 ± 57 mm3 in rats treated with mNBA, 400 mg/kg intraperitoneally, or with Ro 61-8048, 40 mg/kg intraperitoneally, respectively). The administration of mNBA (400 mg/kg intraperitoneally) or Ro 61-8048 (40 mg/kg intraperitoneally) to gerbils with a dialysis probe in their dorsal hippocampus or to rats with a dialysis probe in their parietal cortex significantly increased kynurenic acid concentration in the dialysates. The data suggest that inhibition of kynurenine hydroxylase could be a new avenue to reduce neuronal loss in brain ischemia.
N-methyl-
Kynurenic acid and its derivatives have been shown to reduce neuronal damage in primary neuronal cultures exposed to excitotoxins (Moroni et al., 1992), to reduce the infarct volume after middle cerebral artery occlusion (MCAO) in rats (Chen et al., 1993), and to protect hippocampal pyramidal neurones after transient carotid occlusion in gerbils (Pellegrini-Giampietro et al., 1994). However, KYNA crosses the blood-brain barrier with difficulty, and thus large doses are necessary to significantly increase its concentration in the brain. To obtain a sufficient increase in brain KYNA content, experiments have been performed with suitable KYNA precursors (kynurenine or indolpyruvic acid) or with probenecid, an inhibitor of the transport mechanism, which is responsible for its elimination from the brain (Moroni et al., 1988; Nozaki and Beal, 1992). A large increase in brain KYNA content also has been shown after the administration of inhibitors of kynurenine hydroxylase, a major kynurenine metabolizing enzyme, and this increase has been associated with functional effects, including neuroprotection after ischemic challenge or after excitotoxin injections (Moroni et al., 1991; Speciale et al., 1996a; Miranda et al., 1997). New potent and selective inhibitors of this enzyme recently have been described (Roever et al., 1997), and we have studied their effects on the neuronal damage found in models of focal or global brain ischemia in rodents. We also evaluated the time course and the extent of kynurenine hydroxylase inhibitor-induced changes in KYNA concentrations in the extracellular spaces of brain areas, which are known to be affected by ischemic challenges, to evaluate the mechanism of the neuroprotective effects of these enzyme inhibitors.
MATERIALS AND METHODS
All the experiments were formally approved by an ethical committee and were performed according to the rules of the University of Florence (Florence, Italy). Male Sprague-Dawley rats (body weight 220 to 280 g) and Mongolian gerbils (body weight 60 to 80 g) (purchased from Morini, Reggio Emilia, Italy) were used.
Global forebrain ischemia in gerbils
Gerbils were anesthetized with a mixture of 2% halothane, 75% nitrogen, and 20% oxygen. A ventral midline neck incision was performed to isolate both common carotid arteries, which were occluded with microarterial clips for 5 minutes. At the end of the occlusion period, the clips were released, allowing restoration of carotid blood flow, and the incision was sutured. Halothane administration was discontinued immediately after carotid occlusion, and the animals that remained unresponsive for approximately 20 minutes were considered exposed to forebrain ischemia. Body temperature was monitored and maintained at 37 ± 0.5°C with a rectal thermistor and a heating pad until the animals had fully recovered from the anesthesia. The animals then were placed in a warm environment (30°C), and their rectal temperature was periodically recorded for 3 hours. Gerbils were selected for these experiments because they lack interconnection between the carotid and the vertebrobasilar circulation, so that a complete global forebrain ischemia may be induced by occlusion of the common carotid arteries at the neck (Kirino, 1982). The extent of hippocampal damage found after bilateral artery occlusion was evaluated 7 days after surgery. The animals were killed by decapitation, and the brains were rapidly removed and placed in dry ice. Coronal sections (20 µm) were cut in a cryostat and stained with toluidine blue. The microscopic sections for each animal were analyzed by counting the number of CA1 pyramidal neurones and hilar cells that appeared to be histologically normal (Kirino, 1982; Pellegrini-Giampietro et al., 1994).
Middle cerebral artery occlusion in rats
Anesthesia was induced with 5% halothane in air and maintained with the lowest acceptable concentration of the anesthetic (in most animals, 2%). The left middle cerebral artery was occluded at the proximal portion according to Tamura and coworkers (1981). With the animal placed in the lateral position, a skin and muscle incision between the left eye and the left external ear was made, and the temporalis muscle was retracted. A small burr hole was opened in the basal surface of the temporal bone between the orbita and the foramen ovale. The middle cerebral artery was occluded with a bipolar electrocoagulator in the segment starting near its origin and ending where the artery crosses the inferior cerebral vein; after occlusion, the artery was severed. Body temperature was measured with a rectal probe and kept at 37°C with a negative feedback system (Harvard Homeotermic Blanket Control Unit, Harvard Apparatus, South Natick, MA, U.S.A.) for the duration of the surgical procedure. The animals then were placed in a warm environment for at least 6 hours. Blood pressure and blood gas were not measured, since wounds were sutured quickly (the whole procedure required approximately 15 minutes), and the animal recovered from anesthesia shortly thereafter. Twenty-four hours after the lesion, animals were decapitated, and the brains were frozen in dry ice. Coronal sections 20-µm thick then were prepared in a cryostat and stained with toluidine blue. Lesion area and infarct volume were determined using a computer-assisted image analysis system (Image-Pro Plus, 3.0, Silver Spring, MD, U.S.A.). A small group of animals again was anesthetized 24 hours after MCAO, each chest was opened, and 10 mL of a solution containing 2% 2,3,5-triphenyltetrazolieum chloride (Sigma, Milan, Italy) in saline was slowly injected into the left cardiac ventricle. Twenty minutes later, brains were removed and placed in 4% buffered formalin. Within 2 days, 2-mm thick coronal slices were prepared, and the infarct areas were measured using the computer-assisted image analysis system mentioned earlier.
Administration of mNBA or Ro 61-8048
3,4-Dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide (Ro 61-8048) was solubilized in dimethyl sulfoxide and injected in small volumes intraperitoneally. The maximal amount of dimethyl sulfoxide injected was 100 µL, and controls received an equal amount of solvent. (m-Nitrobenzoyl)-alanine (mNBA) was directly solubilized in Ringer solution. In the dialysis experiments, the compounds were injected once in rats and twice in gerbils.
Implantation of the dialysis membrane
The animals were anesthetized with chloral hydrate (300 mg/kg intraperitoneally) and placed in a stereotaxic apparatus. The skull was opened, and a transverse microdialysis probe (AN 69 membrane, Dasco, Italy; 220-µm internal diameter; 310-µm external diameter, molecular weight cutoff above 15000 d), prepared according to Ungerstedt (1984), was inserted into the rat parietal cortex or the gerbil dorsal hippocampus and fixed with a screw and dental cement as previously reported (Carpenedo et al., 1994). The microdialysis tubing was covered with epoxy glue along its entire length, except for the region corresponding to the cortex or dorsal hippocampus. Dialysis fibers were implanted through small burr holes drilled into the skull and kept in place with screws and dental cement. The coordinates (for fiber inlet and outlet) for the rats were −0.2 mm from the bregma (A-P) and −2.2 mm from the skull surface (H), and for the gerbils, −1.8 (A-P) and −2.4 (H). The length of the exposed membrane surface was 4 mm (for both rats and gerbils).
Ringer solution (in mmol/L: NaCl 122, KCl 3.1, CaCl2 2.3) flowed through the probe at a rate of 3.5 µL/min for 40 minutes. The animals then were placed in individual cages after closing the probe openings. Fifteen hours later, the animals were again connected to the perfusion apparatus. At least 1 hour of stabilization was allowed before collecting the perfusate for KYNA determination.
The recovery of KYNA into the dialysis fluid, measured with the probes used for both rats and gerbils, was 10.5 ± 2%. Data were not corrected for probe recovery.
Measurements of KYNA in the dialysates
The KYNA was measured using HPLC with post-column derivatization and fluorimetric detection (Carpenedo et al., 1994). The dialysate was directly injected into the HPLC apparatus. A reverse-phase column (S 10 ODS2 Spherisorb [Steroglass, Perugia, Italy]) was used, and separation was obtained using 50 mmol/L sodium acetate buffer (pH 6.20) and 8% acetonitrile at a flow rate of 1.0 mL/min as a mobile phase. The post-column derivatizing agent was zinc acetate (0.5 mol/L) at a flow rate of 0.6 mL/min. Fluorimetric detection was obtained using a fluorimeter (Perkin Elmer model LC 240) with an excitation wavelength of 344 nm and an emission of 398 nm.
Statistical analysis
Data were compared using analysis of variance followed by Tukey-Kramer multiple comparison test to determine differences between the various treatment groups. Values were expressed as mean ± SD.
RESULTS
Effects of mNBA and Ro 61-8048 on ischemia-induced neuronal damage in the gerbil CA1 region of the dorsal hippocampus
Saline-treated gerbils subjected to bilateral carotid occlusion for 5 minutes went into a postischemic coma. None of the animal displayed behavioral seizures or other signs of neurologic impairment. Spontaneous motility was recovered approximately 30 minutes after ischemia. Seven days later, histologic examination of the dorsal hippocampus showed that most (92 ± 10%) pyramidal cells in the CA1 region were damaged (Fig. 1). The mean number of intact pyramids counted in a section of the CA1 region of the dorsal hippocampus decreased from 290 ±40 (n = 10, mean ± SD) in nonischemic controls to 23 ± 16 in saline-treated gerbils (n = 18). Giving mNBA (400 mg/kg intraperitoneally) or Ro 618048 (40 mg/kg intraperitoneally), administered three times to gerbils at I, 30, and 360 minutes after reperfusion, induced a dramatic attenuation of postischemic cell loss. Similarly, the mean number of intact hilar cells counted in each section of the dorsal hippocampus decreased from 56 ±8 (n = 10, mean ± SD) to 24 ± 6 (n = 10, mean ± SD) after the ischemic challenge. Animals treated with the doses mentioned earlier of either mNBA or Ro 61-8048 and exposed to ischemia had in each section of their hilus 45 ± 9 and 42 ± 7 intact cells, respectively (n = 12 animals per group, mean ± SD), suggesting that the hilar cells also were protected by treatment with large doses of the kynurenine hydroxylase inhibitors.
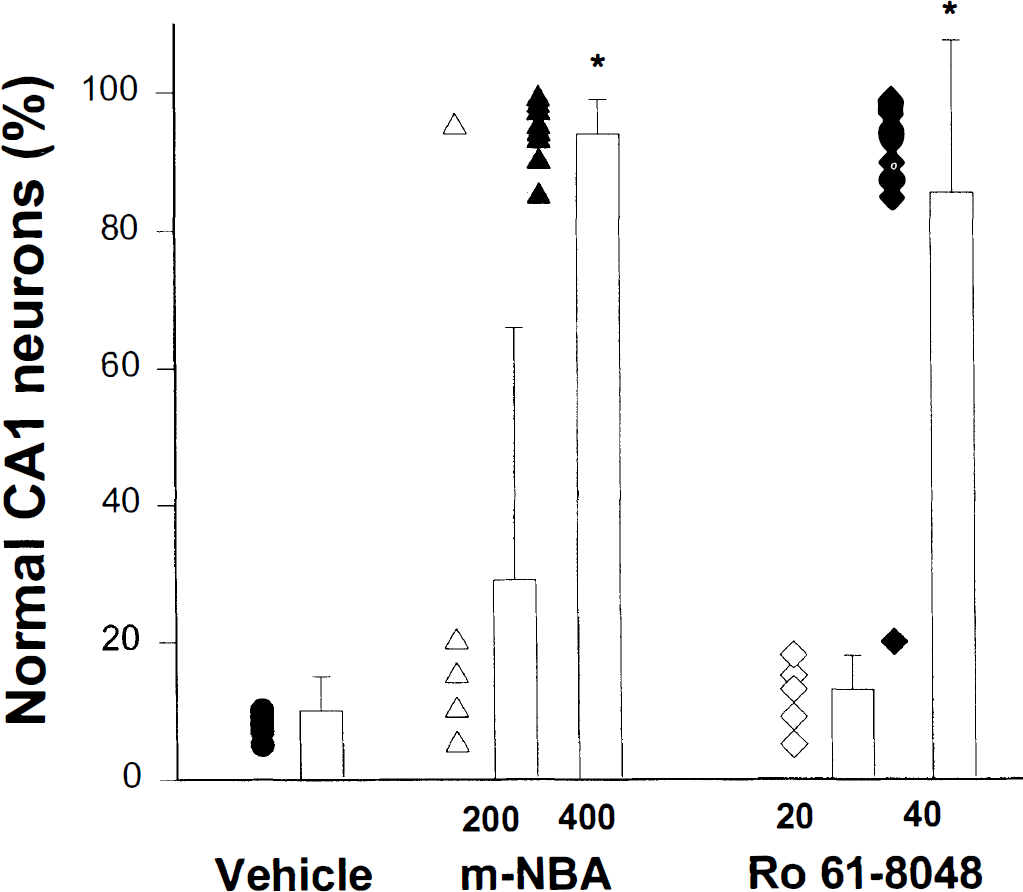
Pyramidal cell damage in the CA1 area in gerbils subjected to transient global ischemia. Each column is the mean percent value ± SD of nonlesioned pyramidal CA1 cells, and each circle (vehicle-treated animals), triangle (mNBA-treated animals), or diamond (Ro 61-8048-treated animals) represents the percentage of nondamaged pyramidal cells found in each gerbil. Sham-operated animals had 290 ± 40 cells in each 20-µm section of the CA1 region of the dorsal hippocampus. P < 0.001 versus vehicle-treated animals.
Intraperitoneal administration of lower doses of the compounds (200 or 20 mg/kg, respectively) given three times according to the schedule reported earlier did not reduce ischemic damage (Fig. 1). Similarly, when the compounds were injected twice (1 and 30 minutes after reperfusion) at 400 and 40 mg/kg, respectively, pyramidal cell loss was no different than that in the controls. The neuroprotective doses of both mNBA and Ro 618048 caused a significant decrease in body temperature in preliminary experiments. In the experiments reported in Fig. 1, body temperature was maintained at 37°C with a heating pad for 3 hours after surgery. We previously reported that in gerbils with bilateral carotid occlusion for 5 minutes, a decrease in body temperature (to 34°C), which started 90 minutes after the ischemic challenge and was maintained at this low level for at least 6 hours, did not affect pyramidal cell loss in the hippocampus (Pellegrini-Giampietro et al., 1994) but caused a slight increase in the number of deaths. In the current experiments, 3 of 15 gerbils treated with mNBA and 3 of 16 animals treated with Ro 61-8048 (400 or 40 mg/kg intraperitoneally, three times, respectively) and only 1 of 18 saline-treated gerbils died on day 2 after surgery.
Effects of mNBA and Ro 61-8048 on the extent of ischemic infarct after middle cerebral artery occlusion in rats
Neuroprotective effects of kynurenine hydroxylase inhibitors in rats with permanent MCAO
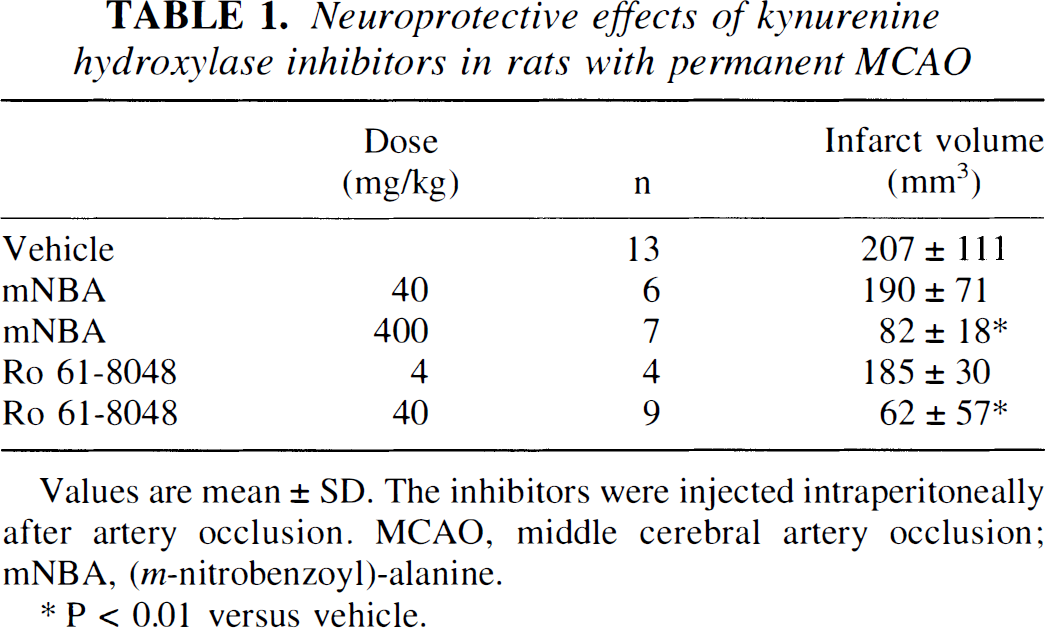
Values are mean ± SD. The inhibitors were injected intraperitoneally after artery occlusion. MCAO, middle cerebral artery occlusion; mNBA, (m-nitrobenzoyl)-alanine.
P < 0.01 versus vehicle.
Effects of mNBA and Ro 61-8048 on extracellular brain kynurenic acid concentrations
It has been previously reported that mNBA administration to rats with a dialysis probe in their hippocampus significantly increases KYNA concentrations in the dialysates (Carpenedo et al., 1994). In the current experiments, we found that mNBA significantly increased KYNA concentrations in the extracellular spaces of the gerbil hippocampus. Similar results were obtained with the new kynurenine hydroxylase inhibitor Ro 61-8048. Figure 2A shows that KYNA concentrations in the hippocampal dialysates increased by approximately 25-fold (from 2.3 ± 1.1 to 56 ± 16 nmol/L, means ± SD) after mNBA (400 mg/kg intraperitoneally, two times in 30 minutes) and from 2.2 ± 0.8 to 48 ± 10 nmol/L after Ro 61-8048 administration (40 mg/kg intraperitoneally, two times in 30 minutes). This increase started 40 minutes after the first administration of the inhibitors, and the maximal dialysate concentration was reached between 2 and 3 hours later.
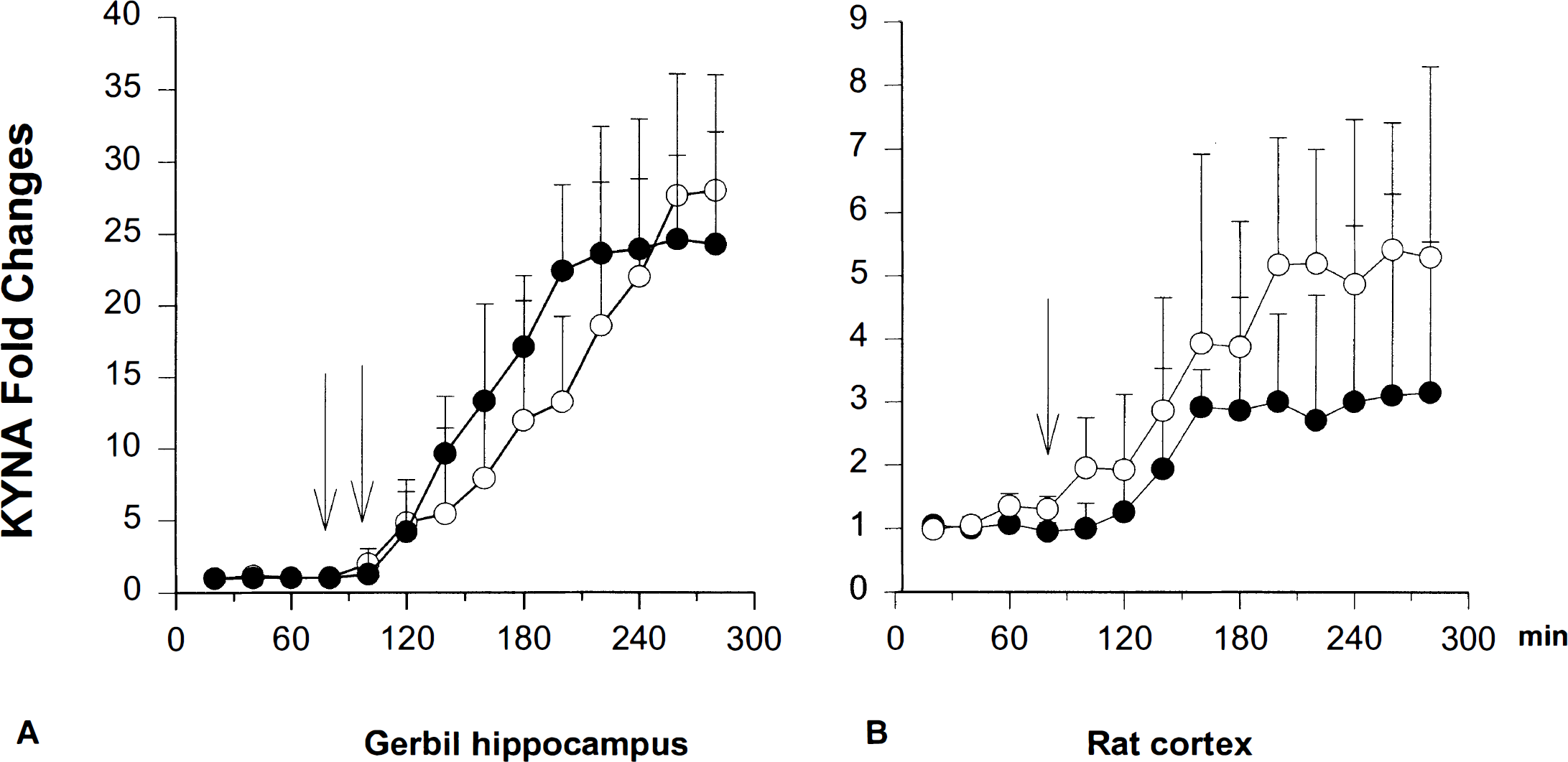
Similar experiments performed in rats with a dialysis probe in their cortex showed that mNBA (400 mg/kg intraperitoneally) increased KYNA concentrations in the dialysis perfusates from 16.5 ± 0.9 to 85 ± 9.6 nmol/L, and Ro 61-8048 (40 mg/kg intraperitoneally) increased it from 15 ± 0.9 to 45 ± 14 nmol/L. The time course of this increase is reported in Fig. 2B.
DISCUSSION
Kynurenine hydroxylase inhibitors have been reported to increase brain kynurenine and KYNA content (Moroni et al., 1991; Speciale et al., 1996b), to reduce spontaneous locomotor activity, and to increase the threshold of electroshock or audiogenic-induced seizures in rats and mice (Carpenedo et al., 1994). Interestingly, qualitatively similar pharmacologic effects have been described after systemic administration of large doses of glycine receptor antagonists (Sing et al., 1990), and it has therefore been proposed that these enzyme inhibitors reduce NMDA receptor function in vivo (Moroni et al., 1991).
Activation of NMDA receptors plays a key role in the neuronal damage found in models of global ischemia in gerbils and focal ischemia in rats (Chen et al., 1993; Pellegrini-Giampietro et al., 1994). The experiments reported here show that inhibition of kynurenine hydroxylase results in strong neuroprotection after ischemic insult and are therefore in line with the hypothesis that by shifting kynurenine metabolism toward KYNA formation, it is possible to reduce glutamate receptor activation and ischemic neuronal damage. Figure 2 shows, however, that KYNA concentrations in the extracellular spaces of the gerbil hippocampus increase from 2.3 ±1.2 to 56 ± 15 and from 2.2 ± 0.8 to 48 ± 10 nmol/L after administration of neuroprotective doses of mNBA or Ro 61-8048, respectively. Similarly, in the rat cortex, neuroprotective doses of mNBA or Ro 61-8048 increase KYNA concentration in the dialysates from 16.5 ± 0.9 to 85 ± 9.6 and from 15 ±0.9 to 45 ± 14 nmol/L, respectively. Considering that probe recovery is approximately 10% and that the affinity of KYNA for the glycine recognition site of the NMDA receptors is 10 to 30 µmol/L (Pellegrini-Giampietro et al., 1989), it can be concluded that the concentrations of KYNA required to reduce the function of ionotropic glutamate receptors should be approximately one order of magnitude larger than those found in brain dialysates. However, it is reasonable to assume that in the vicinity of the synaptic cleft the concentrations of KYNA are significantly larger than those measured in the dialysis fluid. This possibility is supported by immunocytochemical observations showing that the enzyme kynurenine aminotransferase, which is responsible for KYNA synthesis, is located mostly in glial cells in close apposition to the excitatory synapses (Du et al., 1992). It also should be considered that the Km of kynurenine for rat brain kynurenine amino transferase is in the millimolar range, thus suggesting that precursor availability is the major limiting factor for KYNA formation. Since kynurenine production depends on indolamine 2,3-dioxygenase activity and since ischemia strongly induces this enzyme (Saito et al., 1993), it is possible that KYNA formation in animals exposed to brain ischemia and treated with kynurenine hydroxylase inhibitors is larger than that found in the microdialysis experiments reported in Fig. 2. This also could be important in gerbils because kynurenine hydroxylase inhibitors were administered twice in microdialysis experiments, whereas neuroprotection was obtained only when a third administration was given.
The second mechanism possibly involved in the strong degree of neuroprotection of suitable doses of either mNBA or RO 618418 derives from the observation that KYNA synthesis in the brain, but not in the kidney, is significantly reduced by the lack of glucose, lactate, or pyruvate or by modifications of the ionic environment (Hodgkins and Schwarcz, 1998). Modifications in glucose and ionic composition of the extracellular medium qualitatively similar to those able to reduce KYNA synthesis occur in the ischemic brain, and, in preliminary experiments, it has been shown that oxygen and glucose deprivation reduce KYNA synthesis in organotypic hippocampal slice cultures (Peruginelli F., Carpenedo R., unpublished results). It is therefore possible that kynurenine hydroxylase inhibitors antagonize the changes in tryptophan metabolism, which seem to occur in brain tissue as a consequence of ischemic insults. Experiments on the time course of the changes in KYNA synthesis after MCAO or bilateral carotid occlusion in vivo currently are in progress to study this possibility.
Also notice that the direct product of kynurenine hydroxylase activity is 3-OH-kynurenine, a metabolite provided with excitotoxic action in neuronal cell lines (Eastman and Guilarte, 1989) and able to cause, in a concentration-dependent manner, either necrotic or apoptotic types of neuronal death (Okuda et al., 1998). Structure-activity studies show that 3-OH-kynurenine, and other o-aminophenols, may be subject to oxidative reactions initiated by their conversion to quinoneimines, a process associated with concomitant production of oxygen-derived free radicals (Hiraku et al., 1995). The involvement of these reactive species in the pathogenesis of ischemic neuronal death has been widely studied in the last several years, and it has been shown that oxygen-derived free radicals and glutamate-mediated neurotransmission cooperate in the development of ischemic neuronal death (Pellegrini-Giampietro et al., 1990). In particular, kynurenate derivatives provided with the ability to scavenge for free radicals are particularly potent in reducing excitotoxic damage in vitro (Moroni et al., 1992) and ischemic damage in vivo (Pellegrini-Giampietro et al., 1994). It is therefore reasonable to propose that the reduction in 3-OH-kynurenine synthesis and the associated decrease in the production of oxygen-derived free radicals contribute to the strong neuroprotective action exerted by mNBA or Ro 61-8048.
Finally, kynurenine hydroxylation is one of the steps required for the synthesis of quinolinic acid, a compound provided with excitotoxic properties. Either mNBA or Ro 61-8048 is able to reduce blood and brain quinolinic acid content in immune-activated rodents (A. Chiarugi, 1998, unpublished data), and this could contribute to their pharmacologic effects in brain ischemia.
Apart from the relative role that each of the earlier mentioned neurochemical effects of kynurenine hydroxylase inhibitors play in neuroprotection, the current experiments show that the brain damage found after focal or global ischemia may be reduced by appropriate administration of mNBA or Ro 61-8048 and that administration of these agents is effective when performed after the ischemic challenge. A single administration of either mNBA or Ro 61-8048 was sufficient to significantly reduce brain damage in MCAO rats, and at least three administrations (the last performed 6 hours after occlusion of the carotids) were necessary to reduce the death of pyramidal neurones in gerbils.
The kynurenine pathway of tryptophan metabolism has been widely studied in relation to inflammatory, degenerative, or excitotoxic insults. A local accumulation of KYNA associated with a reduction in NMDA receptor function has been obtained by administering indolpyruvate (Russi et al., 1989), kynurenine (Nozaki and Beal, 1992), or enzyme inhibitors (Carpenedo et al., 1994; Speciale et al., 1996b). In particular, it has been shown that large doses of kynurenine reduce NMDA toxicity in newborn rats (Nozaki and Beal, 1992) and that pretreatment with kynurenine hydroxylase inhibitors may reduce ischemic damage in the gerbil hippocampus (Speciale et al., 1996b). Our results further support the concept that kynurenine hydroxylase is an interesting target to indirectly modulate NMDA receptor function in vivo and show that the inhibition of this enzyme is sufficient to reduce neuronal loss in widely accepted models of brain ischemia.