
Abstract
Early postischemic hyperperfusion (EPIH) has long been documented in animal stroke models and is the hallmark of efficient recanalization of the occluded artery with subsequent reperfusion of the tissue (although occasionally it may be seen in areas bordering the hypoperfused area during arterial occlusion). In experimental stroke, early reperfusion has been reported to both prevent infarct growth and aggravate edema formation and hemorrhage, depending on the severity and duration of prior ischemia and the efficiency of reperfusion, whereas neuronal damage with or without enlarged infarction also may result from reperfusion (so-called “reperfusion injury”). In humans, focal hyperperfusion in the subacute stage (i.e., more than 48 hours after onset) has been associated with tissue necrosis in most instances, but regarding the acute stage, its occurrence, its relations with tissue metabolism and viability, and its clinical prognostic value were poorly understood before the advent of positron emission tomography (PET), in part because of methodologic issues. By measuring both CBF and metabolism, PET is an ideal imaging modality to study the pathophysiologic mechanism of EPIH. Although only a few PET studies have been performed in the acute stage that have systematically assessed tissue and clinical outcome in relation to EPIH, they have provided important insights. In one study, about one third of the patients with first-ever middle cerebral artery (MCA) territory stroke studied within 5 to 18 hours after symptom onset exhibited EPIH. In most cases, EPIH affected large parts of the cortical MCA territory in a patchy fashion, together with abnormal vasodilation (increased cerebral blood volume), “luxury perfusion” (decreased oxygen extraction fraction), and mildly increased CMRO2, which was interpreted as postischemic rebound of cellular metabolism in structurally preserved tissue. In that study, the spontaneous outcome of the tissue exhibiting EPIH was good, with late structural imaging not showing infarction. This observation was supported by another PET study, which showed, in a few patients, that previously hypoperfused tissue that later exhibited hyperperfusion after thrombolysis did not undergo frank infarction at follow-up. In both studies, clinical outcome was excellent in all patients showing EPIH except one, but in this case the hyperperfused area coexisted with an extensive area of severe hypoperfusion and hypometabolism. These findings from human studies therefore suggest that EPIH is not detrimental for the tissue, which contradicts the experimental concept of “reperfusion injury” but is consistent with the apparent clinical benefit from thrombolysis. However, PET studies performed in the cat have shown that although hyperperfusion was associated with prolonged survival and lack of histologic infarction when following brief (30-minute) MCA occlusion, it often was associated with poor outcome and extensive infarction when associated with longer (60-minute) MCA occlusion. It is unclear whether this discrepancy with human studies reflects a shorter window for tissue survival after stroke in cats, points to the cat being more prone to reperfusion injury, or indicates that EPIH tends not to develop in humans after severe or prolonged ischemia because of a greater tendency for the no-reflow phenomenon, for example. Nevertheless, the fact that the degree of hyperperfusion in these cat studies was related to the severity of prior flow reduction suggests that hyperperfusion is not detrimental per se. Preliminary observations in temporary MCA occlusion in baboons suggest that hyperperfusion developing even after 6 hours of occlusion is mainly cortical and associated with no frank infarction, as in humans. Overall, therefore, PET studies in both humans and the experimental animal, including the baboon, suggest that hyperperfusion is not a key factor in the development of tissue infarction and that it may be a harmless phenomenon when it occurs in the acute setting. However, an important issue that needs to be resolved by future studies with respect to EPIH relates to the possible occurrence of selective neuronal loss, as opposed to pan-necrosis, in the previously hyperperfused tissue.
Keywords
Clinical studies in which angiography was performed within the first 6 hours of middle cerebral artery (MCA) territory stroke and repeated at follow-up show that about 30% of the initial occlusions have cleared spontaneously by 24 hours and about 70% 1 week later (Fieschi et al., 1989). As reported in a few well-documented clinical cases, early spontaneous MCA recanalization can be associated with immediate, near-complete, or full resolution of neurologic deficits (Taneda et al., 1985; Minematsu et al., 1992; Ringelstein et al., 1992). Accordingly, the goal of thrombolytic therapy is to accelerate clot lysis and to restore CBF in the ischemic penumbra, and in turn reduce infarct size and improve clinical outcome. Although large clinical therapeutic trials conducted in the United States, Europe, and Australia highlight the possible benefits of thrombolysis when performed within the first 3 hours, the increased occurrence of major life-threatening side effects (hemorrhagic transformation) in some patients, however, implies the need “to define and quantify the risks and benefits of thrombolysis much more clearly before it finds its place in the treatment of acute ischemic stroke” (Wardlaw et al., 1997). Thus, the issue of recanalization in terms of clinical benefit remains complex, and this situation may reflect our insufficient knowledge of the pathophysiologic events associated with recanalization of the previously ischemic brain. For instance, recanalization does not necessarily mean adequate tissue reperfusion (von Kummer et al., 1995; Baird et al., 1996). Thus, complete (effective) reperfusion may not occur despite recanalization because of complex and intricate phenomena leading to “no-reflow,” such as hemorrhage, edema, polymorphonuclear leukocytes and platelet plugging, tissue factor-mediated coagulation system activation in the microvessels, and endothelial and subendothelial reperfusion injury (Thomas et al., 1993, del Zoppo, 1997). Conversely, partial, or perhaps complete, reperfusion may occur through an ischemic zone despite persistent middle cerebral artery occlusion (MCAO) as a result of restoration of perfusion pressure through leptomeningeal collateral vessels (Symon et al., 1963; Heiss et al., 1976). Another critical factor for tissue outcome is the time of occurrence of reperfusion after the arterial occlusion, since infarct size increases progressively with longer occlusion periods up to a certain time (Sundt et al., 1969; Weinstein et al., 1986; Kaplan et al., 1991; Memezawa et al., 1992, Minematsu et al., 1992; Wardlaw et al., 1993; Touzani et al., 1997; Young et al., 1997). For instance, Memezawa et al. (1992) found in rats that after 15 minutes of MCAO, selective neuronal necrosis involved the caudoputamen; after 30 minutes of occlusion, infarcts were located to the lateral caudoputamen, and a selective neuronal necrosis involved the cortex in some animals; an occlusion time of 60 minutes induced cortical infarction, and the infarct size increased to a maximum at up to 120 to 180 minutes of occlusion time. These findings therefore suggest that early reperfusion prevents, rather than promotes, extension of the infarct in rodents. However, rodent studies suggest that, at least in some conditions, reversal of arterial occlusion and reoxygenation of brain tissue may be harmful and may lead to edema formation, hemorrhagic transformation, partial neuronal loss, and larger infarcts as a result of so-called reperfusion injury (Dietrich, 1994; Aronowski et al., 1997; Kuroda and Siesjö, 1997).
Postischemic hyperperfusion has long been documented in animal stroke models and is the hallmark of efficient recanalization and reperfusion (Sundt et al., 1969, 1971; Tasdemiroglu et al., 1992). It is interpreted as reflecting the abrupt, or more gradual, restoration of the blood pressure to normal or near-normal values in a cerebrovascular bed dysregulated by prior severe ischemia. Thus, hyperperfusion may be a useful marker of postischemic reperfusion in the clinical setting, where sequential imaging studies are impractical. However, it is unclear whether hyperperfusion plays a harmful, or, conversely, a beneficial specific effect with respect to the reperfused tissue, or else is an epiphenomenon devoid of specific consequences. As a first approach to the problem of recanalization, however, the main question is whether early spontaneous hyperperfusion after ischemic stroke in humans is hazardous to the brain tissue and thus detrimental to clinical outcome. By using combined perfusion and structural imaging techniques with different tracers (e.g., 133Xe, 99mTc-HMPAO single photon emission computed tomography [SPECT]), several authors address this question, but their results, which are briefly summarized later, have been discrepant. Despite its inherent logistic difficulties, positron emission tomography (PET) is the ideal method to study, three-dimensionally, the relations between, on one hand, early postischemic hyperperfusion (EPIH) observed on quantitative CBF images, and, on the other hand, the local cerebral blood volume (CBV; an index of local vasodilation and thus of true hyperemia), the OEF (oxygen extraction fraction, an index of the coupling between flow and oxidative metabolism), and the CMRO2 (Baron et al., 1989), as well as tissue outcome (i.e., pan-necrosis [completed infarction], selective neuronal loss, incomplete infarction, or integrity) and, finally, clinical outcome. These are important goals, since therapeutic implications may result from such observations, especially with respect to thrombolysis and agents directed against reperfusion injury.
Definition of postischemic hyperperfusion
Table 1 lists several words important for the current review and their definitions as they are used here. Regarding hyperperfusion, one practical issue is its operational definition. Its strict definition is an increase in CBF relative to preocclusion values. However, this definition generally is impractical, especially in human studies, because of the lack of prestroke measurement of CBF. In addition, even in the experimental animal, this definition is unreliable at the individual level because of the well-known variability in absolute CBF values from one measurement to the next because of the sensitivity of CBF to general physiologic factors such as Paco2, Pao2 hematocrit, arousal, and possibly also stress hormones. In human studies, one definition that was used in the era of unilateral measurements was the presence of tissue with CBF significantly above the average hemispheric mean (Olsen et al., 1981, 1984), but this is considered over-permissive, since the mere presence of an ischemic focus may result in an area of pseudohyperperfusion in the surrounding tissue. Thus, for instance, the classic notion of a “rim of hyperperfusion” surrounding the ischemic focus was based on data interpreted with the latter definition (Olsen, 1986) but is only exceptionally observed in acute stroke with tomographic methods. Thus, since the advent of the latter methods as well as in the following autoradiographic studies in the laboratory animal (Yamaguchi et al., 1971), the generally used definition for hyperperfusion has been a significant increase in CBF relative to the homologous area of the contralateral hemisphere. This is considered valid, since the general physiologic factors just mentioned should, in most instances, affect both hemispheres to the same extent, whereas the occurrence of reduced CBF in the contralateral (unaffected) hemisphere (so-called transhemispheric diaschisis) does not seem to develop before the subacute stage of stroke (Yamaguchi et al., 1971; Andrews, 1991; Pappata et al., 1993; Iglesias et al., 1996) and, therefore, should not affect the observation.
Definition of terms used in this review
Historical background
Early studies. Several landmark studies document EPIH in both animals and humans. In the 1950s, Meyer et al. (1954) occasionally observed increased tissue oxygen tension in the border zone territories after MCAO in monkeys; after deocclusion, there were instances of prolonged states of abnormally high oxygenation in extensive areas. The late Niels Lassen, in his famous 1966 report published in the Lancet, defined what he called the “luxury-perfusion syndrome” as “overabundant cerebral blood flow relative to the metabolic needs of the brain tissue” with “abnormally small cerebral arteriovenous difference of oxygen”; “after a period of vascular arrest the blood flow is temporarily increased above the resting level in all tissues but more pronounced in the brain”; “a few minutes of hypoxia are followed by reactive hyperemia lasting hours”; and “the hyperemia lasts much longer than would be needed to reoxygenate the brain, thus the cerebral circulation is over-abundantly perfused with bright-red venous blood.”
Frequency and duration of the postischemic hyperperfusion period in experimental stroke
Experimental studies show that, as is the case for other organs, brain hyperperfusion is an almost constant phenomenon after release of arterial occlusion. In rats, numerous studies show that restoration of cerebral perfusion pressure after a period of ischemia consistently results in a marked and variably prolonged hyperperfusion, such that the peak and duration of reactive hyperemia tend to increase with the duration of prior ischemia; this postischemic hyperperfusion phase often is followed by a phase of secondary hypoperfusion generally associated with a poor tissue prognosis (Traupe et al., 1982; Gourley and Heistad, 1984; Todd et al., 1986; Tasdemiroglu et al., 1992).
In ketamine-anesthetized cats (Tamura et al., 1980), postischemic hyperperfusion was observed upon recirculation after 2 hours of MCAO. In this instance, the degree of hyperperfusion in the severely damaged cortex (measured by the hydrogen gas clearance technique) was strongly associated with the leakage of Evans blue and intracerebral petechial hemorrhages. The results suggest that the effect of postischemic hyperperfusion is determined by the severity of tissue injury during the ischemia, which, in turn, is determined both by the degree of CBF reduction and the duration of the ischemic insult (Garcia et al., 1983).
Changes in cortical CBF in response to both permanent and temporary MCAO also were documented by Harada et al. (1988). The authors observed postischemic hyperperfusion in the ectosylvian cortex of the cat but only in the transiently occluded group (l hour).
Postischemic hyperperfusion (or hyperemia) also has been noted in the squirrel monkey (Sundt and Waltz, 1971; Hanson et al., 1975) and in the anesthetized baboon (Symon et al., 1972). After temporary MCAO of 2 hours' duration (Sundt and Waltz, 1971), cortical hyperperfusion was noted in 50% of monkeys immediately after restoration of flow. The authors conclude that this reactive hyperemia developed as a consequence of the preexisting ischemia and, further, that cerebral edema was incompletely correlated with the degree of ischemia or the degree of hyperperfusion.
In the baboon, Crockard et al. (1976) demonstrated that 30% of the electrodes used to measure CBF by the hydrogen clearance technique showed hyperperfusion immediately after removal of the MCA clip in 13 animals. Likewise, in the baboon, Yonas et al. (1990) showed that 20 minutes of vascular occlusion resulted in a brief episode of hyperperfusion with no subsequent alteration seen on computed tomography (CT) and only minimal random neuronal injury.
In summary, the forgoing experimental studies indicate that hyperperfusion is a common finding in models of temporary focal cerebral ischemia whether they are performed in the rat, cat, or a nonhuman primate. As to the consequences of postischemic hyperperfusion, there is no consensus of opinion as to its beneficial or detrimental effects on the size of the eventual infarct.
133Xenon and SPECT studies in humans
By combining single-probe 133Xe-CBF and angiographic studies, Paulson et al. (1970a, b) report hyperperfusion foci in four of five patients without angiographic occlusion studied within 36 hours from onset, compared with one of seven patients in whom MCAO was present. Interestingly, some of the patients with normal findings on angiogram remained symptomatic for a while after the CBF measurement, which in retrospect might suggest “brain stunning” in analogy to the phenomenon of myocardial stunning described in ischemic heart disease.
Olsen et al. (1981), also using the two-dimensional intracarotid 133Xe injection method, found a frequent occurrence of focal hyperperfusion in patients with MCA stroke studied 1 to 4 days after symptom onset. In the acute stage (less than 48 hours), foci of absolute hyperperfusion were found in 4 of 12 patients studied; in only 1 patient, studied 10 hours after symptom onset, did the hyperperfused area correspond to the CT-defined infarct; however, there was no arterial occlusion on the angiogram in three cases, which showed capillary blush and early filling of veins in the hyperperfused area (Hoedt-Rasmussen et al., 1967). In a subsequent report (Olsen and Lassen, 1984), only relative hyperperfusion was described, making interpretations less reliable. The authors conclude that, since early hyperperfusion could be seen both with and without subsequent infarction, it did not have in itself a noxious effect on brain tissue. Studies of CBF reactivity to Paco2 and CBF autoregulation by these authors disclose abnormal regulation in hyperperfused areas associated with final infarction but only little dysregulation in those areas associated with normal findings on late CT scan. However, in these classic studies, the relations between hyperperfusion and infarction were not optimally assessed, mainly because the CBF procedure used was only two-dimensional, and only the affected side was studied, thus limiting the exact delineation of the regions with hyperperfusion (see earlier).
Hartmann (1985), using the bilateral 133Xe probe detector inhalation technique, reports the occurrence of hyperperfusion in the affected hemisphere in two patients who fully recovered in the subsequent hours (“transient ischemic attacks”) consistent with EPIH in a tissue that retained its integrity.
With regard to investigations performed using SPECT, only a few studies have been performed in the acute stage of stroke, and most concerned “metabolic microsphere” tracers. In two patients studied within the first 6 hours of stroke, Shimosegawa et al. (1994, 1995) report an increased fixation of the radiotracer 99mTc-HMPAO in the late-CT hypodense area. Shintani et al. (1995) also report one case of acute stroke in whom an area of massive hyperfixation of 99mTc-HMPAO was found centered in the deep MCA territory and which corresponded to a small infarct in the late MRI scan; the patient made a spectacular recovery. Conversely, the “spectacular shrinking deficit” syndrome, a profound hemispheric stroke syndrome that resolves rapidly over hours to days and leaves only minimal residual deficits, was found to be consistently associated with early tissue reperfusion, with moderate 99mTc-HMPAO hyperfixation observed in one case of five (Baird et al., 1995). Regarding this question of early reperfusion of a previously hypoperfused area, most SPECT studies show clinically beneficial effects (Herderschee et al., 1991, Baird et al., 1996; Barber et al., 1998; Grotta et al., 1998) with rare exceptions (Hanson et al., 1993). In addition to methodologic differences among studies, these discrepant results in the SPECT literature might be related to the tracers used as the metabolic transformation underlying their tissue fixation may be influenced by the local biochemical environment (e.g., tissue acidosis), itself a function of the ischemic cell derangement. Furthermore, all of the tracers used have a limited passage through the blood-brain barrier with, as a consequence, a nonlinear relation between tracer uptake and true CBF at high flow rates and hence a poor sensitivity to mild or moderate hyperperfusion. In addition, 99mTc-HMPAO may show “spurious hyperfixation” in necrotic tissue, which does not exhibit hyperperfusion with 133Xe-SPECT imaging (Sperling and Lassen, 1993), probably as a result of abnormally increased penetration through a disrupted blood-brain barrier and increased retention from a disturbed tissular environment (Shintani et al., 1997). Finally, 99mTc-ECD, although marketed as a perfusion tracer, may reflect cellular functioning more than perfusion (Shishido et al., 1994), which may account for the lack of reported acute stroke cases with increased tracer uptake with this agent (Berrouschot et al., 1998). Overall, therefore, SPECT studies with these kind of tracers do not seem appropriate for the assessment of EPIH. In one study of acute stroke that used 133Xe-SPECT, Nakagawara et al. (1997) report that 2 patients of 14 showed an area of hyperperfusion in relation with the recanalization noted on the angiogram.
CLINICAL PET STUDIES IN THE ACUTE STAGE OF STROKE
Early PET studies
Although PET has led to fundamental new insights into the complex pathophysiologic mechanism of cerebral ischemia in humans (see Baron, 1996 for review), studies in the acute stage of stroke (less than 48 hours) are scarce because of inherent logistic difficulties. Regarding the subacute stage (more than 48 hours), however, several PET studies report that the occurrence of hyperperfusion increased in frequency until about 10 to 15 days after stroke, and that it was consistently associated with low OEF and markedly reduced CMRO2, indicative of luxury-perfusion. The outcome of such areas was almost universally poor, exhibiting necrosis on the late structural imaging procedures (Ackerman et al., 1981; Baron et al., 1981, 1983; Lenzi et al., 1982; see also the SPECT studies of Sugiyama et al., 1986 and Tran Dinh et al., 1997). Based on histopathologic correlations in the cat (Yamaguchi, 1977), this late hyperperfusion in the necrotic core presumably reflects neovascularization with increased capillary density and endothelial hypertrophy.
Two PET studies concerned the period up to 48 hours after stroke onset. Hakim et al. (1987) studied 12 patients and determined the hemodynamic and oxygen and glucose metabolic changes, as well as the cerebral pH, in the severely hypometabolic (presumably infarcted) cortex. They found that this class of tissue exhibited hyperperfusion (i.e., absolute luxury perfusion) in one third of their patients (all were studied later than 23 hours after clinical onset); in these regions, anaerobic glycolysis was enhanced without persistent regional acidosis. Neither tissue nor clinical outcome were prospectively assessed, and the changes in the rest of the ischemic tissue was not reported. Fink et al. (1993) studied 22 patients and defined the probable core of the developing infarct as the most severely affected region on CBF or CMRglc images. They found regions of interest with significant hyperperfusion (CBF greater than the mean + 2 SD of the unaffected hemisphere) in regions surrounding this core in nine patients and always mixed with hypoperfused areas. Hyperperfused regions had a glucose extraction fraction lower than corresponding mirror regions but relatively preserved glucose consumption, and they did not become completely necrotic on follow-up CT or magnetic resonance imaging (MRI) (Fink et al., 1993). Clinical correlations were not discussed. Based on these two studies, it therefore appears that hyperperfusion observed up to 48 hours after stroke can be associated either with early infarction or with essentially preserved tissue.
PET studies of early spontaneous hyperperfusion
The group in Caen, France, prospectively collected a PET stroke data bank according to a strict protocol, which is described in detail elsewhere (Marchal et al., 1993, 1996). A PET study of CBF, OEF, CBV, and CMRO2 with the oxygen 15 steady-state technique (Baron et al., 1989) was performed 5 to 18 hours after onset of symptoms in 30 patients with first-ever MCA territory stroke, that is, with a clinical picture suggesting ischemia in the cortical MCA territory. Patients presenting with early hemorrhagic transformation were excluded. The spontaneous neurologic course was assessed according to Orgogozo's scale (Orgogozo, 1998), which has been validated for MCA stroke, and scoring was obtained at fixed points in time (around days 0, 7, 21, and 60). In survivors, a late CT scan was performed in the chronic stage to delineate the final infarct contours, and a second PET study was done about 1 month after clinical stroke onset. Both PET data sets and the late CT images were coregistered according to a stereotaxic reference.
Analysis of the PET images revealed three distinct patterns of CBF and CMRO2 changes, a finding taken to indicate the existence of considerable pathophysiologic variability within the time interval studied (Marchal et al., 1993, 1995). Pattern I was characterized by a large corticosubcortical area of profoundly reduced CBF and CMRO2 suggesting already irreversibly damaged tissue; the tissue and clinical outcome in each of the nine patients of this category was poor (early death from brain swelling or large infarct with persisting severe neurologic handicap in the survivors). One patient of this category displayed a border zone of hyperperfusion (see later). Pattern II was characterized by a moderate or profound reduction in CBF without an extensive area of severe CMRO2 defect, suggesting ischemia-in-evolution without extensive irreversible tissue damage (i.e., penumbra); the tissue and clinical outcome in the 11 patients of this category varied (from large to small infarcts, and from death to full neurologic recovery), possibly as a result of survival of some of the penumbra from spontaneous restoration of blood flow after the PET study, for example. Pattern III was characterized by focal hyperperfusion without associated hypoperfusion or extensive irreversible tissue damage; it was observed in 10 patients (Fig. 1). The frequency of occurrence of early spontaneous hyperperfusion (less than 18 hours) in this sample (one third of cases) matched the average frequency of early spontaneous recanalization documented by angiography (Fieschi et al., 1989). The following sections describe the characteristics of these 10 patients in terms of clinical, structural imaging, and PET variables in both the acute stage and at follow-up.
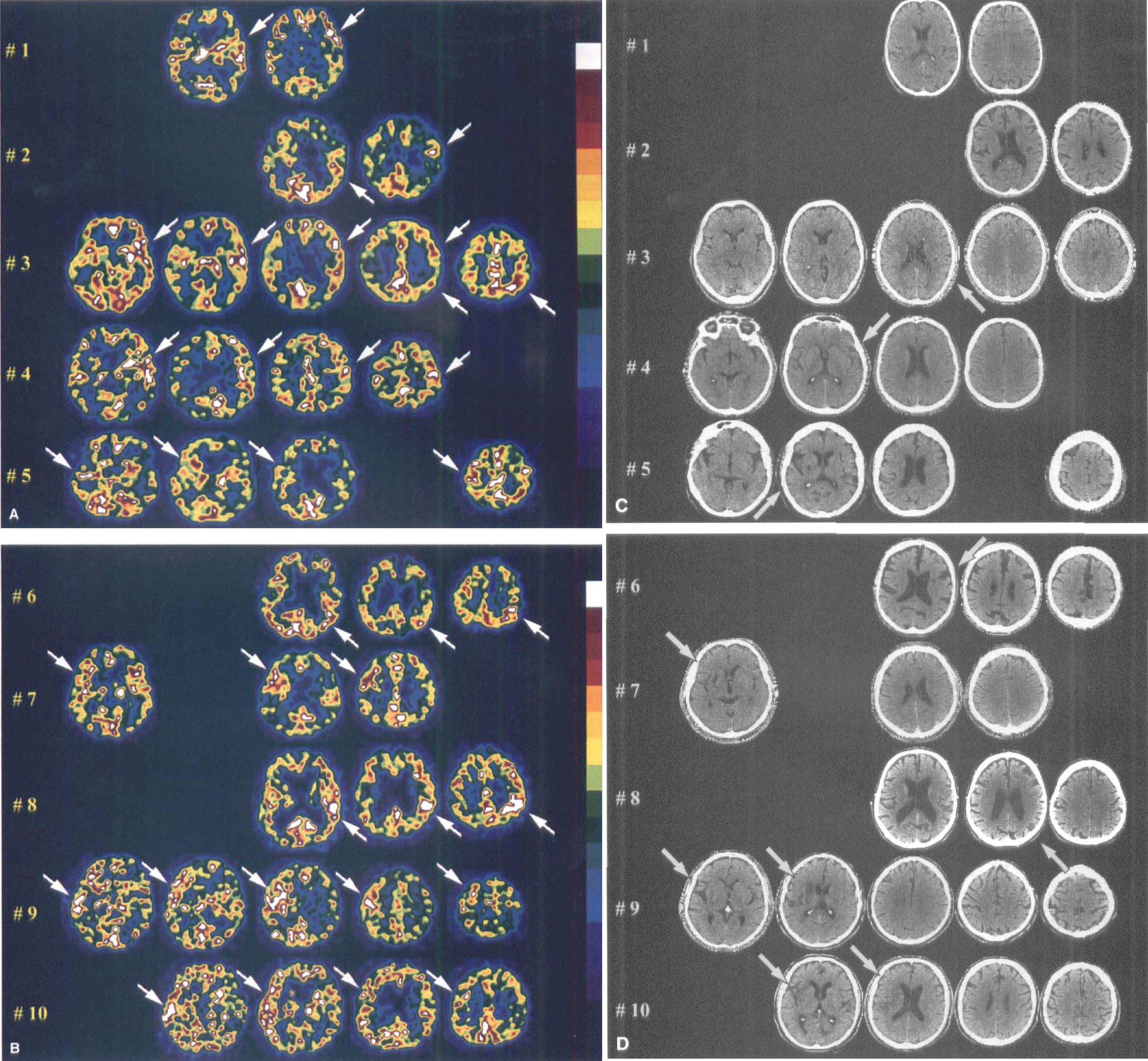
Acute stage CBF positron emission tomography (PET) (performed 5 to 18 hours after symptom onset) showing relevant slices with significant hyperperfusion (
Neurologic impairment and spontaneous clinical outcome in patients with early focal hyperperfusion
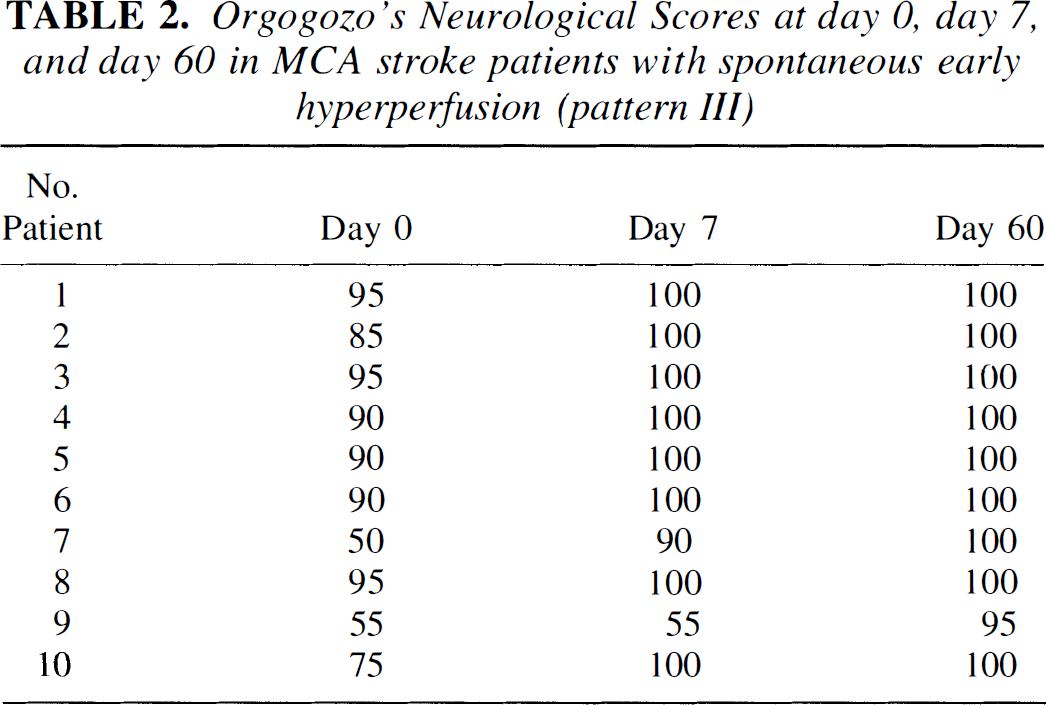
As a group, the pattern III patients were less affected clinically at the time of PET than those of the other PET categories, particularly of pattern I (Orgogozo scores: 82.8 ± 17.5 versus 25 ± 13.9, respectively, P < 0.05) (Fig. 2), which was consistent with the early clinical observations of Hakim et al. (1987). However, taken individually, the day 0 Orgogozo scores overlapped according to the three PET patterns, indicating that the clinical picture by itself is not sufficient to predict the individual pathophysiologic situation. Figure 3 shows the distribution of patients identified into the three PET patterns according to the initial clinical status and the neurologic evolution at 2 months. There was no significant relation between day 0 and day 60 Orgogozo scores for the patients who scored between 20 and 70 at admission, but within this interval the classification of patients according to the three CBF-CMRO2 patterns did have prognostic value (P < 0.05, Fisher's exact test) (Marchal et al., 1995). Neurologic recovery in pattern III patients was remarkably rapid, occurring within 7 days for most (Table 2, Fig. 2). Based on reports about the relations between early artery recanalization and clinical outcome (Ringelstein et al., 1992; von Kummer et al., 1995) and on the lack of persistent MCA stem occlusion at transcranial Doppler ultrasound in all 10 cases, the advanced interpretation was that spontaneous clot dissolution had occurred at some unknown time point before the PET study in these patients and resulted in favorable outcome.
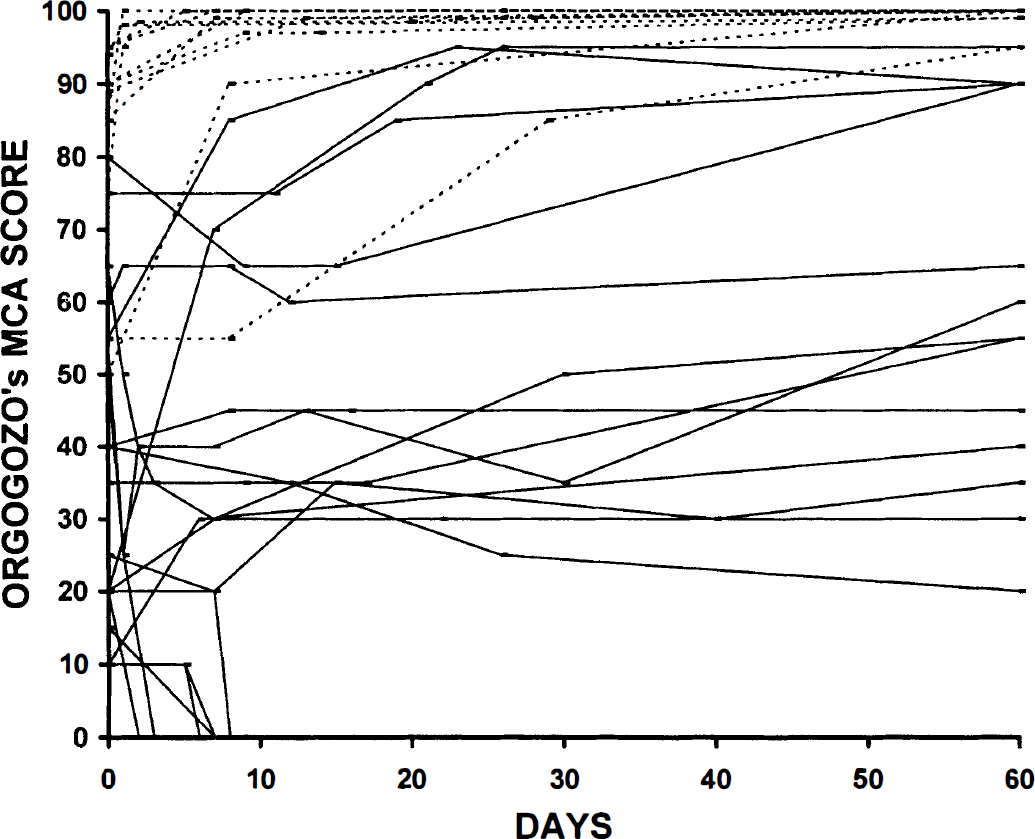
Neurologic status at the day of symptom onset and neurologic evolution over the first 2 months, scored with the Orgogozo's neurologic scale in the population of patients in the study of Marchal et al. (1995) with first-ever MCA territory stroke (n = 30), The 10 pattern III patients generally were less affected at admission than the patients belonging to the two other PET patterns, but overlap with the other PET patterns was present for two patients. However, clinical recovery was excellent in all and rapid (over days only) in all but one. Dashed line represents patients with pattern III (early hyperperfusion).
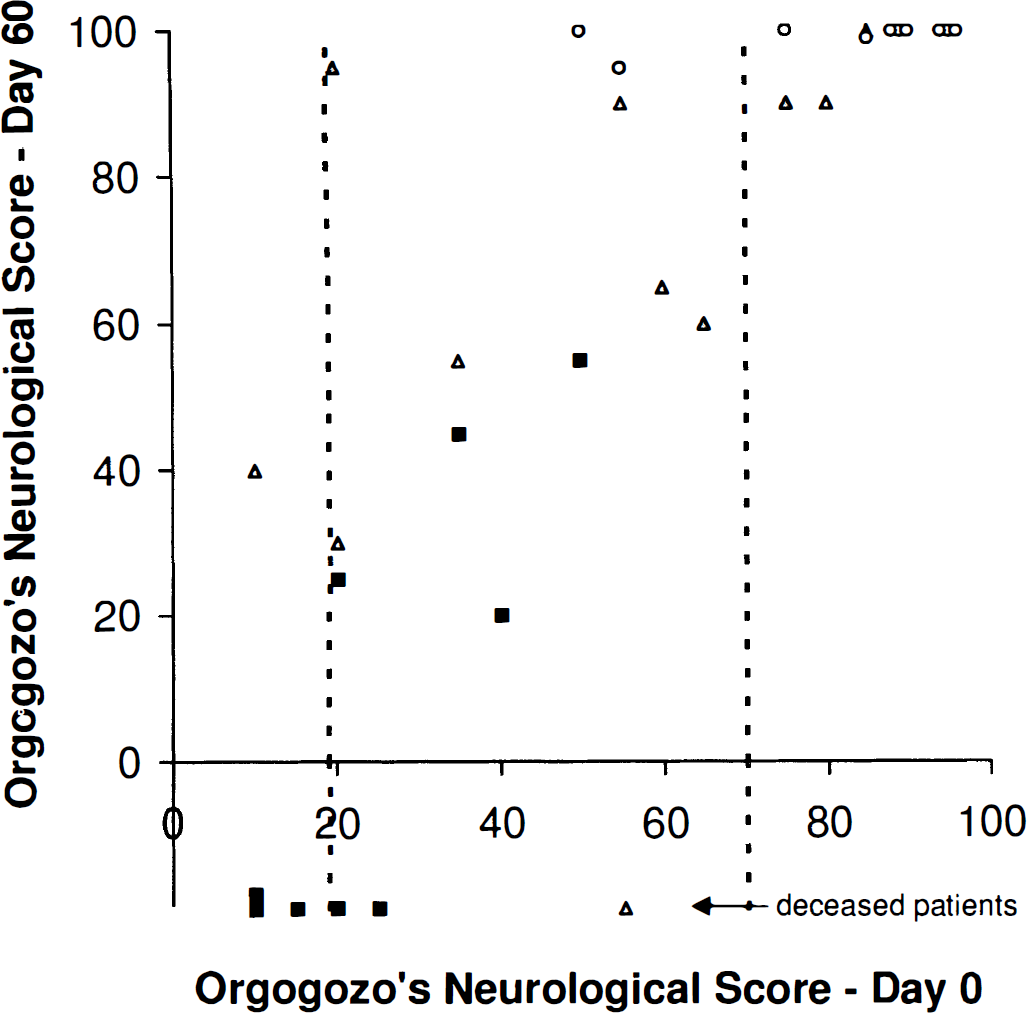
Relationships between day 0 and day 60 Orgogozo scores in the 30 patients of Marchal et al. (1995) identified according to the three CBF-CMRO2 PET patterns, There is a significant positive correlation between day 0 and day 60 clinical scores (P = 0.001, Spearman nonparametric rank correlation), However, in the 20 to 70 interval of day 0 Orgogozo's scores (shown as dotted lines), the PET patterns better predicted day 60 neurologic outcome better than did day 0 Orgogozo scores (P < 0.05, Fisher's exact test, versus not significant, Spearman). Notice that 2 of the 10 pattern III patients belong to this interval. Survivors (n = 24). The six deceased patients were arbitrarily assigned a negative Day 60 Orgogozo score, (■) Pattern I. (▲) Pattern II. (ˆ) Pattern III.
Orgogozo's Neurological Scores at day 0, day 7, and day 60 in MCA stroke patients with spontaneous early hyperperfusion (pattern III)
Structural outcome of the hyperperfused regions
To assess the relations between the hyperperfused areas and the final infarct and to determine their hemodynamic and metabolic status at both PET studies, Marchal et al. (1996) performed a detailed and objective image analysis. To isolate the areas of relative hyperperfusion, isocontours, including all voxels with CBF values above the maximal pixel value in the contralateral homologous mirror regions, were determined. Then, the contours of hypodensities on the late CT scan were delineated and projected onto the corresponding coregistered PET slices. Regions with focally increased CBF were patchy and mainly cortical and spread over most of the affected MCA territory; in contrast, the infarcts, when present, were small and deep seated; in two patients, no infarct was detected (Fig. 1). There was no overlap between the areas of hyperperfusion and the areas of infarction (although questionable overlap was possible in patients 4 and 9, Fig. 1), indicating that hyperperfusion in itself did not cause tissue necrosis. That the hyperperfused regions were mainly cortical and, in most cases, spread over the entire cortical MCA territory, and that the infarcts were small and deep seated was taken as indirect evidence that, early on, the MCA was occluded at the origin of the lenticulostriate arteries. The most likely explanation advanced for the overall lack of hyperperfusion in the infarct itself at the moment of the PET study was partial/patchy no-reflow caused by irreversible tissular and microvascular damage incurred during the phase of ischemia.
However, three uncertainties were pointed out by the authors regarding these correlations with tissue outcome. First, as stated earlier, in one patient classified as pattern I, there was a focus of hyperperfusion bordering an extensive area of near-zero CBF and CMRO2, suggesting recanalization of one branch of the MCA; since this patient died early and no autopsy could be performed, it was impossible to know the eventual status of the hyperperfused tissue. However, because it was associated with normal CMRO2 values, the hyperperfused tissue likely was still viable at the time of PET. Second, cases with cerebral hemorrhage at admission CT were excluded from the study, so that it is unknown whether hyperperfusion in association with acutely hemorrhagic infarction could have been missed. Third, the normal CT appearance rules out pan-necrosis but does not formally eliminate minor damage such as selective neuronal loss (Lassen, 1982; Garcia et al., 1996). However, the hemodynamic and metabolic parameters within the hyperperfused regions were taken as evidence against this latter interpretation (see later).
Hemodynamic and metabolic data in the regions with hyperperfusion
In the hyperperfused areas, and compared with contralateral homologous regions, the CBF was increased by 74% on average, the OEF was significantly reduced (average −22%), and the CBV significantly increased (average +20%) (Table 3). These changes in OEF and CBV indicated the presence of the luxury perfusion type of flow-metabolism uncoupling, together with postischemic “vasoplegia,” consistent with EPIH and supporting the hypothesis of prior ischemia (Marchal et al., 1996). More surprising, the CMRO2 was moderately but significantly increased (average +34%). This unexpected finding was supported by earlier reports in cats and gerbils, showing a delayed increase in CMRO2 after transient global brain ischemia (Hossmann et al., 1976; Nemoto et al., 1981). Two opposing interpretations were given for this postischemic oxygen hypermetabolism: (1) an overshoot of cellular metabolism (i.e., specific protein synthesis, oxidative phosphorylation, ATP formation, synthesis of transcript factors or growth factors) in cells destined to survive; or (2) excessive firing of neurons undergoing irreversible damage from massive release of excitatory amino acids during the period of ischemia (Pulsinelli, 1992), or early noxious inflammatory processes (Stoll et al., 1998). Evidence against the latter mechanisms was the lack of abnormally low CMRO2 at the second PET study (Table 3), since the CMRO2 should have considerably declined had the hyperperfused areas undergone incomplete infarction from excitotoxic mechanisms. This finding also was taken as evidence against the hypothesis of partial neuronal loss occurring as a result of reperfusion injury with oxidative stress, for example. However, in one patient (patient 1, Table 3), there was a reduction of CMRO2 in the chronic stage, which could represent neuronal loss. The slight reduction of CBV in the chronic stage was interpreted as reflecting cortical deafferentation from the deep-seated infarcts (Baron, 1996).
Hemodynamic and metabolic values in the acutely hyperperfused and contralateral mirror regions in the acute and chronic stage PET studies in 10 patients with early post-ischemic hyperperfusion
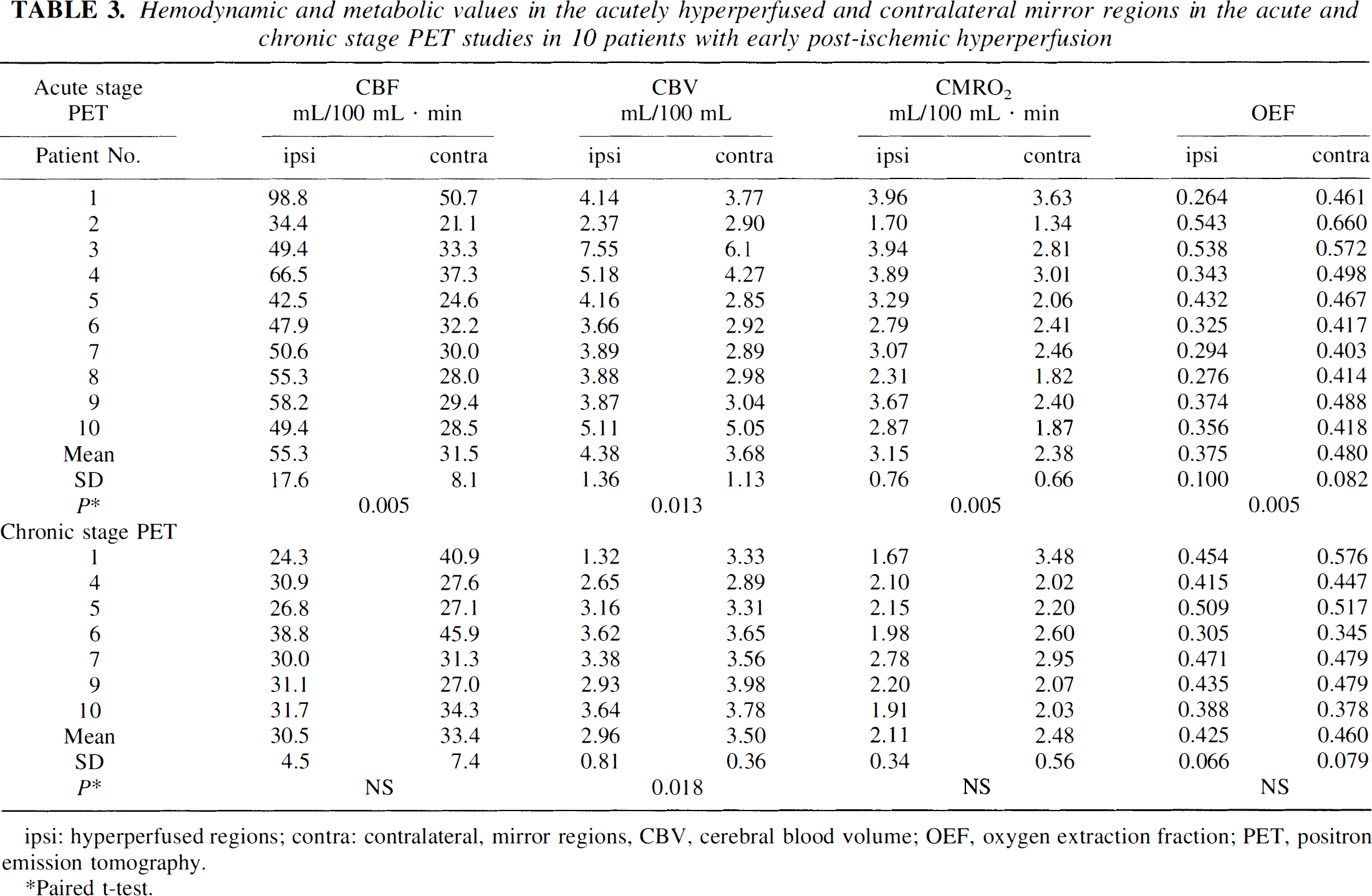
hyperperfused regions; contra: contralateral, mirror regions, CBV, cerebral blood volume; OEF, oxygen extraction fraction; PET, positron emission tomography.
Paired t-test.
In summary, the findings of Marchal et al. (1993, 1995, 1996) suggest that when observed 5 to 18 hours after stroke onset, focal hyperperfusion without already extensive irreversible damage invariably predicted minute (or absent) infarct and excellent spontaneous recovery, so that these patients would not be rational candidates for stroke treatment. Spontaneous arterial recanalization/reperfusion presumably occurred sufficiently early to prevent the ischemic tissue from evolving toward infarction. The good tissular outcome without evidence of selective neuronal loss favors the idea that the phenomenon of reperfusion injury reported in some animal studies did not take place in these patients and that agents to reduce this phenomenon would have been of limited benefit.
PET studies of [11C]-flumazenil binding
A study by Heiss et al. (1998), however, reports data that suggest that hyperperfusion sometimes may be associated with incomplete infarction or selective neuronal loss. They studied the distribution of [11 C]-flumazenil, a PET radioligand selective for the central benzodiazepine receptor (Sette et al., 1993) (which is exclusively borne by neurons) in several patients within the acute stage of ischemic stroke (less than 16 hours from symptom onset) and compared it with CBF and CMRO2, as well as with subacute stage CMRglc and structural MRI findings. In one patient, there was marked hyperperfusion in the clinically affected territory, together with mildly reduced specific binding of the radioligand, but little change in CMRO2. However, in the subacute stage, this area showed reduced repeat [11C]-flumazenil binding and reduced CMRglc despite unaltered MRI. These findings were interpreted as reflecting incomplete cortical infarction, possibly explaining the mild residual language disturbances of this patient. Using SPECT methodology, Nakagawara et al. (1997) also report two patients with extensive hyperperfusion on 133Xe scans in the acute stage who exhibited reduced binding of [123I]-iomazenil in these areas in the chronic stage despite normal findings on late CT scan. There was no clear clinical correlation for this finding. However, histologic proof for the interpretation of selective neuronal loss in these cases is lacking, whereas recent data from baboon studies in our laboratory (Watanabe et al., 1999) indicate that reduced cortical [11C]-flumazenil-specific binding may not always correspond to neuronal loss and perhaps, in some instances, reflects other mechanisms such as deafferentation effects from subcortical infarction (i.e., receptor down-regulation or axon terminal degeneration).
PET studies of hyperperfusion after intravenous thrombolysis
Heiss et al. (1997a), b report on six patients treated acutely with intravenous thrombolysis (recombinant tissue-type plasminogen activator) within 3 hours of symptom onset and studied with H2O−15O PET (nonquantitative) before and after treatment. In four cases, there was residual perfusion in the ischemic area, and recanalization induced a marked hyperperfusion as observed 1 to 3 days later. All of these patients recovered completely from their neurologic deficit, and late structural imaging did not reveal frank infarction in any subject. In two patients, the perfusion was markedly reduced before treatment, and tissue reperfusion after treatment was achieved only at the borders of the ischemic zone without hyperperfusion; both patients experienced extensive infarction and did not recover from their severe neurologic deficit. These findings by Heiss et al. are consistent with, and therefore tend to confirm the validity of, the findings of Marchal et al. (1996): first, they showed that hyperperfusion indeed followed severe ischemia and was induced by recanalization, and, second, hyperperfusion was a marker of good clinical and tissular outcome. Furthermore, they suggest that hyperperfusion develops only when tissue perfusion has not deteriorated to a level below which irreversibility from no-reflow seems inescapable, which explains why EPIH is a marker of good tissue outcome.
Mechanisms and pathophysiologic significance of early postischemic hyperperfusion as observed in humans
The exact mechanisms underlying the transient disruption of CBF autoregulation that causes EPIH still are unknown and probably multifactorial. Many of the molecules released during the phase of ischemia such as potassium, bradykinin, adenosine, arachidonate, nitric oxide, and oxygen radicals are powerful vasodilators (Macfarlane et al., 1991). It is also possible to envisage a neurogenic vasodilation mediated by unmyelinated C fibers surrounding the cerebral vessels and releasing sensory neuropeptides through axon-like reflexes (Macfarlane et al., 1991). However, the findings of Marchal et al. (1996) regarding the CMRO2 in the hyperperfused areas suggest that the focal elevation in CBF might in part reflect passive, physiologic coupling. The relations between tissue reperfusion and hyperperfusion and the development of parenchymal hemorrhage and vasogenic edema are important to consider, especially in the context of the current controversy about thrombolysis therapy (Wardlaw et al., 1997). In the pattern III patients of Marchal et al., early signs of brain edema were not a feature on emergency CT scans (Marchal et al., unpublished data). In animal studies, the reperfusion of ischemic tissues has been associated either with edema or hemorrhage in the cases where the flow was markedly reduced during the ischemic phase (Morawetz, et al., 1978; Ianotti and Hoff, 1983; Heiss et al., 1997a) or with time-related inflammatory changes such as endothelial cell injury, inflammatory cell infiltration, and microvascular dysfunction (so-called reperfusion injury), attributed to polymorphonuclear leukocytes, platelets, and microglia and their ability to increase oxidative stress secondary to the detrimental nitric oxide synthesis and the release of cytotoxic products and cytokines such as interleukin-1β, interleukin-6, tumor necrosis factor-α, or platelet activating factor (del Zoppo, 1997; Kuroda and Siesjö, 1997). However, even in the rat, reperfusion injury is not a universally observed phenomenon after focal ischemia but is a species- and model-related phenomenon, as shown by Aronowski et al. (1997) after extensive experiments where both the species studied and the duration of MCAO were systematically manipulated. In humans, evidence for the reperfusion injury is lacking, and the studies of Marchal et al. tend to dismiss it as marginal at best, at least within the time frame of their investigations relative to onset of symptoms. Indeed, one key to these apparent discrepancies might be related to the time elapsed since the onset of ischemia and to the degree of ischemia during the period of occlusion. Thus, in rats, dogs, and baboons, early reperfusion is accompanied by definite clinical improvement and substantial reduction in the severity of the morphologic lesions that are never hemorrhagic (De Ley et al., 1988; Memezawa et al., 1992; Young et al., 1997). In humans, early recanalization of MCA trunk occlusion is seldom associated either with a worsening of brain swelling or with hemorrhagic transformation (von Kummer et al., 1993; Wardlaw et al., 1993). Conversely, both in humans and in animals, late reperfusion in a territory with damaged blood-brain barrier appears hazardous by promoting hemorrhagic transformation or fatal edema formation, which, however, depends mainly on the size of the irreversibly damaged area, irrespective of whether reperfusion occurs (Irino et al., 1977; Asplund et al., 1991; von Kummer et al., 1993; Wardlaw et al., 1993). In other words, therapeutic reperfusion would be harmless if residual perfusion is above a critical threshold (Hanson et al., 1993; Heiss et al., 1997a). Whether hemorrhage or edema may complicate reperfusion below this as yet putative threshold remains unknown. If the latter was true, however, this would mean that the time window within which to safely reinstate perfusion would be limited, particularly in patients with limited collateral supply. Thus, Ueda et al. (1994) report that hemorrhagic transformation after intraarterial thrombolysis occurred only in patients with pretreatment perfusion markedly reduced below a certain threshold. It is possible, however, that this complication also would have occurred without this treatment, since it is well know that hemorrhagic transformation also occurs in cases with permanent arterial occlusion (Ogata et al., 1989).
EXPERIMENTAL PET STUDIES
The following section addresses the issue of hyperperfusion noted by PET after the reopening of an occluded vessel and hyperperfusion noted during an ischemic episode in two other species, the cat and the nonhuman primate.
Postischemic hyperperfusion after middle cerebral artery occlusion in the cat
Heiss et al. (1997a) published an important study on hyperperfusion observed after temporary MCAO in the cat. The experimental design of this study was chosen to represent closely the clinical situation of stroke in humans. Anesthetized cats were subjected to temporary MCAO of varying duration (30, 60, or 120 minutes). Positron emission tomography then was used to measure CBF and metabolic parameters during the ischemic period and after reopening of the MCA for up to 24 hours. These sequential measurements, which started 5 minutes after MCAO and were repeated at 30-minute intervals during the period of occlusion, were subsequently followed with five measurements made in the reperfusion phase of the protocol.
The results are described briefly as follows. In the 30-minute MCAO group, all animals survived the observation period, and the reperfusion period was characterized by a transient reactive hyperperfusion (relative to preocclusion values) with fast normalization of CBF: no, or only small, infarcts in the deep nuclei were found after histologic analysis. After 60 and 120 minutes of MCAO, the degree of hyperperfusion was related to the severity of prior ischemia (reaching up to 300% of preocclusion values), and cortical/subcortical infarcts of varying sizes developed: approximately 50% of animals survived this duration of ischemia. The animals that eventually died from brain swelling exhibited higher CBF during the hyperperfusion period than those that survived, although this difference was not statistically significant. Based on these observations, it appears that moderate hyperperfusion (reactive hyperemia) in the reperfusion phase was well tolerated when the increase in flow did not exceed approximately 125% of the preischemic control values, whereas more severe periods of hyperperfusion tended to be associated with larger infarcts, brain swelling, and early death. However, since in this study the degree of hyperperfusion was clearly related to the severity of prior ischemia, it cannot be concluded that the more severe hyperperfusion per se was responsible for the worse outcome. Thus, it is likely that severe brain swelling was a consequence of reperfusion in a severely damaged vascular bed, not of the degree of hyperperfusion.
Heiss concludes that when postischemic hyperperfusion is not severe and is of a short duration, late-stage metabolic defects, as well as the final infarction, are smaller. In addition, he correctly states that forced reperfusion by reopening of the MCA cannot salvage already irreversibly damaged tissue but may cause additional damage by inducing edema, and that this latter effect may be aggravated by severe and prolonged hyperperfusion in paralyzed vessels.
The question remains, however: Do the experimental conditions applied to the cat reflect those seen in the clinical situation, or could a species difference explain some of the observed discrepancies? For instance, in the study cited, 30 minutes of temporary MCAO in the cat is well tolerated and shows 100% survival rate (i.e., hyperperfusion is not harmful at this stage); 60 minutes of ischemia gives 50% survival and a similar degree of hyperperfusion (i.e., around 150% of control values on average), which could be detrimental in this situation. This latter situation may be equivalent to late hyperperfusion observed in humans.
Hyperperfusion during permanent middle cerebral artery occlusion
Hyperperfusion during permanent MCAO may be explained by the opening up or widening of a collateral circulation resulting from the development of a pressure gradient toward the ischemic territory or widening of the occluded vessel and, according to at least one investigation, basal values of CBF, ischemic flow, and hyperperfusion are not correlated (Traupe et al., 1982).
In the study by Heiss et al. (1995), six cats were subjected to permanent MCAO, and various hemodynamic and metabolic parameters (CBF, CBV, CMRO2, and CMRglu were measured sequentially over a 24-hour period with PET. In all animals, MCAO reduced immediately ipsilateral CBF to below 30% of the control level. It was observed, in some instances, that during MCAO an effective collateral circulation caused hyperperfusion outside of the ischemic MCA territory. This hyperperfusion was a transient effect and, surprisingly, it was even observed in the contralateral hemisphere and thus may reflect a phenomenon unrelated to prior focal ischemia.
Since less experimental details are given than in the previously cited article by Heiss (see earlier) it is not certain when this period of hyperperfusion was observed. It may be hypothesized that because no evidence of hyperperfusion was observed during temporary MCAO (see aforementioned study), the hyperperfusion took place at least after 2 hours of occlusion. Unfortunately, it was not mentioned if these cats (i.e., those that showed hyperperfusion) had a smaller infarct at histologic analysis.
In earlier studies performed in pentobarbital-anesthetized cats (Yamaguchi et al., 1971, 1977) in which permanent MCAO was induced, hyperperfusion in the ischemic hemisphere was noted in six of seven animals studied within 2 days of occlusion. In this study, which used autoradiographic techniques, hyperperfusion was defined as regions where CBF was above normal and greater than those of the nonischemic hemisphere. Microscopic examination further revealed evidence of mild pathologic changes in these same brain regions. On the contrary, in cats that were studied 8 or 15 days after occlusion, hyperperfusion of gray matter was not accompanied by histopathologic changes, but this finding may be explained by the fact that the regions of hyperperfusion were located outside of the territory previously supplied by the occluded MCA. Notice that no hyperperfusian was observed in animals studied 30 days or more after MCAO. Furthermore, hyperperfusion was not associated with hemorrhagic change or with the formation of edema. Distinct hyperperfusion of white matter was observed in only one animal (15 days after MCAO), which corresponded to necrotic tissue with neovascularization. It is possible, therefore, that hyperperfusion that develops relatively soon in regions with eventual ischemic neuronal damage but without frank infarction after the occlusion of the MCA may reflect reactive hyperemia as a result of increases in CBF to ischemic regions where vascular reactivity was impaired. However, hyperperfusion that develops in brain regions without subsequent pathologic changes, particularly in regions supplied by the anterior cerebral artery, may be attributed to increased flow through collateral vessels as a result of the pressure gradient toward ischemic regions. Yamaguchi (1977) could not clarify the significance of postischemic hyperperfusion in its relation to the subsequent development of an infarct. He postulates that postischemic hyperperfusion may be beneficial if it occurs early enough and lasts long enough to provide sufficient oxygen and glucose and to remove acidic metabolites from the ischemic brain tissue for preservation of normal neuronal function. On the other hand, postischemic hyperperfusion may be harmful if it leads to an increase of cerebral edema or hemorrhagic infarction. Neither of the latter two phenomenon were observed in his study, and he could find no relation between periods of postischemic hyperperfusion and the size of the ischemic or infarcted part of the brain.
Postischemic hyperperfusion after middle cerebral artery occlusion in the baboon
Unlike MCAO in the cat, our own model of focal cerebral ischemia in the baboon results in an infarction that is less severe and permits sequential PET studies to be performed in the subacute, acute, and chronic stages of experimental stroke (Touzani et al., 1995, 1997; Young et al., 1996). Of 28 baboons analyzed (Young et al., ongoing study), only one instance of cortical hyperperfusion has been noted during the period of MCAO (Fig. 4), whereas in 5 baboons of 16, large regions of hyperperfusion were noted 1 hour after reestablishment of flow through the occluded vessel (Fig. 5). During MCAO in the nonhuman primate, we occasionally have observed situations where spontaneous reperfusion has occurred during temporary MCAO (Young et al., 1996) although the values of CBF never exceeded those of the contralateral hemisphere and thus cannot be considered as true hyperperfusion. In the only animal that showed hyperperfusion during MCAO, this region was on the cortical mantle and on the boundary zone between the MCA and the anterior cerebral artery, and this area did not exhibit frank infarction on late MRI (Fig. 4). This spontaneous hyperperfusion could have been related only to the opening of collateral channels, since the microvascular clips were still in place at the time.
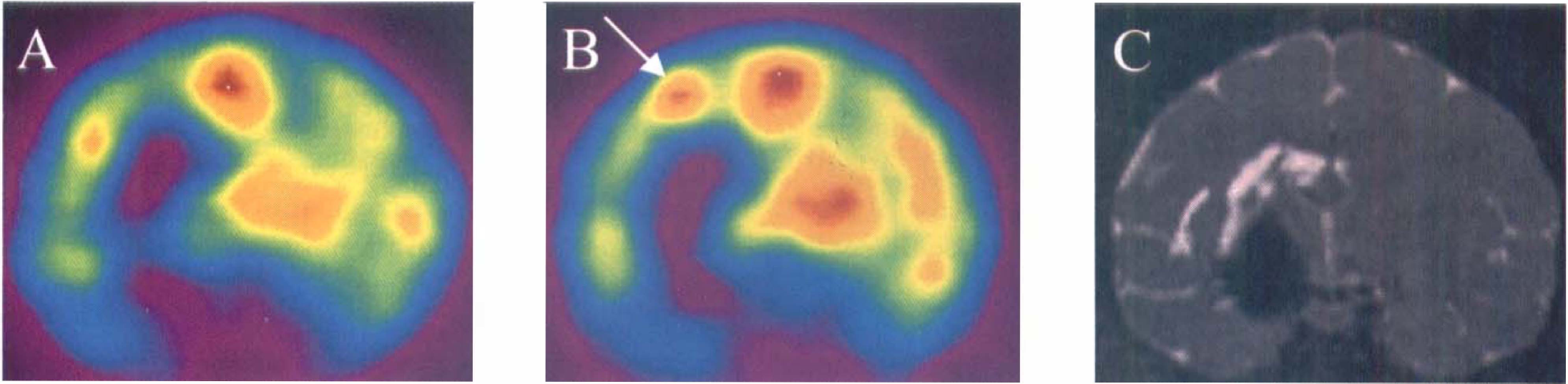
Hyperperfusion during middle cerebral artery occlusion (MCAO) in the anesthetized baboon. The coronal PET images of cerebral blood flow (Siemens ECAT HR+; Knoxville, TN, U.S.A.) were obtained at 1 hour (
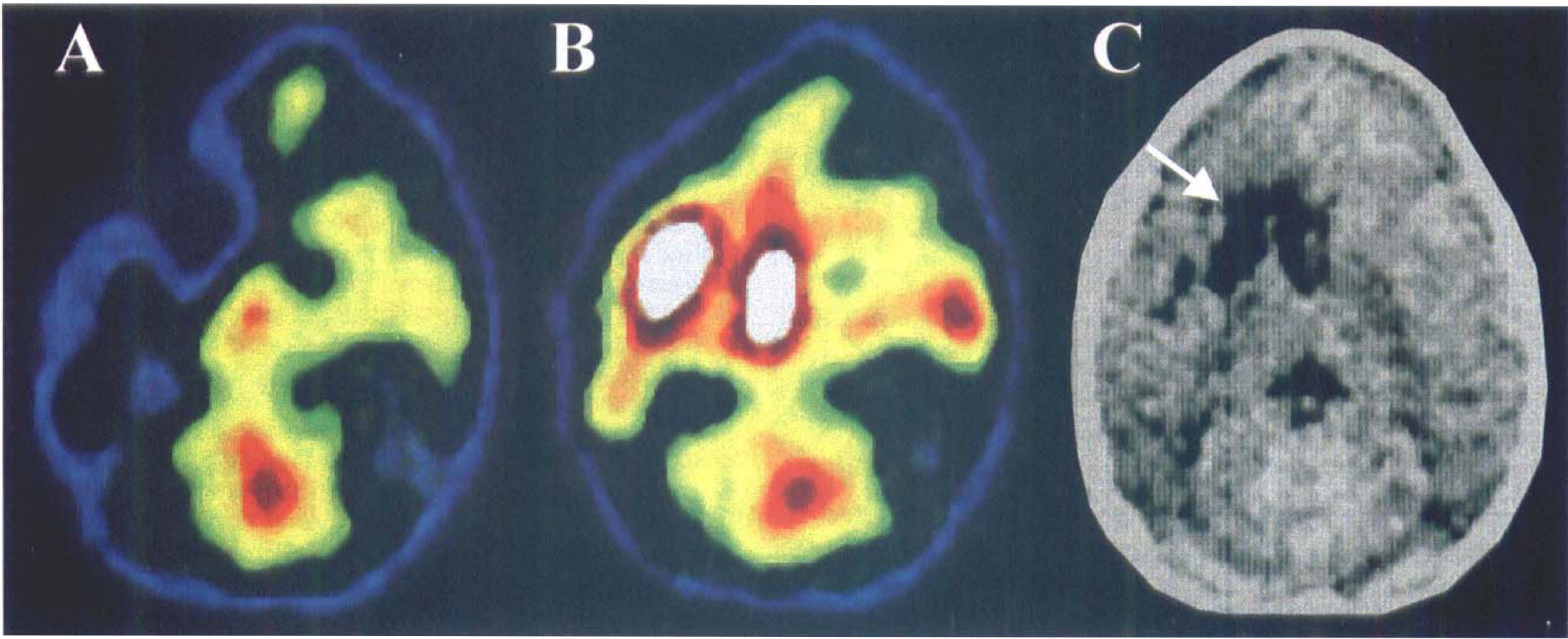
Postischemic hyperperfusion after temporary MCAO (3 hours) in the anesthetized baboon. The axial PET images of cerebral blood flow (LETI TTV03; CEN, Grenoble, France) were obtained during occlusion (MCAO + 1 hour) (
Relation of postischemic hyperperfusion to final infarct topography and volume
No concrete evidence supports the hypothesis that postischemic hyperperfusion is always beneficial to stroke outcome. In the cat model of experimental focal cerebral ischemia, severe hyperperfusion of long duration appears to be detrimental (Heiss et al., 1997a), although this is not clear (see earlier discussion). As in most situations of ischemia, the importance of time plays a cardinal role in functional outcome. Thus, if moderate hyperperfusion is observed at an early stage during stroke onset, prognosis is good. On the other hand, if the phenomenon of hyperperfusion is observed tardively and persists with time, the outcome may be less favorable. Yamaguchi (1977) found no relation between periods of postischemic hyperperfusion and the size of the ischemic or infarcted part of the brain. However, the group of Heiss (1994) reports a finding in one cat where a partial recovery of CBF was observed during the period of MCAO. It was not stated if the values for CBF ever exceeded the baseline levels (i.e., hyperperfusion) in this animal, but it was noted that the cat in question had the smallest infarct when assessed histologically.
From the same laboratory, Graf and colleagues (1997, published in abstract form only) have further studied different patterns of postischemic hyperperfusion after transient MCAO (60 minutes) in a large series of 16 anesthetized cats and compared the observed results with histologic outcome. The authors identified three distinct types of hyperperfusion based on their PET studies. Type 1 showed immediate hyperperfusion lasting for the whole observation period (24 hours) and was associated with a severe reduction in CBF during the ischemic period. At the end of the study, large infarcts were observed. In type 2, the period of hyperperfusion was transient and often followed by hypoperfusion in regions that previously were hyperperfused. In this instance, infarcts were normally restricted to the center of the hyperperfused regions or did not develop at all. In the type 3 pattern, hyperperfused regions grew progressively with time. In this type, CBF reduction also was more gradual and infarcts were small. Again, no correlation with histologic outcome could be made.
Our own unpublished observations in baboons with temporary MCAO showed regions of hyperperfusion immediately after reestablishment of flow through the occluded vessel. We could find no correlation between the presence of hyperperfusion and infarct volume, but the topography of the increased flows always was located on the cortical mantle, whereas infarcts always were in the basal ganglia region (Fig. 5).
CONCLUSIONS AND UNRESOLVED ISSUES
Contradicting the notion of reperfusion injury from the experimental literature, human as well as cat and baboon PET studies show that early hyperperfusion of brain tissue after ischemic stroke appears to be harmless and is associated, in most cases, with rapid and complete clinical recovery and lack of gross infarction on chronic stage structural imaging or histologic examination. However, it is possible that reperfusion-induced injury may develop with late hyperperfusion. It also is possible that selective neuronal loss occurs in areas with EPIH, but the specific role of hyperperfusion, compared with prior ischemia, in this type of cellular death needs to be clarified.
Footnotes
Abbreviations used
Acknowledgments
The authors thank Simon Roussel, PhD, for his helpful comments during the preparation of this review, The authors also thank Mme, Martine Huguet and Ms. Anita Foro for their secretarial expertise and Mr. Dominique Luet for preparation of the figures.