
Abstract
Primary headache syndromes, such as cluster headache and migraine, are widely described as vascular headaches, although considerable clinical evidence suggests that both are primarily driven from the brain. The shared anatomical and physiologic substrate for both of these clinical problems is the neural innervation of the cranial circulation. Functional imaging with positron emission tomography has shed light on the genesis of both syndromes, documenting activation in the midbrain and pons in migraine and in the hypothalamic gray in cluster headache. These areas are involved in the pain process in a permissive or triggering manner rather than as a response to first-division nociceptive pain impulses. In a positron emission tomography study in cluster headache, however, activation in the region of the major basal arteries was observed. This is likely to result from vasodilation of these vessels during the acute pain attack as opposed to the rest state in cluster headache, and represents the first convincing activation of neural vasodilator mechanisms in humans. The observation of vasodilation was also made in an experimental trigeminal pain study, which concluded that the observed dilation of these vessels in trigeminal pain is not inherent to a specific headache syndrome, but rather is a feature of the trigeminal neural innervation of the cranial circulation. Clinical and animal data suggest that the observed vasodilation is, in part, an effect of a trigeminoparasympathetic reflex. The data presented here review these developments in the physiology of the trigeminovascular system, which demand renewed consideration of the neural influences at work in many primary headaches and, thus, further consideration of the physiology of the neural innervation of the cranial circulation. We take the view that the known physiologic and pathophysiologic mechanisms of the systems involved dictate that these disorders should be collectively regarded as neurovascular headaches to emphasize the interaction between nerves and vessels, which is the underlying characteristic of these syndromes. Moreover, the syndromes can be understood only by a detailed study of the cerebrovascular physiologic mechanisms that underpin their expression.
The issue of vascular versus neurogenic mechanisms in primary headaches, such as migraine and cluster headache, still is unresolved and is paralleled somewhat in physiology by arguments about the existence and function of the neural innervation of the cerebral circulation (Heistad and Marcus, 1978; Purves, 1972; Purves, 1978). The pathophysiologic concept of vaseular headaches is based on the idea that changes in vessel diameter or gross changes in CBF would trigger pain and could partly explain the mechanism of action of vasoconstrictor drugs, such as ergotamine (Wolff, 1963). From a physiologic viewpoint, the concept of vascular headache as a pathophysiologic entity implies abnormalities in vessel behavior. Regional cerebral blood flow (rCBF) studies emphasize a dysfunction of the cerebrovascular regulation in headache, whereas little study has been conducted to evaluate the central processing of headache (Olesen, 1991). Insights into the fundamental physiologic mechanisms of these systems has been limited by the lack of methods to visualize the pathophysiologic brain activation and examine its origins. Functional neuroimaging in patients has, however, revolutionized this area and provided unique insights into some of the commonest maladies in humans. In this review, we set into a physiologic construct the advances in the understanding of migraine and cluster headache that recently have accrued from functional neuroimaging, deriving the concept that these headaches are, in essence, neurovascular headaches, an expression of the nerve-blood vessel interaction in cerebrovascular physiology that has been such a subject of controversy. This review concentrates on the trigeminal innervation of the cranial vessels and does not cover the other neuronal systems that are considered in other reviews (Edvinsson et al., 1993).
THE TRIGEMINOVASCULAR SYSTEM
The trigeminocerebrovascular system (Goadsby and Duckworth, 1987) is in a unique, pivotal position in terms of cerebrovascular physiologic influences. It is the sole sensory (afferent) innervation of the cerebral vessels and has, in addition, an efferent potential in pathophysiologic settings. To understand primary headache from a neurovascular perspective, it is necessary to consider the anatomical and physiologic information on the trigeminovascular system.
Anatomy
The trigeminovascular system consists of the neurons innervating the cerebral vessels whose cell bodies are located in the trigeminal ganglion. The ganglion contains bipolar cells, the peripheral fiber making a synaptic connection with the vessel, and other cranial structures, particularly the pain-producing large cranial vessels and dura mater (McNaughton, 1938; McNaughton and Feindel, 1977), and the centrally projecting fiber synapsing in the caudal brain stem or high cervical cord (Goadsby and Hoskin, 1997; Kaube et al., 1993). Tracing studies have identified the trigeminal nerve as the major afferent pathway for pain from the vessels and dura mater (Feindel et al., 1960; Huber, 1899; Penfield, 1932; Penfield, 1934; Penfield and McNaughton, 1940). The innervation of the pial vessels from the trigeminal system has been demonstrated using sensitive tracing techniques (Mayberg et al., 1981). These fibers are predominantly found in the first (ophthalmic) division of the trigeminal nerve and have widely ramifying axons that may innervate several vessels ipsilaterally (Liu-Chen et al., 1983a; Liu-Chen et al., 1983b; Mayberg et al., 1984). These systems are, in evolutionary terms, well conserved and may be found in monkey (Ruskell and Simons, 1987), cat (Keller et al., 1985; Liu-Chen et al., 1984), guinea pig (Yamamoto et al., 1983), gerbils (Matsuyama et al., 1985), and rat (Arbab et al., 1986). The trigeminal innervation is predominantly to the forebrain but extends posteriorly to the rostral basilar artery, whereas the more caudal vessels are innervated by the C2 and C3 dorsal roots, which also synapse with the central trigeminal neurons (Arbab et al., 1988). Some of the projections involve both cerebral (middle cerebral artery [MCA]) and extracerebral (middle meningeal artery) vessels (O'Connor and van der Kooy, 1986).
Transmitters
Several powerful vasodilator peptides are found in cell bodies within the trigeminal neurons that innervate blood vessels. These substances, calcitonin gene-related peptide (CGRP), substance P (SP) and neurokinin A (NKA), are found in various combinations of neurons so that any combination may characterize a particular neuron. The functional consequences of these combinations has not been fully elucidated, but it seems likely that the trigeminocraniovascular innervation is heterogenous in its actions.
Calcitonin gene-related peptide. Calcitonin gene-related peptide is the most potent and perhaps the most interesting of the neuropeptides that have been mapped to the trigeminal system. It is derived by alternative processing of the calcitonin gene messenger RNA (Amara et al., 1982). Biochemical determinations of CGRP by HPLC and radioimmunoassay demonstrate the substance in cat cerebral vessels (Uddman et al., 1985). The trigeminal ganglion contains numerous CGRP immunoreactive cells (Rosenfeld et al., 1983), cells in culture can release CGRP after potassium application (Mason et al., 1984), and CGRP-containing fibers on cerebral vessels are not found after trigeminal nerve section (Uddman et al., 1985). Some of the trigeminal ganglion CGRP-containing cell bodies also contain SP and NKA (Lee et al., 1985; Sundler et al., 1985; Uddman et al., 1985), whereas there is a distinct subpopulation that projects to the cerebral vessels that is enriched with CGRP (O'Connor and van der Kooy, 1988). On the receptor side, there is mRNA for the CGRP-1 receptor on cerebral vessels and in the trigeminal ganglion of humans (Edvinsson et al., 1997). In contrast to SP, CGRP does not dilate cerebral veins (Edvinsson et al., 1986; Edvinsson et al., 1982; McCulloch et al., 1986). Calcitonin gene-related peptide is the most potent vasodilator transmitter identified in the cerebral circulation, and its action is endothelium independent (Hanko et al., 1985) and associated with an increase in vessel wall cyclic AMP (Edvinsson et al., 1985). The endothelium independence suggests that generation of nitric oxide (Moncada et al., 1991) is not involved in CGRP-mediated vasodilation (Baskaya et al., 1995). The increase in vessel wall cyclic AMP is likely to lead to hyperpolarization through potassium channels, which may include activation of large conductance calcium-activated potassium channels (BKCa) (Holland et al., 1996) and ATP-sensitive potassium channels (KATP) (Brian et al., 1996). Given that BKCa activation involves NO generation (Hoang and Mathers, 1998), the KATP mechanism is more likely to be involved.
Substance P. Sectioning of the trigeminal nerve eliminates ipsilateral SP-like immunoreactivity in fibers that are unmyelinated with neuropeptide-containing axonal swellings and an abundance of mitochondria (Liu-Chen et al., 1983a). Substance P-like immunoreactivity has been demonstrated in fibers in the dura mater (Edvinsson and Uddman, 1981) with a marked density around the superior sagittal sinus (Keller et al., 1989). Substance P is a nondecapeptide first described more than 60 years ago (von Euler and Gaddum, 1931) and suggested to be involved in nociceptive transmission (Lembeck, 1953). In many vascular beds, including the cerebral bed, SP is a potent vasodilator, and its effect is endothelium dependent (Zawadzki et al., 1981).
In vitro studies with cow and guinea pig vessels show that SP can be released by potassium, capsaicin, or electrical stimulation and that this is abolished if calcium ions are removed from the superfusion buffer (Edvinsson et al., 1983; Moskowitz et al., 1983). Isolated cerebral vessels from humans (Edvinsson and Uddman, 1982), cat, and guinea pig (Edvinsson et al., 1982) relax in a concentration-dependent manner with SP application, and it also dilates both arteries and veins in situ (Edvinsson et al., 1981). Substance P can induce protein extravasation in the periphery (Gamse and Saria, 1985), and intradermal injection produces wheal, flare, and itching (Hagermark et al., 1978). A similar response is seen in the dura with protein extravasation and mast cell degranulation (Moskowitz and Cutrer, 1993).
Neurokinin A. The β-preprotachykinin that produces SP may be differentially cleaved to yield a decapeptide with a similar profile of action and localization in the trigeminal system, NKA (Nawa et al., 1983). Both SP and NKA coexist in perivascular nerve fibers in peripheral (Sundler et al., 1985) and cerebral vessels (Edvinsson et al., 1988). Neurokinin A can relax cerebral vessels both in vitro and in vivo, although it is only one tenth as potent as SP (Edvinsson et al., 1987; Jansen et al., 1991). The receptor that is activated by NKA has been termed neurokinin-2 (substance K in the old tachykinin terminology) and has been sequenced and cloned (Masu et al., 1987). In this nomenclature, the neurokinin-1 receptor is preferentially activated by SP and the neurokinin-3 receptor by a further product of the preprotachykinins, neurokinin B. This latter peptide does not seem to be located in the trigeminal nerves. Specific neurokinin-2 antagonists will allow a more detailed examination of the role of NKA in cerebrovascular physiology.
Trigeminovascular physiology
Given that the trigeminal nerve supplies a rich network of perivascular fibers to the cranial circulation, which contains powerful vasodilator neuropeptides, what is their physiologic role?
Resting cerebral blood flow. Cerebral blood flow measured with iodoantipyrine and tissue autoradiography is not altered in the cat after trigeminal ganglion section. After unilateral section, flow is identical to homologous contralateral cortex (Edvinsson et al., 1986). Furthermore, glucose utilization is not affected by trigeminal ganglion section, and thus the usual close relation between flow and metabolism is not disturbed (Edvinsson et al., 1986).
Hypercapnia and hypoxia. In a series of well-conceived studies in the rat, it has been shown using the radiolabeled microsphere technique that unilateral trigeminal ganglionectomy does not alter cerebrovascular responsiveness to hypercapnia (Sakas et al., 1989). However, in the cat, the hyperemia after temporary occlusion of the brachiocephalic and left subclavian vessels is partly mediated (60%) by a trigeminal mechanism (Macfarlane et al., 1991).
Autoregulation. Induction of hypertension outside of the autoregulatory range or induction of a tonic/clonic seizure with bicuculline in trigeminal ganglionectomized animals leads to a reduced vasodilator response when compared with the nonganglionectomized side (Sakas et al., 1989). The effect is seen in the cortical gray matter but not in the diencephalon or white matter. These experiments delineate that the responses observed resulted from the axon-reflex part of the trigeminovascular system, since root section does not eliminate the effect whereas ganglionectomy does. Similarly, trigeminal fibers appear to limit noradrenaline-induced constrictor response in the pia (Moskowitz et al., 1988) and play a role in restoring constricted vessels to their normal caliber (McCulloch et al., 1986). Normal perfusion pressure range autoregulation is unimpaired by trigeminal root section.
TRIGEMINAL SYSTEM STIMULATION
In vitro
There is good evidence that stimulation of trigeminal neurons in vitro can result in the release of SP from nerve terminals in the pia (Moskowitz et al., 1983). This is likely to represent depolarization-dependent activation of trigeminal neurons and thus satisfy one of the criteria for SP being a neurotransmitter in this system.
In vivo
Craniovascular circulation. Stimulation of the trigeminal ganglion in humans by either thermocoagulation (Onofrio, 1975; Sweet and Wepsic, 1974) or injection of alcohol (Oka, 1950) can cause facial flushing, usually in the division or divisions appropriate to the manipulation. In addition, this flushing is accompanied by an increase in facial temperature of 1 to 2°C (Drummond et al., 1983). Corresponding with this flush, there is an increase in the dilator peptides SP and CGRP in the external jugular (Goadsby et al., 1988) but not in the peripheral circulation (Schon et al., 1987). Such changes also are seen in the cat (Goadsby et al., 1988). In addition, trigeminal ganglion stimulation in the cat (Lambert et al., 1984) or monkey (Goadsby et al., 1986) leads to a diminution of carotid resistance, with increased flow and facial temperature predominantly through a reflex mechanism. The afferent limb of this arc is the trigeminal nerve, and the efferent limb is the facial/greater superficial petrosal nerve (parasympathetic) dilator pathway. About 20% of the dilation seen remains after facial nerve section and probably is mediated by antidromic activation of the trigeminal system directly. The portion running through the parasympathetic outflow traverses the sphenopalatine (pterygopalatine) and otic ganglia (Goadsby et al., 1984) and uses vasoactive intestinal polypeptide as one of its transmitter (Goadsby and Macdonald, 1985).
Cerebrovascular circulation. Turning to studies that focus on CBF in the isolated cross-perfused dog brain, it has been shown that stimulation of the trigeminal ganglion can increase CBF by 19%, a response not altered by distal nerve section (Lang and Zimmer, 1974). Measured as total internal carotid blood flow in the monkey, trigeminal ganglion stimulation does not alter total flow (Goadsby et al., 1986), whereas specific regional flows are altered. Unilateral ganglion stimulation increases frontal and parietal cortical blood flows bilaterally without affecting other cortical areas, white matter, or brain stem flow. These flow changes in the cerebral circulation can be largely blocked by facial nerve section (Goadsby and Duckworth, 1987). Similar changes have been observed in humans undergoing trigeminal ganglion thermocoagulation (Tran-Dinh et al., 1992). Stimulation of the superior sagittal sinus, a pain-producing centrally placed intracranial structure, leads to a quantitatively greater increase in CBF than does trigeminal ganglion stimulation, as measured with iodoantipyrine and brain dissection in the cat (Lambert et al., 1988). The trigeminal neural innervation of the cerebral circulation is somatotopically selective for the structure activated. Stimulation of the superior sagittal sinus increases CBF, as measured by laser Doppler flowmetry, but does not alter carotid flow in the same manner, in contrast to the effect of trigeminal ganglion stimulation, which increases both cerebral and noncerebral cranial blood flow (Goadsby et al., 1997). It is likely that primary headache syndromes entrain these craniovascular responses in their expression, and this is the physiologic basis for the changes described later in cluster headache and experimental trigeminal head pain.
Dura mater and dural mast cells. The dura and covering vessels of the cerebral circulation are the protection and a portal of entry to the CNS. The dura mater, as one of the important pain-producing intracranial structures (Ray and Wolff, 1940), has been the subject of detailed study (Moskowitz, 1990). The dura mater is innervated by branches of the trigeminal nerve, whose cell bodies are to be found in the trigeminal ganglion (Mayberg et al., 1984). The innervation is to some extent somatotopic, with the first division giving rise to fibers that innervate the anterior fossa and tentorium cerebelli, the second division innervating the orbital roof, and the third, structures in the middle fossa (Steiger et al., 1982). The tentorial nerve arises from the ophthalmic nerve proximal to the superior orbital fissure and runs along the edge of the tentorium (Feindel et al., 1960). The supply from the mandibular nerve enters the cranial vault by the foramen spinosum arising from an extracranial portion of the nerve. The supratentorial dural sympathetic innervation arises from the ipsilateral superior cervical ganglion (Keller et al., 1989), as does the dura of the posterior fossa (Keller et al., 1985). In the posterior fossa, the dural innervation also has important contributions from the trigeminal ganglion and from the upper two dorsal root ganglia (Keller et al., 1985). There is a dense plexus of SP- and CGRP-containing nerves around the superior sagittal sinus that is equally distributed anteroposteriorly, whereas the distribution of vasoactive intestinal polypeptide varies from dense anteriorly to sparse posteriorly, as it does for the vessels (Keller and Marfurt, 1991). In terms of the reflex connections that exist between the trigeminal and parasympathetic innervation, the vasoactive intestinal polypeptide-ergic nerves arise from the same site as those for the cerebral circulation, the sphenopalatine ganglion (Uddman et al., 1989).
Ultrastructural evidence demonstrates a neuroexocrine role for the neural innervation of the dura mater in that the sympathetic nerves arising from the superior cervical ganglion make functional connections with the mast cells in the walls of large cerebral vessels (Dimitriadou et al., 1987). In the rat, unilateral trigeminal ganglion stimulation can cause mast cell degranulation in the dura mater and tongue (Dimitriadou et al., 1992). The mast cells are located randomly over the rat dura, with some concentration at the vessels, but in monkey, humans, and cat, they are only located at the branching of vessels and thus are well placed for some regulatory role. Certainly, changes in the vessels have been directly demonstrated in humans in cluster headache (Dimitriadou et al., 1990). The localization of various mediators of inflammation within the influence of the trigeminal innervation (Dimitriadou et al., 1991) may explain the release of some mediators and probably platelet changes reported in migraine and cluster headache. In experimental meningitis, there is a demonstrated interaction between changes in brain blood flow and the trigeminovascular innervation of the rat (Weber et al., 1996).
CEREBRAL BLOOD FLOW, FUNCTIONAL IMAGING, AND PRIMARY HEADACHE SYNDROMES
Observations reporting changes in CBF in association with migraine attacks (Olesen, 1991) have driven an effort to understand the cerebrovascular physiologic mechanisms in the context of head pain. In this section, we try to integrate this relationship by exploring recent data from functional imaging methods.
From blood vessels to neurons: migraine
The pioneering work of Olesen and colleagues (Friberg et al., 1994; Olesen et al., 1990; Olesen et al., 1981) revealed a focal reduction of rCBF for migraine attacks with aura, usually in the posterior parts of one hemisphere. These data have been reproduced and are robust. Using single photon emission computed tomography (SPECT) in migraine without aura, no blood flow changes were noticed (Olesen et al., 1990; Olesen et al., 1982). Since this pioneering work, several studies have used different techniques to demonstrate gross changes in cortical perfusion to explain either the aura or the headache in migraine.
Friberg and colleagues (1994) demonstrated with SPECT that interictally almost 50% of those with migraines had abnormal interhemispherical asymmetries in rCBF. These asymmetries were discrete compared with those seen during the aura phase of a migraine attack. The authors conclude that, at least interictally, a cerebrovascular dysregulation existed. In retrospect, given that in the experimental animal, stimulation of the trigeminovascular system leads to a pronounced increase in cortical blood flow, such changes would not be surprising (Goadsby and Duckworth, 1987). In an elegant study, the same group (Friberg et al., 1991) combined the measurement of rCBF and blood flow velocity in the MCA using transcranial Doppler sonography. The MCA velocity on the headache side was significantly higher than that on the nonheadache side, returning to normal values after treatment with sumatriptan. No change was seen in the rCBF in the MCA supply territory. The authors conclude that in the headache phase, there might be a dilation in the MCA on the headache side that was reversed by the vasoconstrictor action of the 5HT1B/1D receptor agonist sumatriptan (Henkes et al., 1996; Humphrey et al., 1991). However, since the CBF was unaffected, no evidence for an important role for nutrient flow was established. Consistent with the lack of a correlation, it has been shown using transcranial Doppler that the vasoconstrictor effect of sumatriptan is not temporally related to headache relief (Limmroth et al., 1996).
Woods and colleagues (1994) published the first report of positron emission tomography (PET) measurements in a patient from the start of a spontaneous migraine attack without aura, observed while lying in the PET scanner for another purpose (Fig. 1). The patient was studied while she was participating in a visual activation paradigm and was scanned with 12 successive measurements of rCBF. After the sixth scan, she developed unilateral headache, nausea, photophobia, and phonophobia. The first decrease in rCBF, noted during the seventh scan, was found bilaterally in the visual association cortex. In each subsequent scan, every 12 minutes, the decrease in rCBF spread contiguously across the cortical surface at a relatively constant rate, sparing the cerebellum, basal ganglia, and the thalamus. The hypoperfusion involved the middle and posterior cerebral artery territories. The authors estimated the maximal decrease of rCBF to be about 40%, which probably is not at an ischemic level (Astrup et al., 1981). Most of the changes were relatively short lasting, with substantial recovery by the time of the next measurement 12 to 15 minutes later. This case report is remarkable for two reasons. First, it illustrates for the first time a bilateral spreading hypoperfusion in a spontaneous migraine attack measured with PET. Even more remarkable is the fact that this patient experienced visual blurring only and thus had migraine without aura (The Headache Classification Committee of The International Headache Society, 1988). These findings are not in line with the SPECT studies (Lauritzen and Olesen, 1984; Olesen, 1991; Olesen et al., 1982) in which no changes in rCBF in migraine attacks without aura has been observed although the attack timing is different.
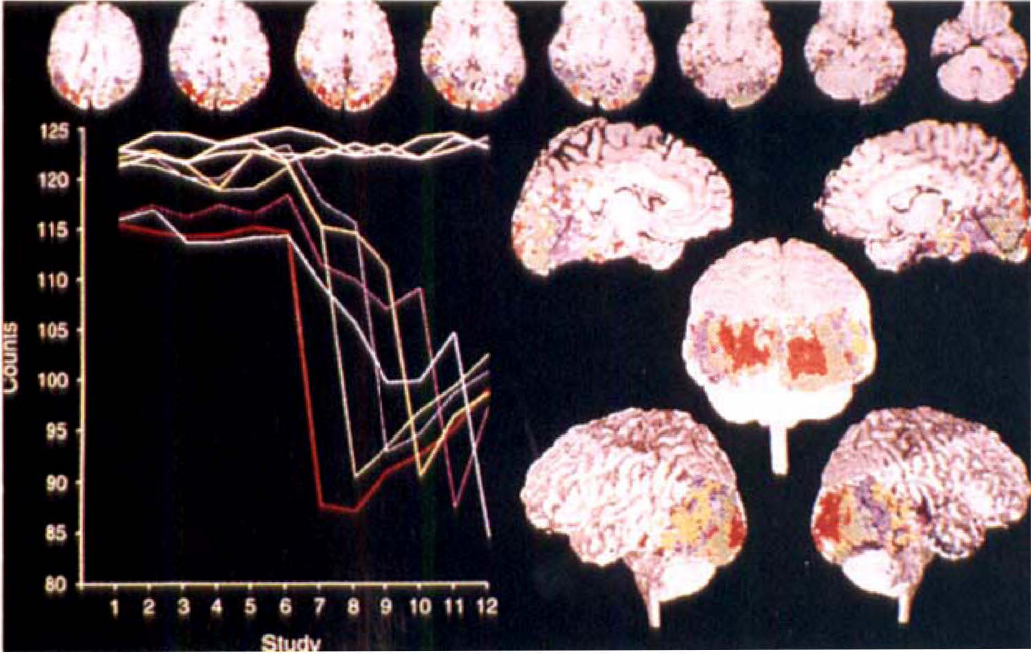
Time activity profiles of decreases of CBF during a spontaneous migraine attack. The patient was studied while she was participating in a visual activation paradigm and was scanned with 12 successive measurements of rCBF. The colors of the time-activity curves correspond to areas of the brain that correlate significantly with the various time-activity curves. The first decrease in rCBF, noted in the seventh scan, was found in the visual cortex. In each subsequent scan, the involvement spread continuously across the cortical surface and involved the middle as well as the posterior cerebral artery territory (Woods et al., 1994). Copyright ©1994 Massachusettes Medical Society. All rights reserved.
It has been suggested that the underlying pathophysiologic mechanism in migraine without, as well as with aura, is the spreading depression of (Leao 1944a, 1944b, 1947), occurring in different depths of the cortex, as suggested by Richter and Lehmenkühler (1993). However, the typical hyperperfusion at the front of the wave that has been described in animal experiments (Duckrow, 1991; Duckrow, 1993; Goadsby et al., 1992; Lauritzen, 1994; Piper et al., 1991; Wolf et al., 1996) was not detected in this patient. One explanation is the spatial and temporal resolution of about 70 seconds of PET-rCBF measurements. Previous studies have been few, and the attacks had already commenced (Herold et al., 1985; Weiller et al., 1995), so that changes in CBF taking place early in the attack would have been missed. Despite all of the logistic problems, there is a clear need for further studies to investigate migraine attacks as early as possible to confirm these results. At this point, neither Doppler nor measurements of global CBF suggest that changes in vessel diameter or gross changes in CBF are the primary cause or account adequately for the head pain of primary headaches. Thus, attention recently has been focused on possible CNS structures as an explanation for many of the facets of migraine.
Positron emission tomography is the method of choice to quantitatively measure rCBF as an index of regional neuronal activity (Fox et al., 1987; Frackowiak and Friston, 1994). In a recent PET study in nine right-sided patients with migraine without aura (Weiller et al., 1995) significantly higher rCBF values (+11%) were found during the acute attack compared with the headache-free interval in brain stem structures over several planes. These structures were toward the midline but contralateral to the headache side. We have speculated that the contralateral changes may represent rostral rather than caudal projection systems (Goadsby and Fields, 1998). Increased activation also was found in the inferior anterocaudal cingulate cortex, as well as in the visual and auditory association cortices during the attack, but was not detectable in these areas in the interval scan or after relief from headache and migraine-related symptoms through treatment.
The consistent increases in rCBF in the brain stem (dorsal midbrain [periaqueductal gray] and dorsolateral pontine tegmentum [locus ceruleus]) persisted even after sumatriptan had induced complete relief from headache, nausea, phonophobia, and photophobia. This increase was not seen outside of the attack. It can be concluded that the observed activation was unlikely to be just a result of pain perception or increased activity of the endogenous antinociceptive systems. It is beyond the resolution of the PET scanner to attribute foci of rCBF increases to distinct brain stem nuclei. However, the foci of maximum increase coincided, in the Talairach space (1988), with the anatomical location of the dorsal raphe nucleus and the locus ceruleus. Dysfunction of the regulation of these brain stem nuclei involved in antinociception and extracerebral and intracerebral vascular control provides a far-reaching explanation for many of the facets in migraine (Goadsby, 1997; Goadsby et al., 1991; Lance et al., 1983). The importance of the brain stem for the genesis of migraine is further underlined by the presence of binding sites for specific antimigraine compounds on these structures (Goadsby and Gundlach, 1991). The best direct clinical evidence for the brain stem as primum movens in migraine was reported by Raskin (1987) on nonheadache patients who developed migraine-like episodes after stereotactic placement of electrodes in the periaqueductal gray for treatment of chronic pain. Interestingly, these headaches responded to antimigraine treatments and this observation has been reproduced (Veloso et al., 1998).
From blood vessels to neurons: cluster headache
Previous studies of CBF in cluster headache are few. Most have been done with SPECT, and the results of this semiquantitative method have been heterogeneous, some reporting an increase (Norris et al., 1976; Sakai and Meyer, 1978), some a decrease (Nelson et al., 1980), and some no differences in cortical blood flow (Henry et al., 1978; Krabbe et al., 1984), probably because of methodologic differences. Spontaneous and glyceryl trinitrate (nitroglycerin)-provoked attacks are reported to be accompanied by a bilateral decrease in MCA blood flow velocities, implying vasodilation (Dahl et al., 1990; Tegeler et al., 1996). A recent PET study by Di Piero and coworkers (1997) considered cluster headache patients out of the active period and normal volunteers using the cold water pressor test. They demonstrated changes in pain transmission systems, which bear more detailed examination. The fact that the alterations also are present out of the active period of the disease suggests a possible involvement of central tonic pain mechanisms in the pathogenesis of cluster headache.
In 1996, the first PET study in cluster headache was reported (Hsieh et at, 1996a). Although the authors investigated only four patients, their findings supported their earlier work (Hsieh et al., 1995), suggesting a preference of the nondominant hemisphere, especially for the anterior cingulate cortex, in affective processing of chronic ongoing pain syndromes. These results contribute to understanding central pain transmission systems, but given the small numbers, require confirmation.
Cluster headache, which has been regarded as a vascular headache, has been attributed to an inflammatory process in the cavernous sinus and tributary veins (Hardebo, 1994). Inflammation has been thought to obliterate venous outflow from the cavernous sinus on one side, thus injuring the traversing sympathetic fibers of the intracranial internal carotid artery and its branches. According to this theory, the active period ends when the inflammation is suppressed and the sympathetic fibers partially or fully recover. This theory is based substantially on abnormal findings using orbital phlebography in cluster headache patients (Hannerz, 1991; Hannerz et al., 1987b; Sjaastad, 1992) and the fact that nitroglycerin and other vasodilators can induce a cluster attack (Ekbom, 1968).
However, in a study on cluster patients using magnetic resonance imaging, no definite pathologic changes were found in the area of the cavernous sinus (Sjaastad and Rinck, 1990). Using SPECT, parasellar hyperactivity was present in 50% to 80% of cluster patients and in 70% of those with migraine (Sianard-Gainko et al., 1994). Comparable findings on orbital phlebography can be seen in the cavernous region in patients with Tolosa-Hunt syndrome (Hannerz, 1988); hemicrania continua (Antonaci, 1994); short-lasting, unilateral, neuralgiform headache attacks with conjunctival injection and tearing (SUNCT) syndrome (Hannerz et al., 1992; Kruszewski, 1992); and chronic paroxysmal hemicrania (Antonaci, 1994; Hannerz et al., 1987a), suggesting that the changes are not specific for cluster headache. Furthermore, the frequency of pathologic findings at orbital phlebography in cluster headache was not higher than in cervicogenic headache, migraine, and tension-type headache, and the pattern in each disease was generally the same (Bovim et al., 1992). Moreover, given the circadian rhythmicity of attacks and cycling of bouts, a purely vasogenic cause cannot explain the entire picture of cluster headache (Goadsby and Lance, 1988). Clinical and experimental data show nitroglycerin-provoked (Ekbom, 1968) and spontaneous cluster attacks to be comparable (Fanciullacci et al., 1995; Goadsby and Edvinsson, 1994): nitroglycerin does not alter rCBF significantly (Hsieh et al., 1996a; Krabbe et al., 1984), and the headache can be rapidly and effectively aborted with sumatriptan (Ekbom, 1991). This approach therefore was used to study brain activation during nitroglycerin-induced cluster attacks (May et al., 1998a).
In this study, which included nine patients with cluster headache during the acute attack compared with the headache-free state, we observed areas of activation that fell into two broad groups: areas known to be involved in pain processing or responses to pain, such as cingulate and insula cortex and contralateral thalamus, and areas activated specifically in cluster headache but not in other causes of head pain, notably the hypothalamic gray (Figs. 2 and 3). These data taken together with that from migraine and experimental head pain, demonstrate that primary headache syndromes share some processing pathways, as might be expected, but may be distinguished on a functional neuroanatomical basis by areas of activation specific to the clinical syndrome.
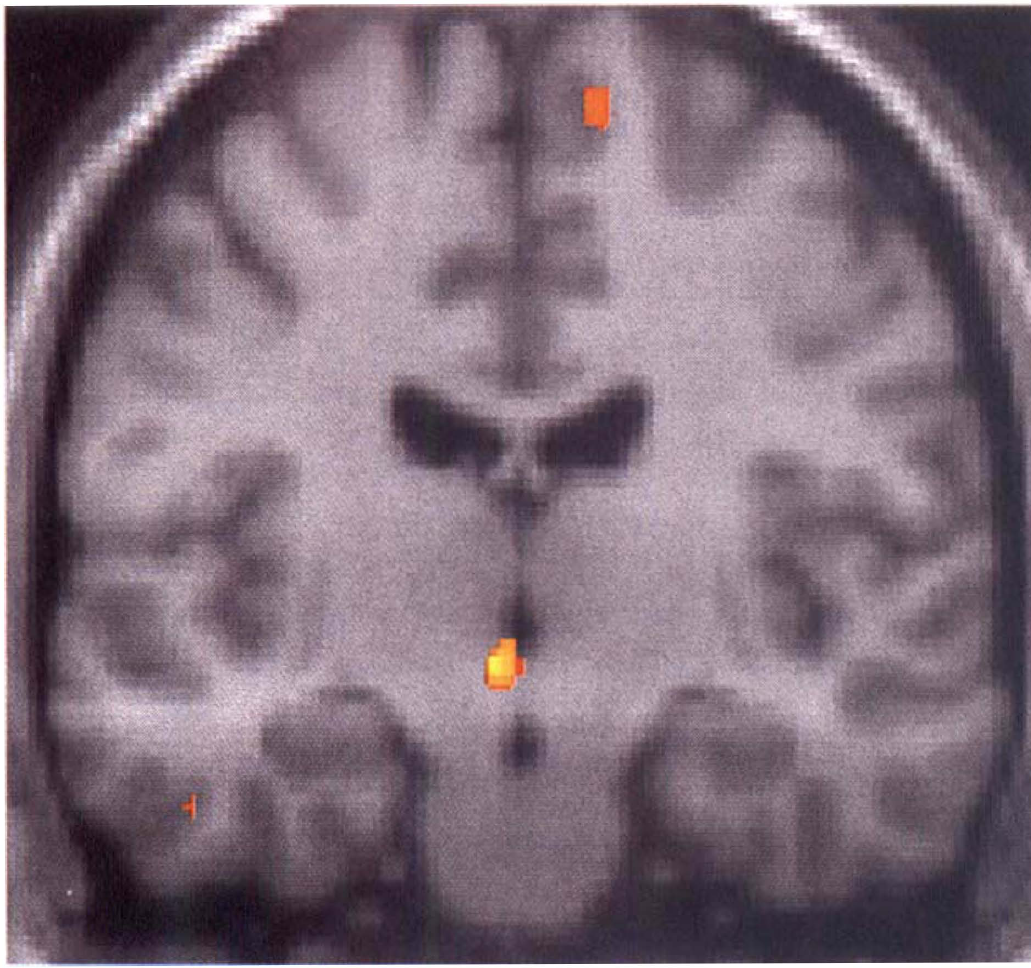
Comparison of nitroglycerin-induced acute cluster headache attack and rest (no pain) condition in nine patients with active chronic cluster headache. The activations during the attack are shown as statistical parametric maps that show the areas of significant rCBF increases (P < 0.001) in color superimposed on an anatomical reference derived from a T1-weighted magnetic resonance (MR) image. The left side of the picture is the left side of the brain. Significant activation was detected next to the third ventricle, slightly lateralized to the left and rostral to the aqueduct. The activation is ipsilateral to the pain side, lies in the diencephalon, and coincides, in the atlas (Talairach and Tournaux, 1988), with the hypothalamic gray matter (May et al., 1998a).
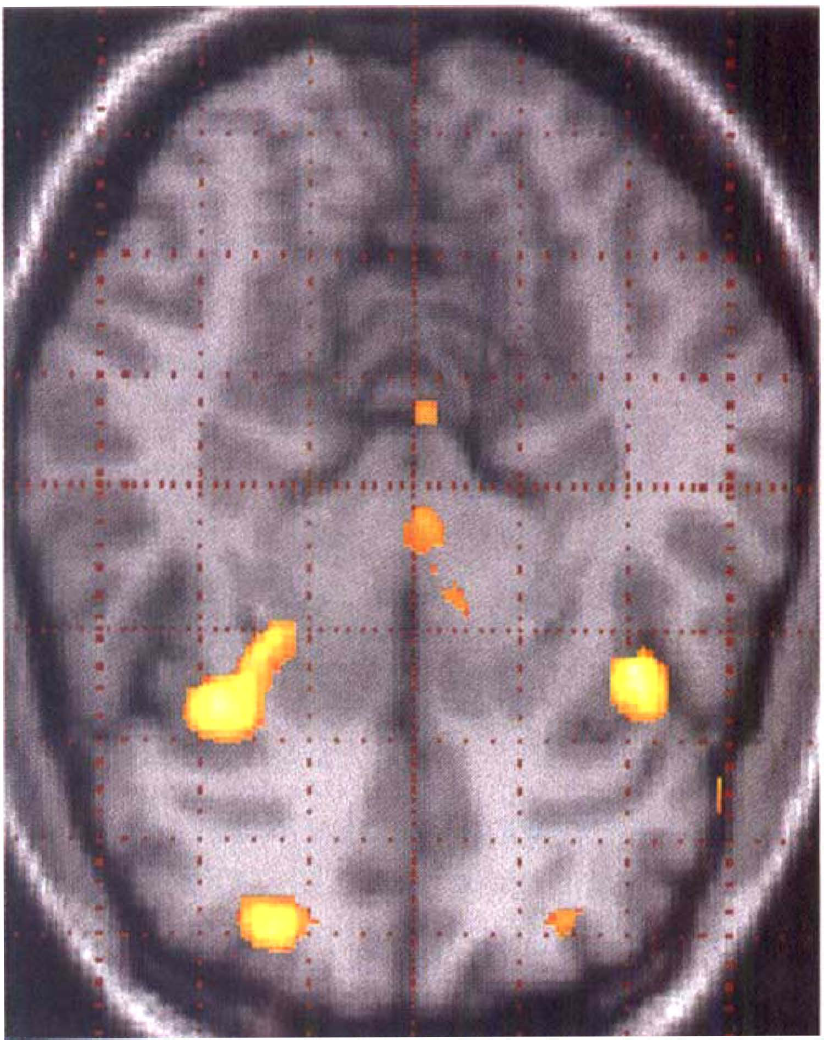
Comparison of nitroglycerin-induced acute cluster headache attack and rest (no pain) condition in nine patients with active chronic cluster headache. The activations during the attack are shown as statistical parametric maps that show the areas of significant rCBF increases (P < 0.001) in color superimposed on an anatomical reference derived from a T1-weighted MR image. The anterior part of the brain corresponds to the right side of the picture, the posterior parts to the left side, and the left side of the brain corresponds to the top of the image, the right side to the bottom. Significant activation was detected in the right frontal lobe (BA 10), bilaterally in the insula, in the cerebellum/vermis and in the hypothalamic gray matter (May et al., 1998a).
Capsaicin-induced experimental head pain
In a PET study on experimental head pain (May et al., 1998b), seven healthy male volunteers without a history of headache were studied during an acute experimental pain state. Since craniovascular pain is transmitted predominantly through the ophthalmic division of the trigeminal nerve, the other divisions being involved to a lesser extent (McNaughton, 1966), a small amount of capsaicin was administered subcutaneously to the forehead to evoke a painful sensation in the first division of the trigeminal nerve.
During the acute pain state compared with the resting state, increases in rCBF were found bilaterally in the anterior insula (Brodman area, BA 13), the contralateral thalamus, the ipsilateral anterior cingulate cortex (BA 24/32), and in the cerebellum, bilaterally (May et al., 1998b) (Fig. 4). Activation of the anterior cingulate cortex has been repeatedly reported in PET studies on the sensation of somatic or visceral pain and attributed to the emotional response to pain (Casey et al., 1994; Hsieh et al., 1996a; Jones et al., 1992; Rosen et al., 1994). Activations in the insula have been demonstrated in previous studies after application of heat (Casey et al., 1994; Coghill et al., 1994; Minoshima et al., 1995), subcutaneous injection of ethanol (Hsieh et al., 1996), somatosensory stimulation (Burton et al., 1993), and during cluster headache (Hsieh et al., 1996a) and atypical facial pain (Derbyshire et al., 1994). Given its anatomical connections, the insula has been suggested as a relay of sensory information into the limbic system and is known to play an important role in the regulation of autonomic responses (Mesulam and Mufson, 1985). The thalamus is a site where activations would most be expected in the acute pain state. Activation of the contralateral thalamus resulting from pain is known from experimental animals (Goadsby, 1997) and functional imaging studies in humans (Casey et al., 1994; Rosen et al., 1994). Furthermore, vasodilation of the carotid artery also was demonstrated with capsaicin-induced experimental head pain (May et al., 1998b), which we consider in detail below.
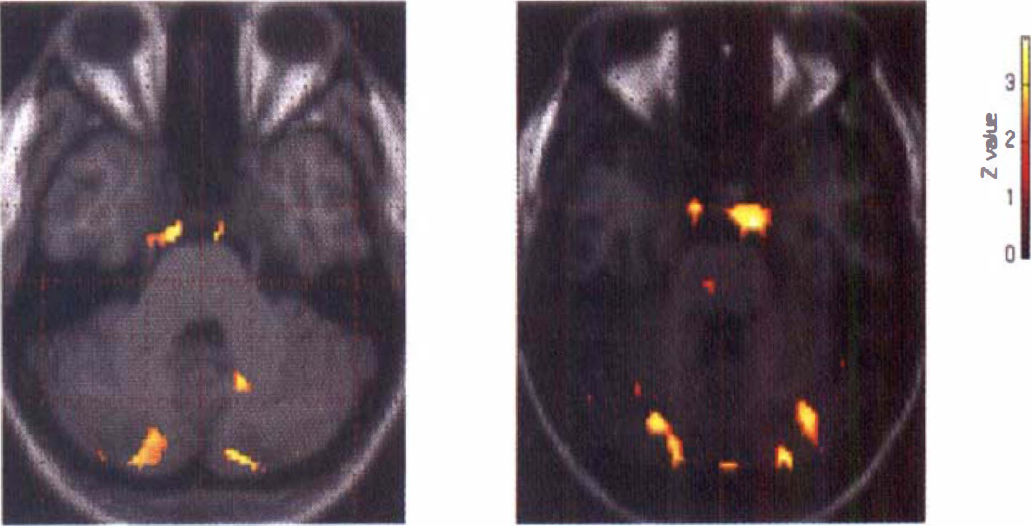
Activation in intracranial vessels in acute cluster headache compared with rest state (left side of the figure) and in capsaicin-induced head pain (right side of the figure) compared with the resting state. The activations during the attack are shown as statistical parametric maps that show the areas of significant rCBF increases (P < 0.001) in color superimposed on an anatomical reference derived from a T1-weighted MR image. The right side of the picture is the right side of the brain (May et al., 1998b).
WHAT IS SPECIFIC ABOUT THE OBSERVED POSITRON EMISSION TOMOGRAPHY ACTIVATIONS IN HEAD PAIN?
Just as it is striking that no brain stem activation occurs in cluster headache, in contrast to acute migraine (Weiller et al., 1995), similarly neither the areas specifically active in migraine or in cluster headache were seen in experimental pain induced by capsaicin injection into the forehead (May et al., 1998b). This is remarkable because, as suggested by the anatomy of the trigeminovascular system, both of these primary headaches largely involve the first, ophthalmic, division of the trigeminal nerve. Injection of capsaicin into the forehead would activate first-division afferents of the trigeminal nerve, which must share much of the nociceptive anatomy of the primary neurovascular headaches, yet the patterns of activation are unique in each setting. Considering cluster headache specifically, these data clearly imply that the activation observed in the posterior hypothalamic gray matter is involved in the pathogenesis, probably in a permissive or triggering manner, rather than simply as a response to first-division nociception per se. Furthermore, given that this area is involved in circadian rhythm and sleep-wake cycling (Moore, 1997; Nitz and Siegel, 1996), the data establish an involvement of this hypothalamic area as a primum movens in the acute cluster attack. In comparison with the PET study in spontaneous migraine (Weiller et al., 1995), no brain stem activity was found during the acute pain state when compared with the resting state. Moreover, no hypothalamic activation has been seen in either migraine (Weiller et al., 1995) or capsaicin-induced (May et al., 1998b) experimental head pain, in contrast to nitroglycerin-induced cluster headache (May et al., 1998a). This confirms that the activations seen in these primary headache syndromes are specific to the disease.
In summary, the findings from functional imaging support the idea that the pathogenesis of migraine is related to an imbalance of activity between brain stem nuclei regulating antinociception and vascular control (Goadsby et al., 1991; Lance et al., 1983), respectively. The pathogenesis of cluster headache is most likely to be found in the CNS in pacemaker or circadian regions of the hypothalamic gray matter (Goadsby and Lance, 1988).
RECENT OBSERVATIONS ON THE CEREBRAL VESSELS
In the study of experimental head pain, there was a bilateral activation pattern in midline structures over several planes (from −32 to −20 mm with respect to the anterior commissure-posterior commissure line), slightly lateralized to the left, anteriorly to the brain stem, and posteriorly to the chiasmatic region with the focus of maximum significance located at +2, +2, −22 mm (May et al., 1998b) in Tailarach coordinates (Talairach and Tournaux, 1988). Superimposed on a magnetic resonance imaging template, the location of the activation most likely covers intracranial arteries and the region of the cavernous sinus bilaterally, but more marked on the ipsilateral side. This holds true in the group study and in five of seven single-subject studies.
Similarly, in the cluster headache study, there was a strong activation observed in the same region, the cavernous sinus. This change might be interpreted as an increased venous inflow from the superior ophthalmic vein draining the ophthalmic artery, or a longer transit time for the tracer in this region, possibly resulting from an impeded venous drainage. Another more exciting possibility is that the observed increase in activation might result from a bilateral dilation of the internal carotid artery, since such a change suggests a vasodilation mediated by the ophthalmic division of the trigeminovascular system, a neurally driven vasodilation. It is difficult to assess the contribution of these two sources to the measured activity using PET.
To further address this question, magnetic resonance angiography was performed using the same study design as in the PET study. Using an image calculation tool, the angiographies were subtracted from each other, leaving only structural changes between conditions. It was demonstrated that in the condition of nitroglycerin inhalation without headache, there was dilation of the basilar artery and the internal carotid arteries bilaterally compared with the rest state (May et al., unpublished data). These vessels stayed dilated during the third condition (cluster headache attack), some 20 to 30 minutes later.
Using PET, significant activity in the region of the cavernous sinus was previously described in cluster headache patients (Hsieh et al., 1996a). An inflammatory process in the cavernous sinus and tributary veins has been proposed as being primarily responsible for cluster headaches (Hardebo, 1994). Given that we have observed vasodilation in large vessels in cluster headache and increased signal in the region of the cavernous sinus after capsaicin injection to the forehead again in a PET study (May et al., 1998b), it seems clear that the vascular changes are an epiphenomenon of activation of the trigeminovascular system (Goadsby and Duckworth, 1987), and that cluster headache is not a disorder of the carotid vasculature or cavernous sinus. Our data suggest that activation of the trigeminal system, as such, triggers the impeded arterial or venous drainage or increase in flow in the region of these vessels. At a physiologic level, the common link is the involvement of the ophthalmic division of the trigeminal nerve by a neurally driven dilation of the carotid vessels (Fig. 5). On this background, a reappraisal of the pathophysiologic mechanism of cluster headache is demanded. The data establish that cluster headache, far from being a primarily vascular disorder, is a condition whose genesis is to be found in the CNS in pacemaker or circadian regions of the hypothalamic gray matter and expression involves neurovascular dilator mechanisms.
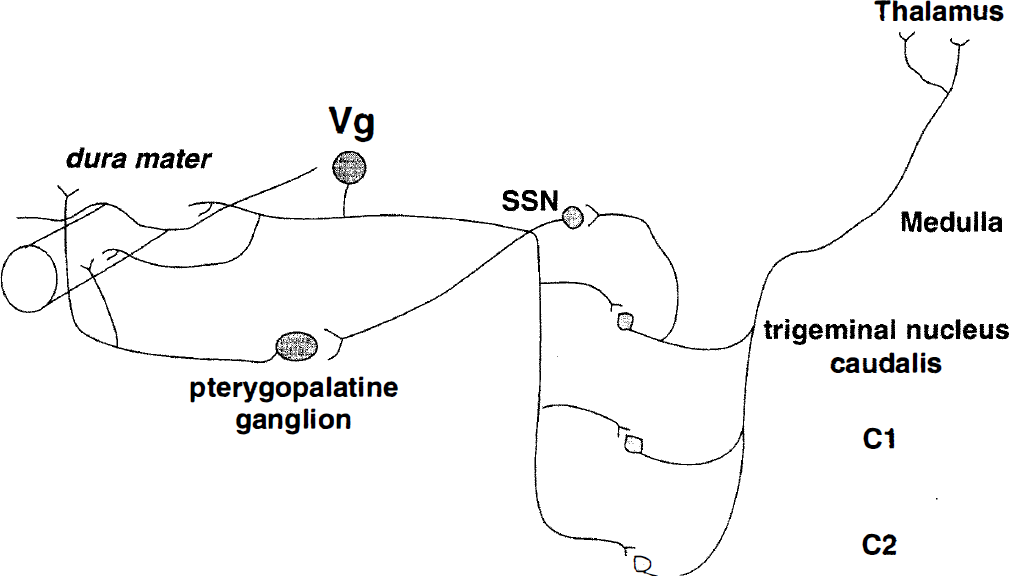
Outline of the components of the trigeminal-autonomic reflex relevant to both diseases, such as cluster headache, and with more general interest in cerebrovascular physiologic mechanisms. The ophthalmic (first) division of the trigeminal nerve with cell bodies in the trigeminal ganglion (Vg) innervates pain-producing structures in the head such as the dura mater. These bipolar neurons project to second-order neurons in the trigeminocervical complex (trigeminal nucleus caudalis and dorsal horns of C1, and C2) with a reflex connection to the superior salivatory nucleus (SSN). Preganglionic parasympathetic autonomic neurons project from the superior salivatory nucleus through the facial (seventh) nerve passing through the geniculate ganglion and synapsing in the pterygopalatine (sphenopalatine), otic, and internal carotid miniganglia with classic hexamethonium-sensitive nicotonic receptors. These neurons project to cranial vessels and to the dura mater. Pain signals from the trigeminocervical complex ascend to thalamus through the quintothalamic tract, which decussates in the brain stem.
CONCLUSION
In conclusion, advances in functional brain imaging clearly suggest that vascular changes are not the primary cause for head pain in migraine. In migraine and cluster headache, experimental and clinical data suggest an activation of the trigeminal innervation of the cranial circulation with involvement of vasoactive neuropeptides such as CGRP (Edvinsson and Goadsby, 1995; Goadsby and Edvinsson, 1993; Goadsby et al., 1988) to explain the peripheral pain mechanisms. To account for the periodicity and other clinical features in migraine, a primary dysfunction of the midbrain endogenous antinociceptive system (periaqueductal gray and dorsal raphe nucleus) and the neural control of CBF (dorsal raphe nucleus and locus ceruleus) (Goadsby and Lance, 1988) seems most likely. The cluster headache data implicate the region of the hypothalamic gray matter as being involved in the fundamental disease process, whereas both PET and magnetic resonance angiography have shown vascular changes which, taken with the experimental head pain data, are indicative of trigeminovascular activation in those conditions. These clinical conditions are extremes of physiology and as such contributes to understanding what has been the subject of argument since Willis (1664); there is a functional neural innervation of the cerebral circulation, and it can be entrained during certain pathophysiologic states. If we are to understand the cerebral circulation and treat maladies related to, or expressed by, its neural innervation, we must continue to explore the normal role of these nerve systems and their role in disease states.
Footnotes
Acknowledgments
The authors acknowledge the important contributions to our recent functional imaging studies from Drs. A. Bahra, C. Buchel, R. Turner, and R.S.J. Frackowiak.