
Abstract
Glutamate receptor-mediated responses were investigated by using a whole-cell recording and an intracellular calcium ion ([Ca2+]i) imaging in gerbil postischemic hippocampal slices prepared at 1, 3, 6, 9, 12, and 24 hours after 5-minute ischemia. Bath application of N-methyl-D-aspartic acid (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), and kainate showed that NMDA-, AMPA- and kainate-induced currents were enhanced in postischemic CA1 pyramidal neurons at 1 to 12 hours after 5-minute ischemia. NMDA and non-NMDA receptor-mediated excitatory postsynaptic currents (EPSC) were examined in postischemic CA1 pyramidal neurons at 3 hours after 5-minute ischemia to confirm whether synaptic responses are enhanced in the postischemic CA1 pyramidal neurons. The amplitudes of NMDA- and non-NMDA-receptor-mediated EPSC were enhanced in the postischemic CA1 pyramidal neurons. NMDA-, AMPA-, and kainate-induced [Ca2+]i elevations were also examined to determine whether the enhancement of currents is accompanied by the enhancement of [Ca2+]i elevation. The enhancements of NMDA-, AMPA-, and kainate-induced [Ca2+]i elevations were shown in the postischemic CA1. These results indicate that NMDA and non-NMDA receptor-mediated responses are persistently enhanced in the CA1 pyramidal neurons 1 to 12 hours after transient ischemia, and suggest that the enhancement of glutamate receptor-mediated responses may act as one of crucial factors in the pathologic mechanism responsible for leading postischemic CA1 pyramidal neurons to irreversible neuronal injury.
Keywords
Transient ischemia damages specific populations of central neurons. Pyramidal neurons in the CA1 of the hippocampus are particularly vulnerable and show delayed neuronal death after transient ischemia (Kirino, 1982; Pulsinelli et al., 1982). It has been reported that the ischemic damage in the CA1 is reduced by the prior administration of glutamate receptor antagonists (Gill et al., 1987), and also importantly that the delayed neuronal death in the CA1 is effectively prevented by glutamate receptor antagonists even when they were administered after the ischemic insult (Gill et al., 1988; Sheardown et al., 1990; Li and Buchan, 1993). These observations indicate that the condition leading to CA1 neuronal death is reversible after ischemia, and also that glutamate receptors are involved in the pathologic mechanism responsible for the CA1 delayed neuronal death during a postischemic period.
In the present study, we investigated glutamate receptor-mediated responses in postischemic CA1 pyramidal neurons using a whole-cell recording and an intracellular calcium ion imaging in gerbil hippocampal slice preparations to determine whether functional changes of glutamate receptors are involved in postischemic CA1 pyramidal neurons.
MATERIALS AND METHODS
Chemicals
N-methyl-D-aspartic acid (NMDA) and (+)-bicuculline were obtained from Sigma Chemical Co. (St Louis, MO, USA). D(−)-2-amino-5-phosphonopentanoic acid (D-APV), (S)-a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX) were purchased from Tocris Cookson Ltd. (Bristol, UK). Other agents came from Wako Pure Chemical Industries Ltd. (Osaka, Japan).
Preparation
The following experiments were conducted in accordance with the Guidelines for Animal Experimentation at Ehime University School of Medicine.
Male Mongolian gerbils (weighing 60 to 80 g; Seiwa, Fukuoka, Japan) were used. Ischemia was induced essentially as described in previous studies (Mitani et al., 1991; 1994b). In brief, gerbils were anesthetized and maintained with a mixture of 2.5% halothane in nitrous oxide/oxygen (7:3). Through a ventral midline cervical incision, the bilateral common carotid arteries were exposed and then a 4-0 silk suture was looped around each artery. The head of the animal was held in a stereotaxic apparatus (type 1430; David Kopf, USA). Then, the animal was turned over and held in a supine position. Thermocouple needle-probe (0.4 mm in diameter, TN-800; Unique Medical Co., Japan) and thermocouple meter (TME-300; Unique Medical Co., Japan) assemblies were used to monitor brain (striatum) and rectal temperatures. Halothane administration was decreased, and the animals were maintained with a mixture of halothane (1%) in nitrous oxide/oxygen (7:3). The sutures around the two common carotid arteries were pulled by 12-g weights to occlude the circulation (Mitani et al., 1990b). After ischemia, the sutures were cut and removed to restore the blood flow. The vessels were visually inspected for the absence of blood clots and the recovery of blood flow under an operative microscope. The brain and rectal temperates were maintained at 37 ± 0.3°C during ischemia and for 30 minutes after the onset of recirculation. Then, the thermocouple probes were gently pulled out, and all surgical incisions were carefully sutured. Gerbils were treated with antibiotics, removed from the stereotaxic apparatus, and brought into a comfortable position on a warming blanket. After awakening, the animals were returned to individual cages in a room maintained at constant temperature (26°C) and allowed access to food and water ad libitum.
The procedures used for preparation of hippocampal slices were essentially the same as those described in previous studies (Mitani et al., 1993; 1994a). In brief, the hippocampal slices were prepared from postischemic gerbils at 1, 3, 6, 9, 12, and 24 hours after 5-minute ischemia and from control gerbils that were subjected to the identical surgical procedures except for the clamping of the common carotid arteries at 3 hours before the slice preparation. The gerbils were anesthetized with ether and decapitated. Their brains were rapidly removed, and transverse slices (300 μm thick) were cut with a vibrating slicer (DTK-1000; Dosaka CO., Kyoto, Japan). Slices were incubated at 36°C for 1 hour and then maintained at room temperature (23 to 25°C) in a standard Ringer solution containing (mmol/L): NaCl, 124; KCl, 2; KH2PO4, 1.5; NaHCO3, 26; MgCl2, 1; CaCl2, 2.5; glucose, 10; pH 7.4, when equilibrated with 95% O2/5% CO2 gas mixture. Usually six to 10 hippocampal slices were obtained from an animal. All solutions including perfusing fluids used in the present study were equilibrated with 95% O2/5% CO2 gas mixture.
Measurements
All measurements in the slices were made by observers who were blinded to the experimental conditions.
Whole-cell recording. The slice was transferred into a recording chamber (volume about 0.5 mL) mounted onto the stage of a microscope (Axioskop; Zeiss, Germany) and held down with a nylon net stretched out on a U-shaped piece of flattened platinum wire (Edwards et al., 1989). Pyramidal neurons in the CA1 were visualized using Nomarski optics with a water-immersion × 40 objective (Zeiss, Germany) at a magnification of × 640 and also visualized using a charge coupled device camera and television monitor system (XC-77; Sony, Japan) at a magnification of × 1000. The slice was perfused with a standard Ringer solution (34°C) at a flow rate of 3 mL/min.
Whole-cell pipettes were pulled from thin-walled borosilicated glass capillaries (GC150TF-15; Clark Electromedical Instruments, UK) and were filled with an intracellular solution containing (mmol/L): CsCl, 130; MgCl2, 3; CaCl2, 1; Na-ATP, 2; Na-GTP, 2; ethylene glycol-bis(b-aminoethyl ether) N, N, N′, N′-tetraacetic acid, 10; N-2-2-hydroxyethylpiperazine N″ 2-ethanesulfonic acid, 10 (adjusted to pH 7.2 with CsOH). The direct current resistance of the pipettes was about 5 MΩ. Whole-cell currents were recorded using a patch clamp amplifier (EPC-7; List Electronics, Germany). Liquid-junction potential between the perfusion fluid and the pipette solution was corrected. The seal resistance was usually more than 10 GΩ. Signals were low-pass filtered at 10 kHz (3-pole Bessel filter) and stored on a pulse code modulation video tape recorder for later off-line analysis.
Glutamate agonist (12.5-μmol/L NMDA, 12.5-μmol/L AMPA, or 50-μmol/L kainate)-induced currents were investigated in CA1 pyramidal neurons at 1, 3, 6, 9, 12, and 24 hours after 5-minute ischemia and control neurons (8 to 10 animals at each time point) to examine the time-course changes in postischemic glutamate receptor-mediated responses. Tetrodotoxin (1 μmol/L) and bicuculline (20 μmol/L) were added routinely to a standard Ringer solution to block Na+ currents and spontaneous synaptic activities mainly mediated by γ-aminobutyric acid, respectively. To release NMDA responses from a Mg2+ block (Nowak et al., 1984; Mayer et al., 1984), Mg2+ was omitted from the standard Ringer solution (we defined this solution as a nominally Mg2+-free Ringer solution in the present study), and glycine (10 μmol/L) was also added to the solution to ensure a constant, saturating concentration of glycine for the NMDA receptor (Johnson and Ascher, 1987; Hestrin et al., 1990; Takahashi et al., 1996). The glutamate agonists were bath applied to a slice for 60 seconds by switching the superfusion line at the inlet of the recording chamber with magnetic pinch valves. The dead-space time for a complete exchange of the solution was about 20 seconds. Usually, a single trial of the agonist application was performed in each slice. NMDA receptor antagonist D-APV (25 μmol/L) and AMPA receptor antagonist NBQX (25 μmol/L) were used to test their blocking effects on the appropriate agonist-induced currents.
Twenty-four postischemic animals at 3 hours after 5-minute ischemia and 24 control animals were used to examine the changes in dose-response curves for each glutamate agonist. Glutamate agonist (1 to 500 μmol/L of NMDA, 0.5 to 200 μmol/L of AMPA, or 1 to 500 μmol/L of kainate) was bath applied to the slice and the whole-cell recordings were made in CA1 pyramidal neurons with the same procedure as described above.
Intracellular calcium ion imaging. Intracellular calcium ion ([Ca2+]i) imaging was performed to determine whether changes in the glutamate agonist-induced [Ca2+]i elevation are involved during a postischemic period. The procedures used were essentially the same as described in previous studies (Mitani et al., 1990a; 1994a). Twelve gerbils were used (4 post-ischemic animals at 3 hours after 5-minute ischemia, 4 postischemic animals at 24 hours after 5-minute ischemia, and 4 control animals). The slices were loaded with a fluorescent indicator, rhod-2 acetoxymethyl ester (Minta et al., 1989) (5 μmol/L, Dojin, Japan) diluted in a standard Ringer solution for 45 minutes at 26°C after a 1-hour incubation period within a standard Ringer solution. After the loading incubation, the slices were incubated in a standard Ringer solution for at least 30 minutes at 26°C to allow hydrolysis of the ester.
Changes in levels of [Ca2+]i were measured using an inverted fluorescence microscope, a high performance video camera, and an image processor setup. A side illumination system with a low magnification objective lens (×4) was used to visualize the fluorescence image of a whole slice, and the image was subjected to the image analysis (Kudo et al., 1991). The slice was transferred to a flow-through chamber (volume about 0.1 mL) mounted on an inverted fluorescence microscope (Olympus IMT-2, Japan), and was submerged and perfused with a standard Ringer solution (34°C) at a flow rate of 3 mL/min. Tetrodotoxin (1 μmol/L) and bicuculline (20 μmol/L) were added routinely to the standard Ringer solution. The slice was excited at 550 nm using an ultraviolet lamp (100 W, Osram, Germany) with an interference filter (550 nm, slit width < 16 nm). Signals of the fluorescence (> 580 nm) were captured on a silicon intensified target camera (C2400-8; Hamamatsu, Japan), then stored and processed using an image processor (Argus-100; Hamamatsu, Japan). Before measurement of changes in [Ca2+]i the slice loaded with rhod-2 was excited with a 550-nm light and the image on a television monitor was examined to confirm that the dye was uniformly distributed throughout the slice. Then, the measurement of changes in [Ca2+]i was performed. The slice was excited with 550-nm light every 5 seconds. During the measurement, the glutamate agonist (12.5 μmol/L of NMDA, 25 μmol/L of AMPA, or 50 μmol/L of kainate) was bath applied for 60 seconds to the slice. A single trial of the agonist application was performed in a slice. For NMDA application, Mg2+ was omitted from the standard Ringer solution and 10 μmol/L of glycine was added.
The fluorescence ratio was calculated as follows: the numerical value of element (pixel) of each image taken every 5 seconds during measurement of [Ca2+]i was divided by the value of the corresponding element in the 550-nm image that was taken before the measurement of changes in [Ca2+]i. The ratio images were displayed on a high-resolution television monitor (Sony, Japan), photographed, and analyzed.
Data analysis
Data obtained by whole-cell recordings were analyzed using the pCLAMP software package (Axon Instruments, version 6.0) on an IBM AT. All data are presented as mean ± SD, and the statistical significance of difference (at P < .01 and P < .05) was assessed using Student's t-test or analysis of variance (ANOVA) that included Dunnett's multiple-comparison procedure. All analyses were performed using the StatView (version 4.02) and Super ANOVA (version 1.11) statistical packages (Abacus Concepts Inc., Berkeley, CA, USA).
RESULTS
Agonist-induced currents
The time-course changes in postischemic glutamate receptor-mediated responses were studied in 324 postischemic CA1 pyramidal neurons at 1, 3, 6, 9, 12, and 24 hours after 5-minute ischemia (n = 54 neurons from each group) and in 54 control CA1 pyramidal neurons. NMDA (12.5 μmol/L) evoked inward currents (peak current = 2.62 ± 0.37 nA, mean ± SD, n = 15) in control CA1 pyramidal neurons held at −80 mV in a nominally Mg2+-free Ringer solution (Fig. 1A-1). The responses were almost completely blocked by 25 μmol/L of D-APV (Fig. 1A-2) and by 1 mM Mg2+ (Fig. 1A-3). The blockage of the response by Mg2+ was shown in a voltage-dependent manner: The NMDA-induced inward currents appeared even in the presence of 1 mmol/L Mg2+ when the membrane potentials were held more positive (−20 mV) (Fig. 1A-4). The NMDA-induced currents were significantly enhanced in postischemic CA1 pyramidal neurons 1 to 12 hours after 5-minute ischemia compared with those in controls (Fig. 1B-1). Mean peak currents at 1, 3, 6, 9, and 12 hours after 5-minute ischemia (holding potentials = −80 mV) were 3.41 ± 0.44 (1.30-fold increase, P < .01, Dunnett's multiple-comparison procedure), 3.47 ± 0.41 (1.32, P < .01), 3.28 ± 0.41 (1.25, P < .01), 3.31 ± 0.62 (1.26, P < .01) and 3.22 ± 0.58 nA (1.23, P < .01), respectively (Fig. 2). The responses were almost completely blocked by 25 μmol/L of D-APV (Fig. 1B-2) and by 1 mmol/L Mg2+ (Fig. 1B-3). The voltage-dependent nature of NMDA responses in the presence of Mg2+ was also observed in postischemic CA1 pyramidal neurons 1 to 12 hours after 5-minute ischemia. The NMDA response appeared at −20 mV (Fig. 1B-4). The significant enhancement of NMDA-induced currents was not observed in postischemic CA1 pyramidal neurons at 24 hours after 5-minutes of ischemia (Fig. 1C-1). Mean peak current at −80 mV was 2.80 ± 0.55 nA (1.07-fold increase) (Fig. 2). The responses were almost completely blocked by 25 μmol/L of D-APV (Fig. 1C-2). In the presence of 1 mmol/L Mg2+, the NMDA-induced currents at 24 hours after ischemia were voltage-dependently blocked by Mg2+ in 11 of 15 postischemic CA1 pyramidal neurons; however, the remaining 4 postischemic pyramidal neurons showed small but apparent NMDA-induced inward currents even at −80 mV (Fig. 1C-3). The NMDA-induced currents obviously appeared at −20 mV (Fig. 1C-4).
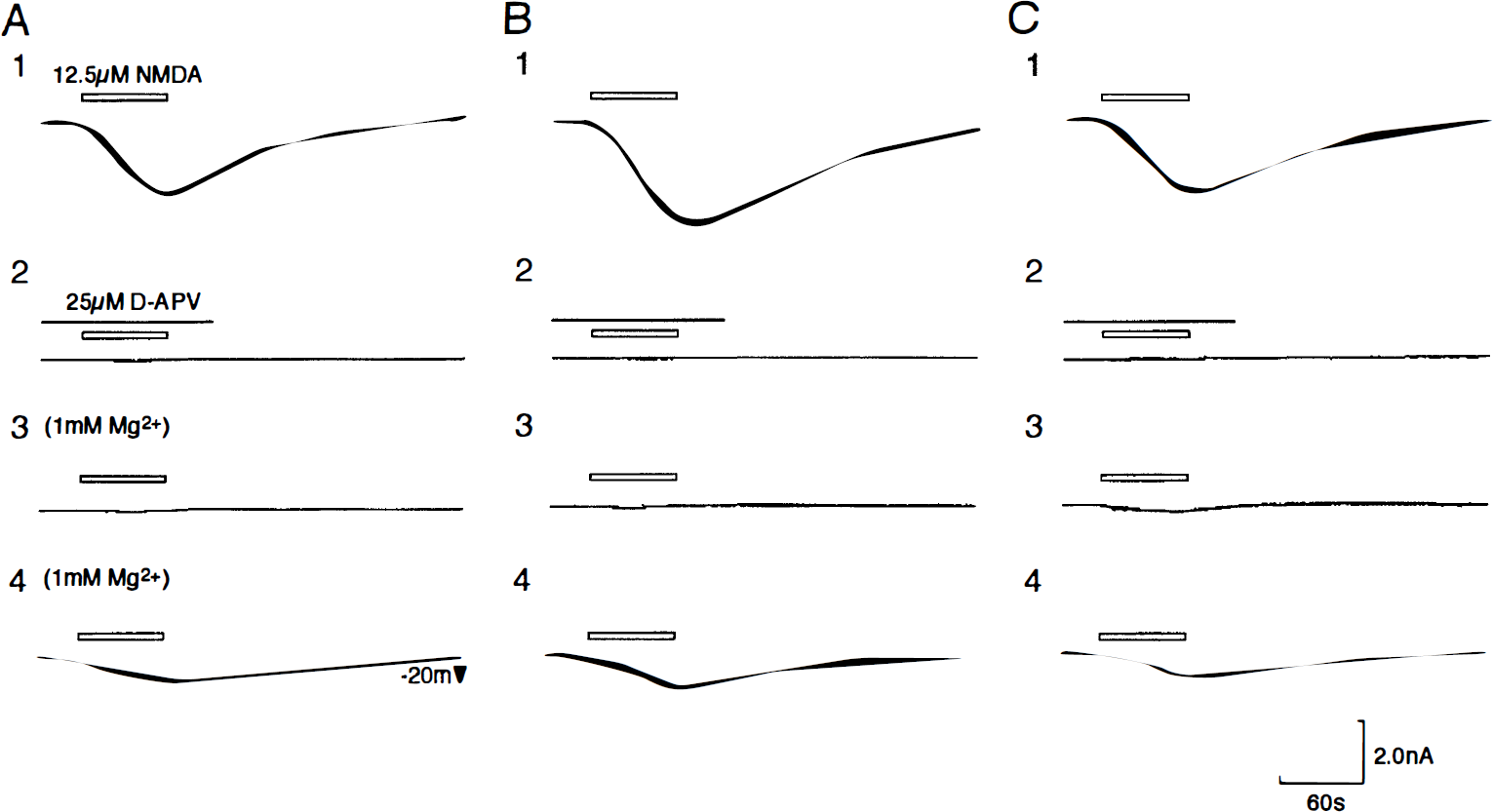
Whole-cell responses of postischemic CA1 pyramidal neurons to bath application of NMDA.
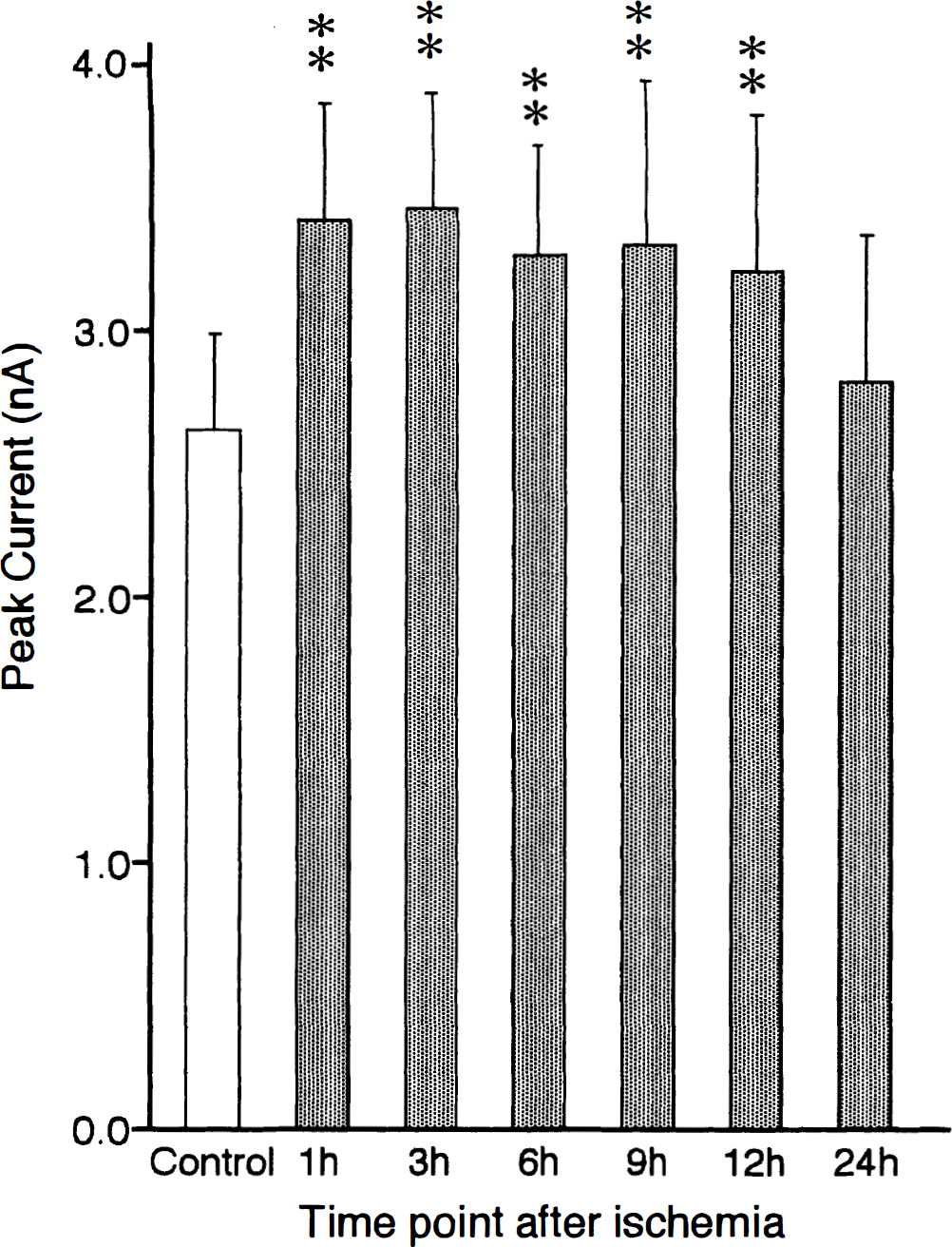
The time-course changes in the peak amplitude of NMDA-induced inward currents in postischemic CA1 pyramidal neurons (15 neurons at each time point) are represented. Responses to NMDA were measured in a nominally Mg2+-free condition (holding potential: −80 mV). The NMDA-induced currents were recorded from control CA1 pyramidal neurons (open column) and postischemic CA1 pyramidal neurons 1, 3, 6, 9, 12, and 24 hours after 5-minute ischemia (dark columns). The ordinate represents peak amplitude of NMDA-induced currents; the abscissa represents time point after 5-minute ischemia. The values are mean ± SD. Significances are **P < .01 (one-way analysis of variance, Dunnett's multiple-comparison procedure).
AMPA (12.5 μmol/L) evoked inward currents (peak current = 3.04 ± 0.34 nA, n = 15) in control CA1 pyramidal neurons held at −80 mV (Fig. 3A-1). The responses were almost completely blocked by the AMPA receptor antagonist NBQX (25 μmol/L) (Fig. 3A-2). The AMPA-induced currents were significantly enhanced in postischemic CA1 pyramidal neurons 1 to 12 hours after 5-minute ischemia compared with those in controls (Fig. 3B-1). Mean peak currents at 1, 3, 6, 9, and 12 hours after 5-minute ischemia (holding potential = −80 mV) were 3.66 ± 0.58 (1.20-fold increase, P < .01), 4.02 ± 0.51 (1.32, P < .01), 3.68 ± 0.36 (1.21, P < .01), 3.58 ± 0.34 (1.18, P < .01) and 3.48 ± 0.26 nA (1.14, P < .05), respectively (Fig. 4). The enhanced AMPA-induced inward currents in postischemic CA1 pyramidal neurons were almost completely blocked by 25 μmol/L of NBQX (Fig. 3B-2). The significant enhancement of AMPA-induced currents was not observed in postischemic CA1 pyramidal neurons at 24 hours after 5-minute ischemia (Fig. 3C-1). Mean peak current at 24 hours after 5-minute ischemia (holding potential = −80 mV) was 3.01 ± 0.51 nA (0.99-fold increase) (Fig. 4). The responses were almost completely blocked by 25 μmol/L of NBQX (Fig. 3C-2).
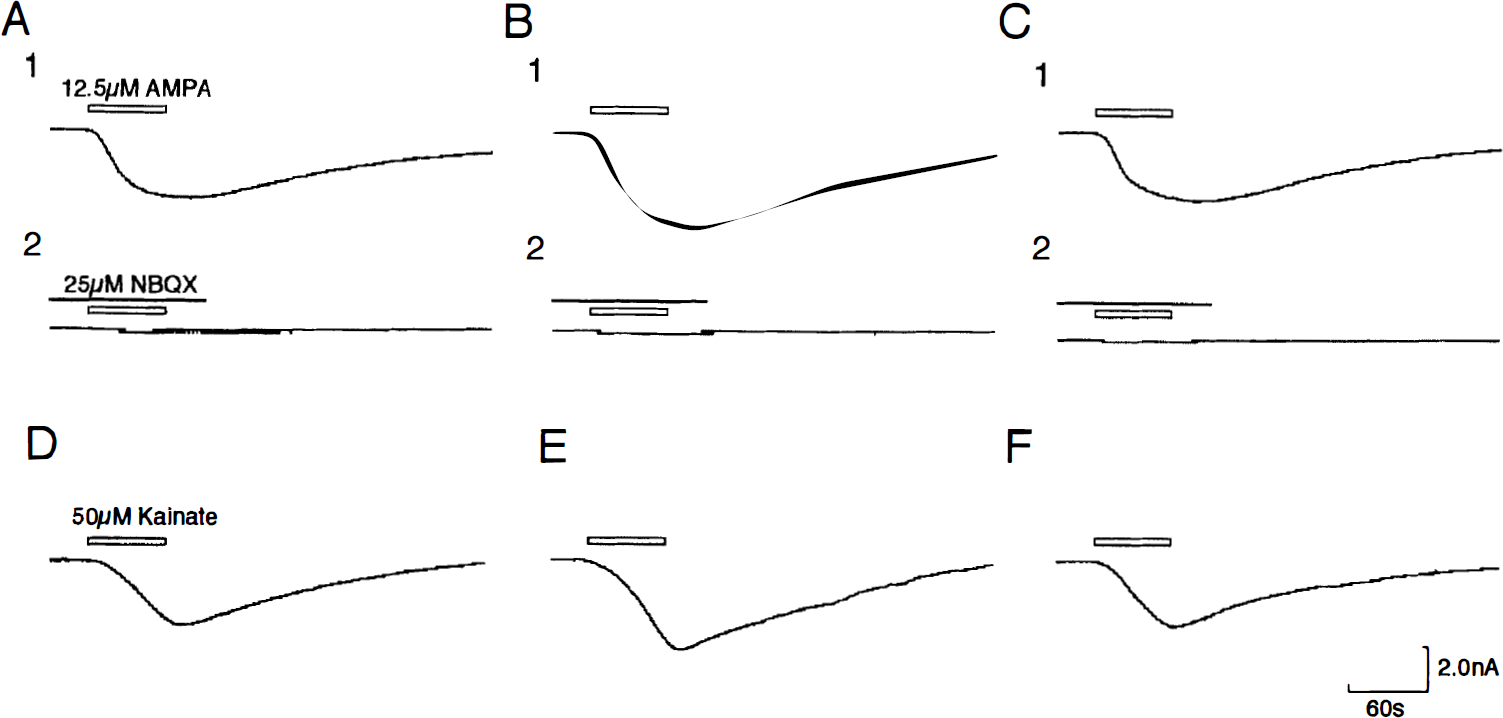
Whole-cell responses of postischemic CA1 pyramidal neurons to bath application of AMPA and kainate. All recordings were obtained at the membrane potential of −80 mV. The external solution contained 1 μmol/L of tetrodotoxin and 20 μmol/L of bicuculline.
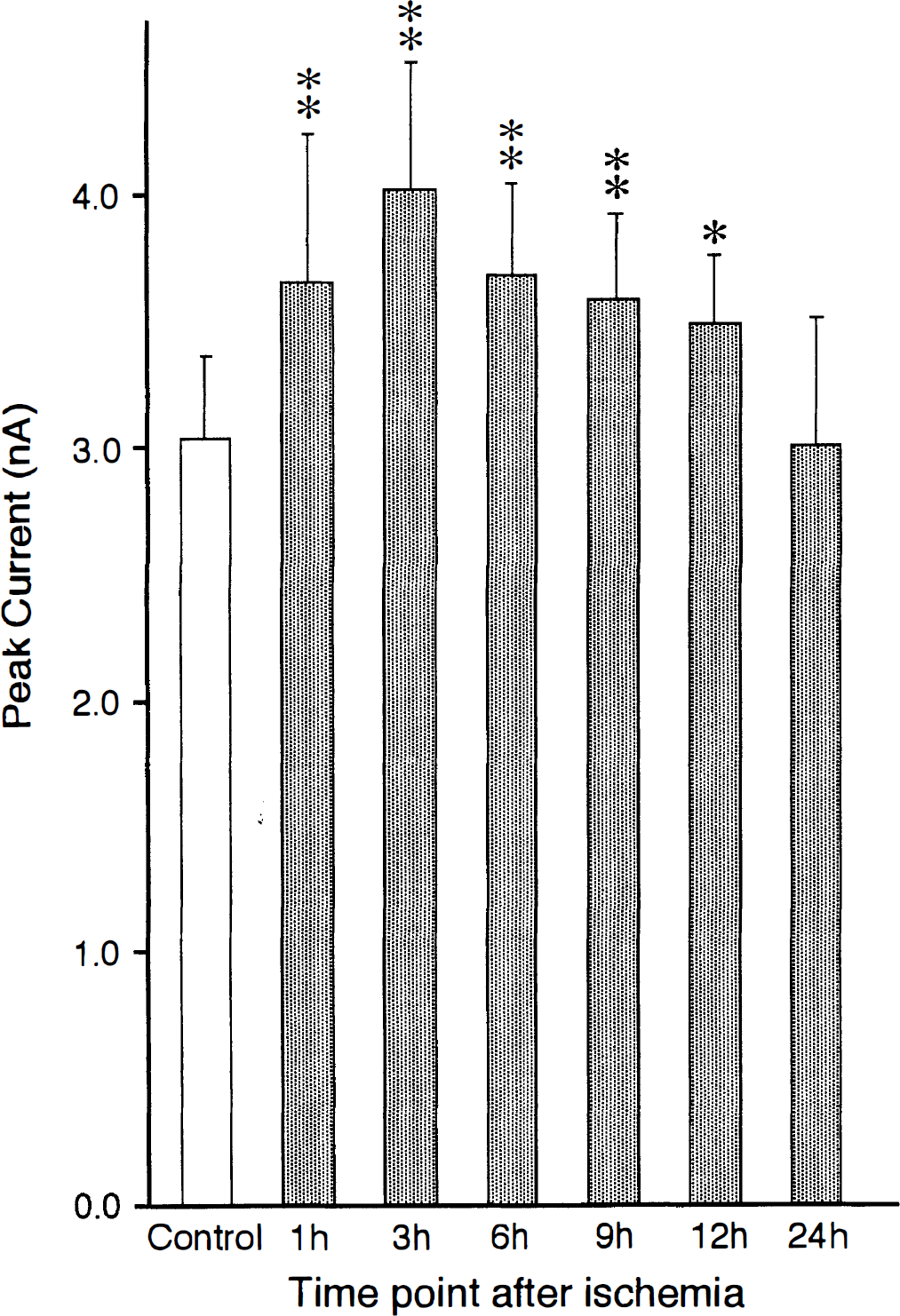
The time-course changes in the peak amplitude of AMPA-induced inward currents in postischemic CA1 pyramidal neurons (15 neurons at each time point) are represented. The AMPA-induced currents were recorded from control CA1 pyramidal neurons (open column) and postischemic CA1 pyramidal neurons 1, 3, 6, 9, 12, and 24 hours after 5-minute ischemia (dark columns). The ordinate represents peak amplitude of AMPA-induced currents; the abscissa represents time point after 5-minute ischemia. The values are mean ± SD. Significances are ** P < .01, * P < .05 (one-way analysis of variance, Dunnett's multiple-comparison procedure).
Kainate (50 μmol/L) evoked inward currents (peak current = 2.93 ± 0.32 nA, n = 15) in control CA1 pyramidal neurons held at −80 mV (Fig. 3D). The kainate-induced currents were significantly enhanced in postischemic CA1 pyramidal neurons 1 to 12 hours after 5-minute ischemia compared with those in controls (Fig. 3E). Mean peak currents at 1, 3, 6, 9, and 12 hours after 5-minute ischemia (holding potentials = −80 mV) were 3.61 ± 0.32 (1.23-fold increase, P < .01), 3.87 ± 0.46 (1.32, P < .01), 3.66 ± 0.31 (1.25, P < .01), 3.51 ± 0.33 (1.20, P < .01) and 3.33 ± 0.34 nA (1.14, P < .01), respectively (not illustrated). The significant enhancement of kainate-induced currents was not observed in postischemic CA1 pyramidal neurons at 24 hours after 5-minute ischemia (Fig. 3F). Mean peak current at 24 hours after 5-minute ischemia (holding potential = −80 mV) was 2.99 ± 0.26 nA (1.02-fold increase).
The dose-dependency of agonist-induced currents was studied in 225 postischemic CA1 pyramidal neurons 3 hours after ischemia and 225 control CA1 pyramidal neurons. No significant differences were observed in the maximal response for each agonist (NMDA, AMPA, and kainate) between postischemic CA1 pyramidal neurons and control CA1 pyramidal neurons (Fig. 5). The concentration needed to evoke 50% of the maximal response (EC50) for NMDA in postischemic CA1 pyramidal neurons (11 μmol/L) was approximately half of that observed in control CA1 pyramidal neurons (EC50 = 20 μmol/L) (Fig. 5A). EC50 for AMPA (5 μmol/L) and for kainate (31 μmol/L) in postischemic CA1 pyramidal neurons were also approximately halves of those observed in control CA1 pyramidal neurons (EC50 for AMPA = 10.5 μmol/L, for kainate = 57 μmol/L), respectively (Fig. 5B, C). These results suggest that the expression of the enhancement of NMDA- and non-NMDA-induced currents is mediated by upregulation of postsynaptic NMDA and non-NMDA receptors but not by an increment in the number of NMDA and non-NMDA receptors.
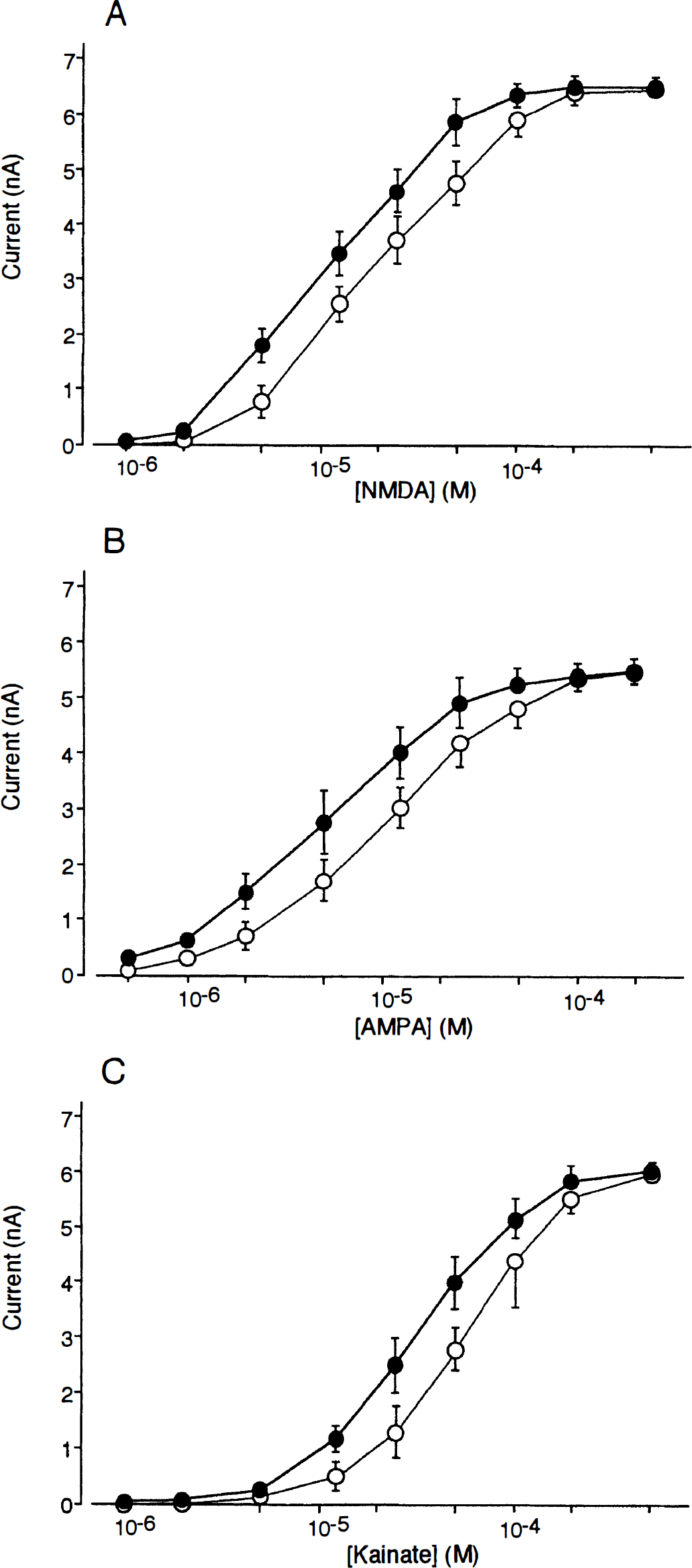
Dose-response curves for peak inward currents induced by bath applications of NMDA
EPSC
EPSC were recorded from 16 postischemic CA1 pyramidal neurons at 3 hours after 5-minute ishcemia and 16 CA1 control pyramidal neurons.
In control CA1 pyramidal neurons, the NMDA receptor-mediated EPSC were evoked by stimulation of the Schaffer collateral-commissural fibers (mean peak currents: 115 ± 43 pA at −70 mV) and the peak current-voltage (I-V) relation was nearly linear over the almost whole range of membrane potentials in a nominally Mg2+-free Ringer solution (n = 8) (Fig. 6A, D). The reversal potential was about 0 mV. The addition of 1 mmol/L Mg2+ markedly decreased the inward currents at membrane potentials more negative than −30 mV (not illustrated). In postischemic CA1 pyramidal neurons, the NMDA receptor-mediated EPSC were enhanced in a nominally Mg2+-free Ringer solution (n = 8) (Fig. 6B). Mean peak current of NMDA receptor-mediated EPSC was 162 ± 44 pA at −70 mV (1.41-fold increase; P < .05, Student's t-test). The peak I-V relation was nearly linear over the almost whole range of membrane potentials (Fig. 6D). The reversal potential was about 0 mV. The addition of 1 mmol/L Mg2+ markedly decreased the inward currents at membrane potentials more negative than −30 mV (Fig. 6C). The peak I-V relation after the addition of Mg2+ was nonlinear with a region of negative slope resistance in the range −90 to −30 mV (Fig. 6D).
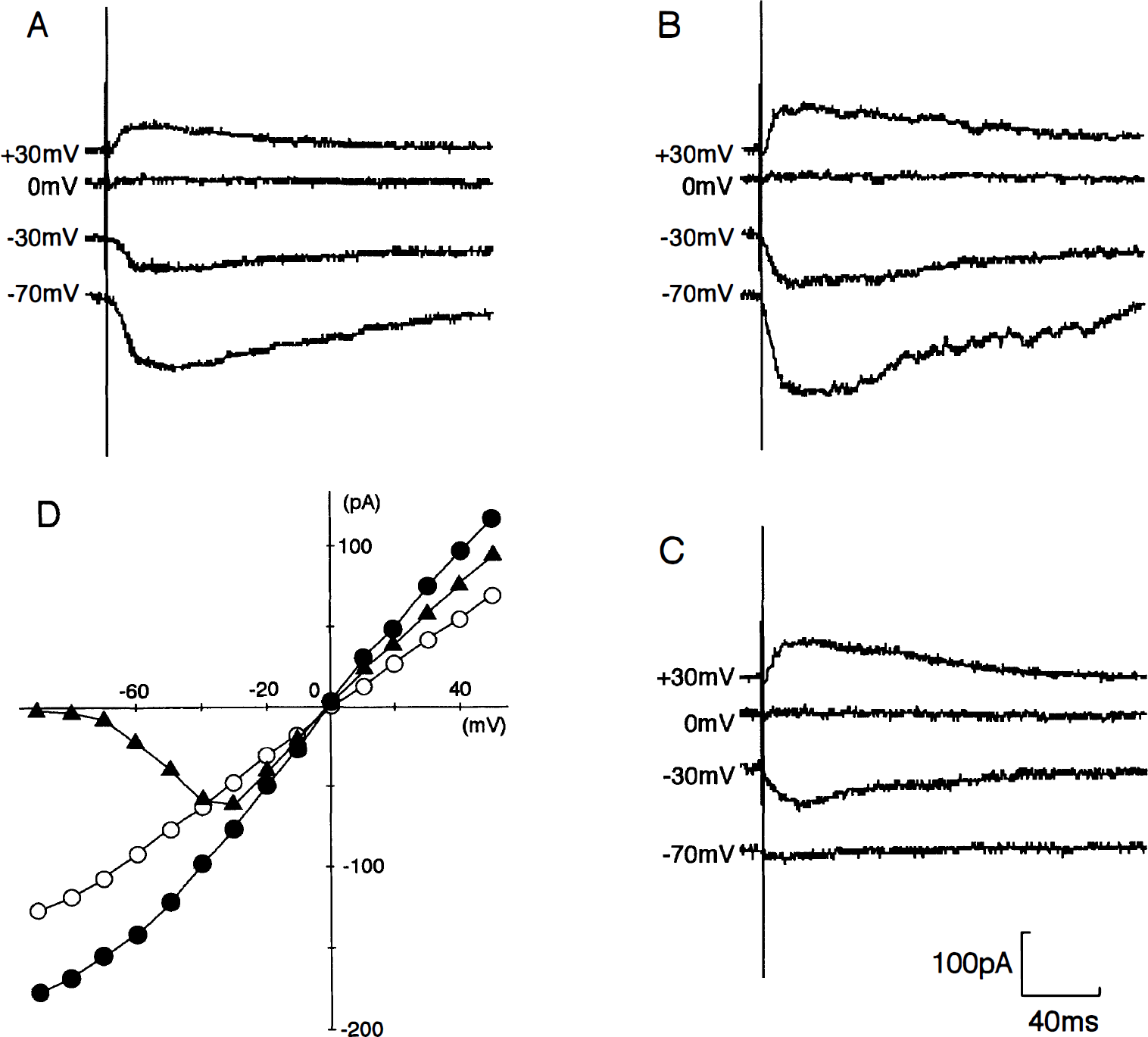
NMDA receptor-mediated excitatory postsynaptic currents (EPSC) recorded from a control CA1 pyramidal neuron
In control CA1 pyramidal neurons, the non-NMDA receptor-mediated EPSC were evoked by stimulation of the Schaffer collateral-commissural fibers (mean peak currents: 118 ± 40 pA at −70 mV) and the peak I-V relation was linear over the whole voltage range with a reversal potential of about 0 mV (n = 8) (Fig. 7A, C). In postischemic CA1 pyramidal neurons, non-NMDA receptor-mediated EPSC were enhanced (n = 8) (Fig. 7B). Mean peak current of non-NMDA receptor-mediated EPSC was 163 ± 45 pA at −70 mV (it was 1.38-fold increase; P < .05, Student's t-test). The peak I-V relation was linear over the whole voltage range with a reversal potential of about 0 mV (Fig. 7C).
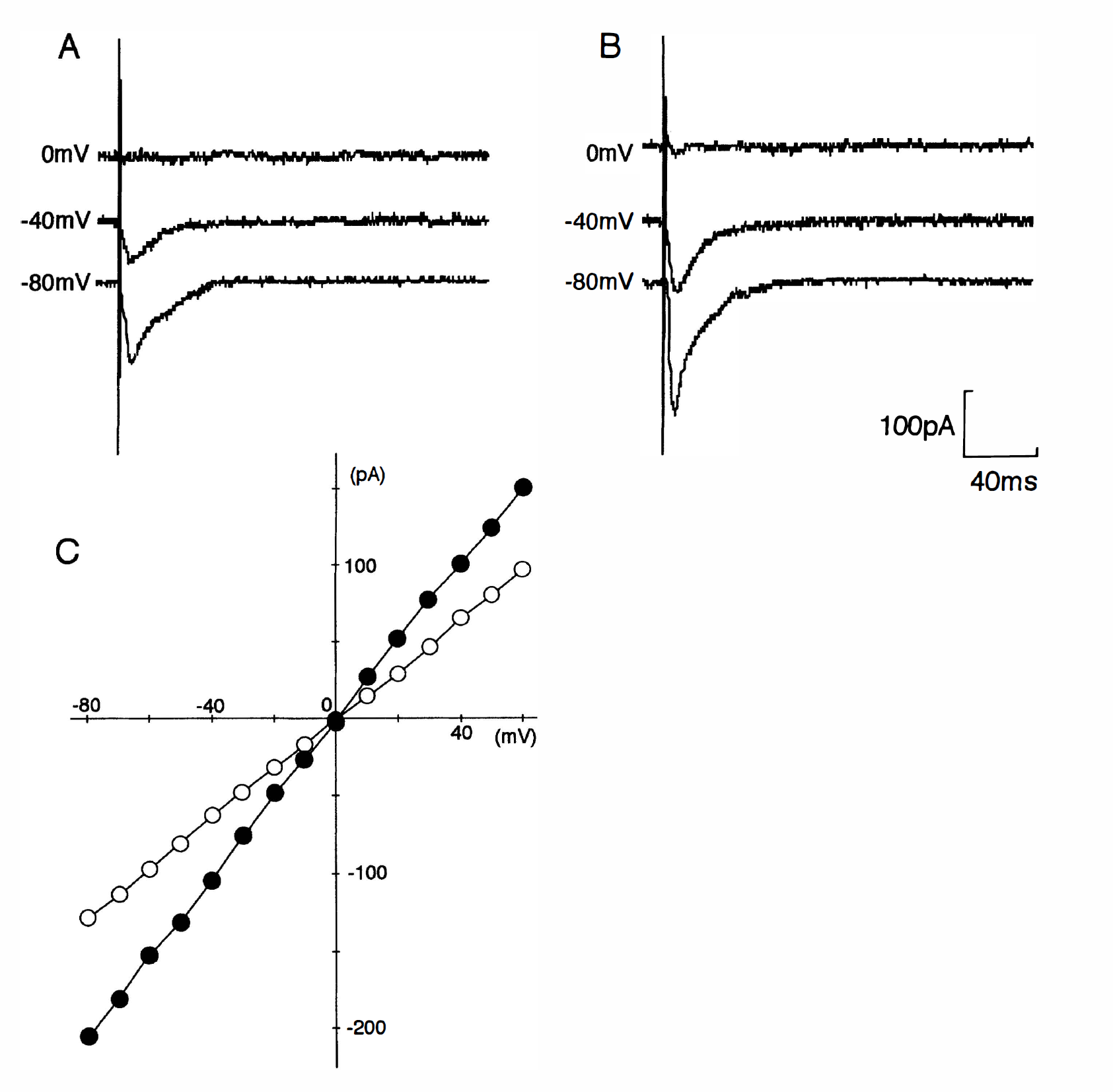
Non-NMDA receptor-mediated excitatory postsynaptic currents (EPSC) recorded from a control CA1 pyramidal neuron
No significant changes were detected in the rise-time and the decay time constant of the NMDA receptor- and non-NMDA receptor-mediated EPSC between postischemic CA1 pyramidal neurons and control CA1 pyramidal neurons.
[Ca2+]i elevation
[Ca2+]i elevations induced by glutamate agonists were measured in 24 postischemic hippocampal slices 3 hours after 5-minute ischemia, 24 postischemic hippocampal slices 24 hours after 5-minute ischemia, and 24 control hippocampal slices. Before application of agonists, [Ca2+]i levels were low in all hippocampal regions (Fig. 8A). There were no significant differences in resting [Ca2+]i levels between postischemic hippocampal slices and control slices.
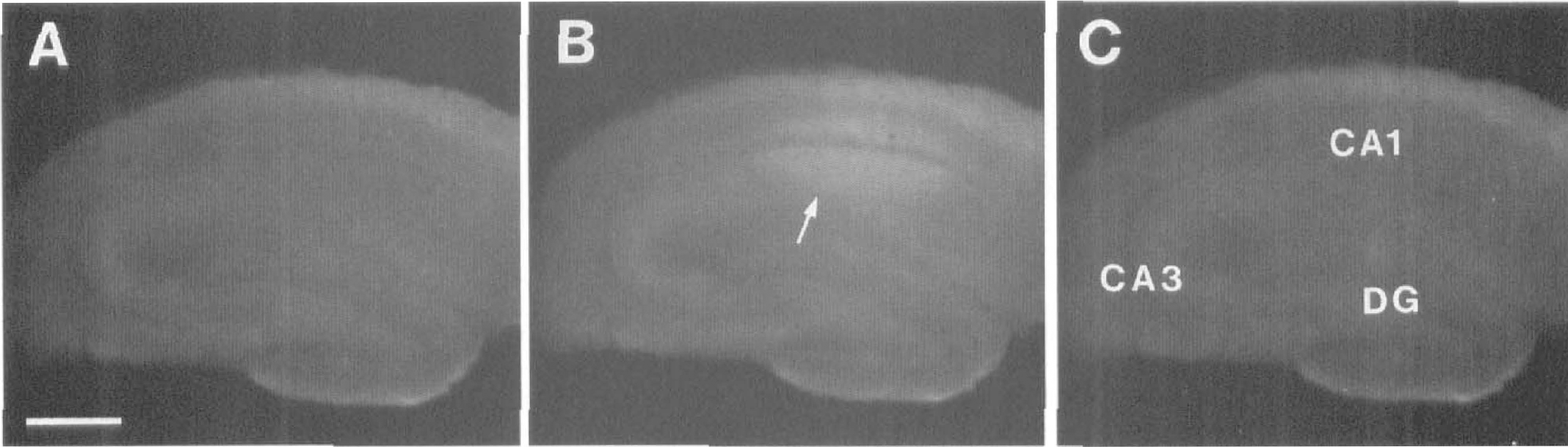
Photographs showing NMDA-induced [Ca2+]i elevation in a postischemic hippocampal slice 3 hours after 5-minute ischemia. The slice was perfused with a nominally Mg2+-free Ringer solution containing 10 μmol/L of glycine. The images were obtained immediately before the application of 12.5 μmol/L of NMDA
NMDA (12.5 μmol/L) evoked a region-specific increase in [Ca2+]i fluorescence in the CA1 of control slices in a nominally Mg2+-free Ringer solution (n = 8): The [Ca2+]i elevation in the CA1 was much larger than that in the CA3 (Figs. 9A, 10A). The mean peak ratio of [Ca2+]i elevation evoked by 12.5 μmol/L of NMDA was 1.16 ± 0.02 in the CA1 and 1.06 ± 0.02 in the CA3. Small [Ca2+]i elevation was also observed in the dentate gyrus; however, it was not analyzed in the present study. In postischemic hippocampal slices at 3 hours after 5-minute ischemia, NMDA-induced [Ca2+]i elevation was significantly enhanced in the CA1 but not in the CA3 (n = 8) (Figs. 8, 9B, 10A). The mean peak ratio of [Ca2+]i elevation evoked by 12.5 μmol/L of NMDA was 1.33 ± 0.04 in the CA1 (P < .01, Dunnett's multiple-comparison procedure) and 1.06 ± 0.03 in the CA3. The significant enhancement of NMDA-induced [Ca2+]i elevation in the CA1 was not observed in postischemic hippocampal slices at 24 hours after 5-minute ischemia (Fig. 9C): The mean peak ratio of [Ca2+]i elevation evoked by 12.5 μmol/L of NMDA was 1.18 ± 0.04 in the CA1 (n = 8).
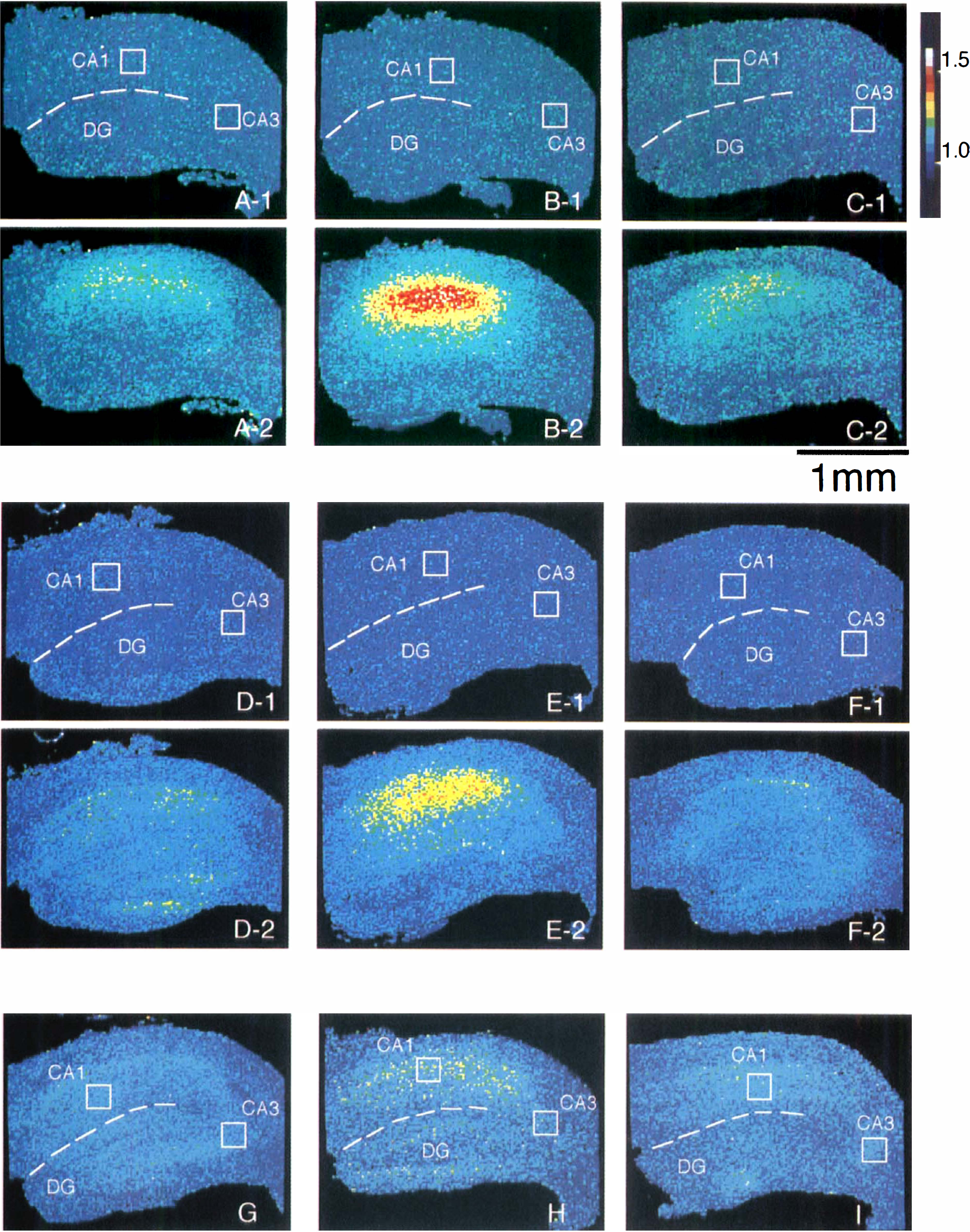
Fluorescence ratio images of [Ca2+]i elevations evoked by bath applications of NMDA, AMPA, and kainate. Pseudocolor images are shown using a [Ca2+]i image processor, in which the hues represent the fluorescence ratio (calibration is indicated by the color bar). NMDA-induced [Ca2+]i elevation obtained in a control hippocampal slice
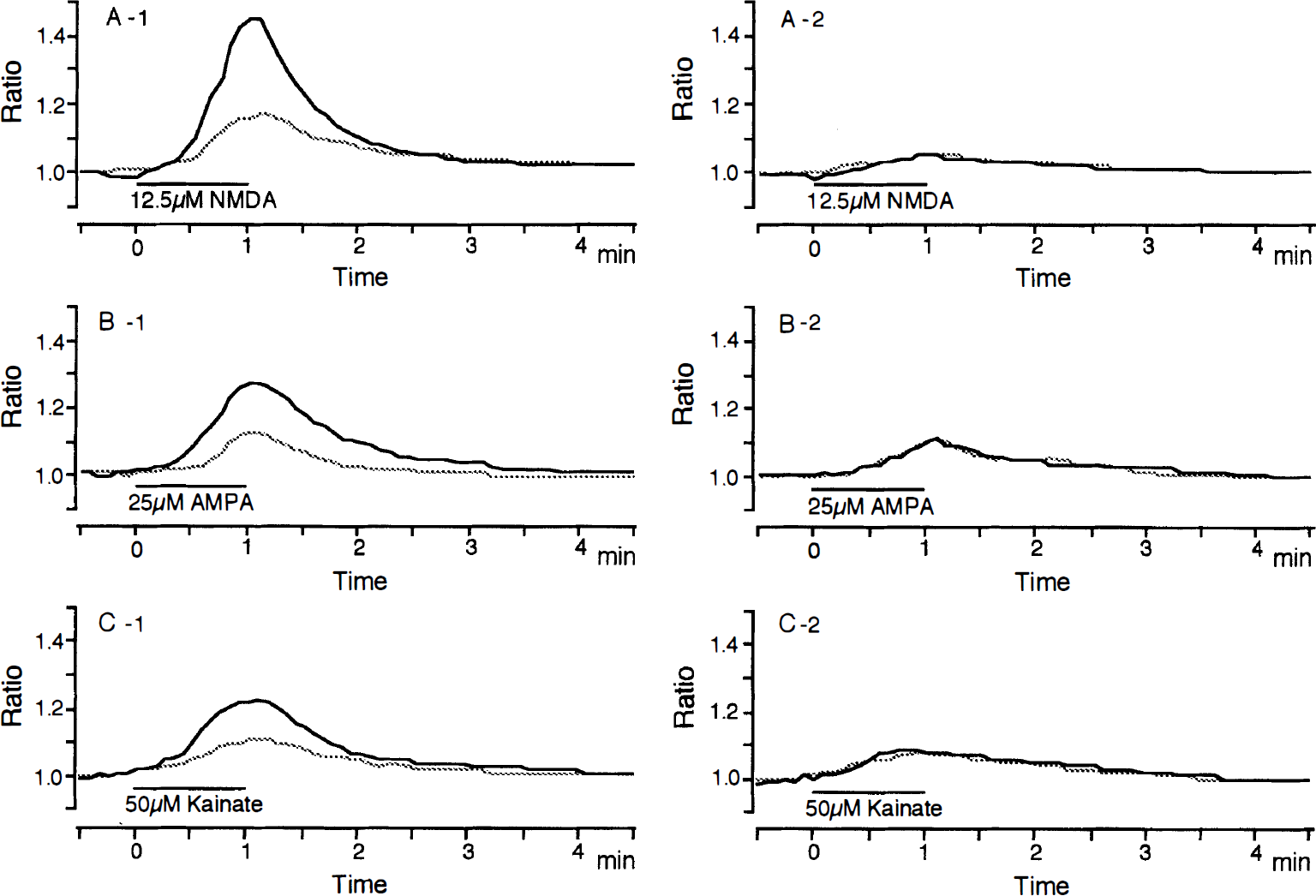
Temporal patterns of changes in [Ca2+]i elevations evoked by bath applications of 12.5 μmol/L of NMDA
The AMPA (25 μmol/L)- and kainate (50 μmol/L)- induced [Ca2+]i elevations did not show an apparent region-specific increase in control hippocampal slices (n = 8 each): The [Ca2+]i elevations showed comparatively uniform increases throughout all areas (Fig. 9D, G). The mean peak ratio of [Ca2+]i elevation evoked by 25 μmol/L of AMPA was 1.14 ± 0.03 in the CA1 and 1.13 ± 0.02 in the CA3, and the mean peak ratio of [Ca2+]i elevation evoked by 50 μmol/L of kainate was 1.11 ± 0.03 in the CA1 and 1.09 ± 0.03 in the CA3. In postischemic hippocampal slices at 3 hours after 5-minute ischemia, significant enhancements of AMPA- and kainate-induced [Ca2+]i elevations were observed in the CA1 but not in the CA3 (n = 8 each) (Fig. 9E, H). The mean peak ratio of [Ca2+]i elevation evoked by 25 μmol/L AMPA at 3 hours after 5-minute ischemia was 1.28 ± 0.06 in the CA1 (P < .01) and 1.12 ± 0.04 in the CA3. The mean peak ratio of [Ca2+]i elevation evoked by 50 μmol/L of kainate at 3 hours after 5-minute ischemia was 1.16 ± 0.04 in the CA1 (P < .05) and 1.09 ± 0.05 in the CA3. The significant enhancement of AMPA- and kainate-induced [Ca2+]i elevations in the CA1 was not observed in postischemic hippocampal slices at 24 hours after 5-minute ischemia (Fig. 9F, I). The mean peak ratios of [Ca2+]i elevation in the CA1 evoked by 25 μmol/L of AMPA and 50 μmol/L of kainate were 1.18 ± 0.04 and 1.13 ± 0.03, respectively.
DISCUSSION
The present study showed that both NMDA and non-NMDA (AMPA and kainate) receptor-mediated currents were persistently enhanced in CA1 pyramidal neurons 1 to 12 hours after transient ischemia and also that the enhancement of currents was accompanied by the enhancement of [Ca2+]i elevation. As shown in the present [Ca2+]i imaging study, transient ischemia did not alter the responses of the CA3 to glutamate agonists. Thus, the enhancement of glutamate agonists-induced responses may be specific to regions undergoing ischemic damage. The activation of non-NMDA receptors will induce influx of Na+ and depolarization of membrane potentials, and then will induce [Ca2+]i elevation which is secondarily caused by the opening of voltage-gated Ca2+ channels after the membrane depolarization. The depolarization of CA1 pyramidal neurons will also induce activation of NMDA receptors by the reduction of Mg2+ block of NMDA receptor channels (Nowak et al., 1984; Mayer et al., 1984). The activated NMDA receptors will enhance Ca2+ and also Na+ influx through NMDA-receptor channels. Thus, the enhancement of [Ca2+]i and [Na+]i elevations will be induced during 1 to 12 hours after 5-minute ischemia when the postischemic CA1 pyramidal neurons respond to glutamate released as a transmitter, as suggested in the experiment 2. The enhancement of intracellular elevation of these cations will continually activate Ca2+ and Na+ pumps at the expense of metabolic energy during 1 to 12 hours after 5-minute ischemia, and will waste neuronal energy that is needed for repair from ischemic damage. These intracellular processes may lead postischemic CA1 pyramidal neurons to irreversible neuronal injury.
The enhancement of glutamate receptor-mediated responses was observed 1 to 12 hours after 5-minute ischemia. Li and Buchan (1993) have observed that delaying administration of a non-NMDA receptor antagonist NBQX 12 hours after transient ischemia results in dramatic CA1 neuroprotection, but delaying treatment to 24 hours postischemia results in the loss of the neuroprotective effect, and then they have estimated that the critical interval for reversibility of neuronal death is between 12 and 24 hours. This estimation suggests that the enhancement of glutamate receptor-mediated responses during 1 to 12 hours after 5-minute ischemia observed in the present study is the response of CA1 pyramidal neurons at a reversible stage. It has not been reported that delayed treatment to 24 hours or more after transient ischemia can remarkably protect CA1 neuronal degeneration. This implies that the responses of glutamate receptors recorded at 24 hours or more after transient ischemia may be those of CA1 pyramidal neurons at an irreversible stage. Hori and Carpenter (1994) have observed that the Mg2+ block of the NMDA response is reduced in CA1 pyramidal neurons 24 to 48 hours after transient ischemia. In the present study, a few of CA1 pyramidal neurons at 24 hours after 5-minute ischemia showed small but apparent NMDA-induced currents at −80 mV even in the presence of 1 mmol/L Mg2+. These results may indicate that the normal voltage-dependent blockade of the NMDA channels by Mg2+ is maintained when the condition leading to CA1 neuronal death is reversible and it is reduced when the condition leading to CA1 neuronal death is irreversible.
The present study also showed that enhancement of NMDA and non-NMDA (AMPA and kainate) receptor-mediated responses was induced by the direct application of glutamate agonists. It indicates that the site responsible for generating the enhancement is, at least in part, in postsynaptic structures, i.e., NMDA and non-NMDA receptors. The postischemic intracellular processes producing the activation of NMDA and non-NMDA receptors are unknown; however, the functional change of the CA1 pyramidal neurons may be induced by an excessive increase in [Ca2+]i during transient ischemia. It has been observed that an excessive increase in [Ca2+]i is induced in the CA1 but not in the CA3 during in vitro ischemic condition (Mitani et al., 1990a, 1993, 1994b). The increase in [Ca2+]i will activate various intracellular calcium-dependent enzymes including calcium-dependent protein kainase C (PKC) and calcium/calmodulindependent protein kainase II (CaM-KII). The activation of PKC and CaM-KII can produce persistent enhancement of NMDA and non-NMDA receptor-induced responses (Urushihara et al., 1992; Kelso et al., 1992; McGlade-McCulloh et al., 1993), and several in vivo studies have revealed that PKC and CaM-KII are activated in postischemic brain (Picone et al., 1989; Cardell et al., 1990; Wieloch et al., 1991). These reports present the possibility that the excessive increase in [Ca2+]i during 5-minute ischemia activates PKC and CaM-KII in the CA1 pyramidal neurons, and then the activated enzymes produce persistent enhancement of NMDA and non-NMDA receptor-induced responses during the postischemic period.
Footnotes
Acknowledgements
The authors thank Dr. T. Takahashi, Institute for Brain Research, University of Tokyo for advice and Mr. T. Yagi and Mr. K. Ohno for their assistance.