
Abstract
Excitatory stimulation in hippocampal slices results in biphasic NAD(P)H fluorescence transients. Previous studies using differing stimulus protocols agreed that the oxidation phase is a consequence of mitochondrial metabolism, but the reduction phase has been attributed to (1) mitochondrial nicotinamide adenine dinucleotide (NADH) generation or (2) astrocytic glycolysis triggered by glutamate uptake. In an attempt to reconcile these two views, the present study examined NAD(P)H signals evoked by a wide range of stimulus durations (40 ms to 20secs). A combination of ionotropic glutamate receptor (iGluR) antagonists (6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), 2-amino-5-phosphonopentanoic acid (APV)) virtually abolished responses to brief stimuli (40 to 200 ms, 50 Hz), but a significant fraction of the signal elicited by extended stimulation (20 secs, 32 Hz) was resistant to CNQX/APV. Glycolysis was inhibited by removal of glucose and addition of 2-deoxyglucose (2DG) (10 mmol/l) or iodoacetic acid (IAA, 1 mmol/l). Pyruvate was provided as an alternative substrate for oxidative phosphorylation and the A1 receptor antagonist 1,3-Dipropyl-8-cyclopentylxanthine (DPCPX) included to prevent decreases in synaptic efficacy. If sufficient pyruvate was supplied, responses to brief and extended stimuli were unaffected by glycolytic inhibition and not significantly reduced by an inhibitor of glucose uptake (3-O-methyl glucose, 3 mmol/l). When timed to arrive at the peak of overshoots generated by extended synaptic stimulation, brief pyruvate applications (10 mmol/l, 2mins) had little effect on evoked NAD(P)H increases. Flavoprotein autofluorescence transients after extended stimuli matched (with inverted sign) NAD(P)H responses. Responses to extended stimuli were not reduced by a nonselective inhibitor of glutamate uptake DL-Threo-β-benzyloxyaspartic acid (TBOA). These results suggest that NAD(P)H transients report mitochondrial dynamics, rather than recruitment of glycolytic metabolism, over a wide range of stimulus intensities.
Introduction
The nicotinamide adenine dinucleotide molecules nicotinamide adenine dinucleotide (NADH) and nicotinamide adenine dinucleotide phosphate (NADPH) (together abbreviated NAD(P)H) are fluorescent in their reduced forms and excited by light in the wavelength range of 340 to 390 nm; Chance et al, 1962). The intrinsic fluorescence of these important metabolic intermediates as well as that of flavoprotein cofactors such as flavin adenosine dinucleotide (FAD) have been used for over 40 years to follow changes in the metabolic state of central nervous system tissue under a wide range of physiologic and pathologic challenges (Chance, 2004; Jobsis et al, 1971; Mayevsky et al, 1988; Schuchmann et al, 2001; Sick and Perez-Pinzon, 1999; Strong et al, 1996). In at least one early study, an evaluation of NAD(P)H changes resulting from extended electrical stimulation in brain slice tissue was made, which showed an early transient decrease in NAD(P)H fluorescence and a later and much longer, but still transient, increase (Lipton, 1973). Studies since then have confirmed the biphasic nature of the NAD(P)H signal after bursts of electrical discharge in single neurons (Duchen, 1992) and in hippocampal slices (Foster et al, 2005; Schuchmann et al, 2001; Sick and Perez-Pinzon, 1999). More recently, we have shown that quite robust (> 1%) fluorescence signals from NAD(P)H and flavoproteins can be elicited in hippocampal slices by a small number of action potentials evoked in a time window of ~ 100 ms to 1 secs (Shuttleworth et al, 2003). Owing to the signal size and the rapid onset of the initial phases of the signals, these activity-coupled fluorescence changes have considerable potential for dynamic mapping of electrical activity patterns in the cortex (see also Murakami et al, 2004; Reinert et al, 2004).
Despite the extensive history of NAD(P)H fluorescence measurements in brain preparations, the relative contributions of mitochondrial and glycolytic metabolism and the cell types that may be responsible for compound fluorescence signals in brain remain unclear. We recently evaluated mechanisms underlying NAD(P)H changes evoked by brief stimulus trains in hippocampal slices and concluded that transients (initial NAD(P)H decrease and subsequent longer-lasting increase) were because of mitochondrial metabolism that was largely confined to neurons (Shuttleworth et al, 2003). For the shortduration stimuli used in our previous study (1 to 25 pulses, 50 Hz), NAD(P)H responses were reduced by >90% in the presence of iGluR blockers (APV and CNQX), implying that electrical activity in postsynaptic neurons was in large part creating the metabolic demand underlying the signals (Shuttleworth et al, 2003). Under this stimulus protocol then, factors such as glutamate re-uptake by neurons and glia or presynaptic activity appeared minor. The time courses of the NAD(P)H and flavoprotein responses were of opposite sign and had very similar spatial and temporal characteristics (Shuttleworth et al, 2003) as would be expected if the signals were of mitochondrial origin, as initial phases may arise primarily from NADH and flavin adenine dinucleotide (reduced form; FADH2) oxidation in the respiratory transport chain, followed by regeneration in the trichloroacetic acid (TCA) cycle, where they share obligatory coupling. The activity of TCA cycle enzymes can be stimulated by Ca2+ elevations (McCormack and Denton, 1993; Rutter and Rizzuto, 2000) and mitochondrial Ca2+ increases have been implicated as key contributors to NAD(P)H transients after neuronal stimulation (Duchen, 1992; Duchen and Biscoe, 1992; Kann et al, 2003; Reinert et al, 2004; Schuchmann et al, 2001). However, a significant portion of evoked responses appear resistant to Ca2+ removal (Kann et al, 2003; Reinert et al, 2004; Shuttleworth et al, 2003), emphasizing the role that other mechanisms (including ATP depletion because of Na+ extrusion; Lewis and Schuette, 1975) may play as possible triggers for mitochondrial metabolism. The ionic mechanisms responsible for triggering mitochondrial NADH production have been discussed recently (Kann et al, 2003; Reinert et al, 2004; Shuttleworth et al, 2003).
Although recent work has generally emphasized the contribution of mitochondrial mechanisms in brain slice, earlier work concluded that glycolytic NADH production could directly contribute to transient stimulus-coupled NAD(P)H increases. High concentrations of exogenous pyruvate were found to selectively decrease stimulus-induced NAD(P)H increases in brain slice (Lipton, 1973; see also Foster et al, 2005) and this could be because of depletion of cytosolic NADH levels. A recent report of NAD(P)H fluorescence transients in acute hippocampal slices concluded that overshooting NAD(P)H increases after electrical stimulation were entirely because of glycolysis (not mitochondrial activity) that occurs in astrocytes as a consequence of glial glutamate uptake (Kasischke et al, 2004). Using the resolving power of two-photon microscopy (Zipfel et al, 2003), it was established that the delayed overshooting phase of the response arose in astrocytes and was postulated, but not directly shown, to arise from transiently increased NAD(P)H from glycolysis. The temporal relationship between signals in presumed neuronal and glial compartments was taken as novel evidence supporting an important role of a lactate shuttle between astrocytes and neurons, after synaptic activation in acute hippocampal slices (Kasischke et al, 2004; Pellerin and Magistretti, 2004).
The reasons for very different conclusions concerning the roles of mitochondrial and glycolytic metabolism in synaptically driven NAD(P)H transients are not readily apparent. One significant difference between recent studies in acute hippocampal slices was the duration of the presynaptic stimulation, as our work involved brief stimuli (200 to 500 ms; Shuttleworth et al, 2003), whereas evidence for astrocytic NAD(P)H increases was obtained after much longer stimulus trains (typically 20 secs duration; Kasischke et al, 2004). However, neither study directly evaluated the effects of inhibitors of glycolysis on these responses.
Owing to the potential utility of NAD(P)H signals in mapping neuronal activity as well as in assessing mitochondrial state in neuropathologies, we sought to clarify better their metabolic bases and whether there might be a significant shift with the degree of imposed electrical activity. We have directly evaluated the role of glycolysis as well as the contribution of glutamate receptor activation and re-uptake after a range of stimulus conditions. We find, contrary to what has been proposed, that responses elicited by a range of stimulation, including brief stimulation (50 Hz, 40 to 200 ms in bicuculline), a standard long-term potentiation (LTP) induction protocol (100 Hz, 1 sec), and extended stimulation (32 Hz, 20 secs) were all unaffected by inhibition of glycolysis. This suggests that the overshoot in NAD(P)H transients is not normally because of neuronal-glial signaling, but can be accounted for by mitochondrial metabolism. Some of the results have been presented in abstract form (Brennan et al, 2004).
Materials and methods
Slice Preparation
Male mice (C57BL/6) were obtained from Harlan Laboratories (Indianapolis, IN, USA) at 4 to 6 weeks of age and were housed in standard conditions (12 h light/dark cycle) for up to 2 weeks before euthanasia. Numbers in the study refer to the number of slices with a maximum of two slices from an individual animal used for each data set. Mice were deeply anesthetized with a mixture of ketamine and xylazine (85 and 15mg/mL, respectively, subcutaneously) and decapitated. Brains were rapidly removed and placed in ice-cold cutting solution (see below for composition). Coronal sections (350 μm) were cut on a Vibratome (Technical Products Internation, St Louis, MO, USA) and slices were subsequently transferred to oxygenated room temperature artificial cerebrospinal fluid (ACSF) (see below). Cutting and recording solutions were both 300 to 305 mOsm/L. After warming to 34°C for 1 h, the ACSF was exchanged again and slices were then held at room temperature. Individual slices were then transferred to a recording chamber and perfused with oxygenated ACSF at 2 mL/min. Recording temperature was either 25°C or 35°C, as noted for specific experiments.
NADH and Flavoprotein Fluorescence Measurements
Detection of NAD(P)H and flavoprotein autofluorescence was performed as described recently (Shuttleworth et al., 2003), with minor modifications. All imaging was performed after focusing onto the surface of the slice with an × 10 water-immersion objective (numerical aperatures 0.3 Olympus) and fluorescence collected after 2 × 2 binning of the 640 × 480 line image. An acquisition rate of 0.33 Hz was used for all experiments, except where noted in the studies shown in Figure 7. The reduced form of NADH is fluorescent (Ex ~360nm, Em ~450nm; Aubin, 1979) and the oxidized form is nonfluorescent. For NADH imaging, 360 nm excitation was delivered via a fiber optic/monochromator system (Polychrome IV; Till Photonics, Grafelfing, Germany) and reflected onto the slice surface using a dichroic mirror (DMLP 400 nm, Chroma Technology, Brattleboro, VT, USA). Fluorescence emission (> 410 nm) was collected with a cooled interline transfer CCD camera (IMAGO; Till Photonics). Image data were background-subtracted to account for camera noise and presented as the changes in fluorescence intensity/prestimulus fluorescence intensity (ΔF/F0) from stratum radiatum. ΔF measurements were based on total tissue fluorescence. The contribution of NAD(P)H fluorescence to total tissue fluorescence, as assessed by total measured decreases after prolonged carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) exposure (5 μmol/L, 40mins), was approximately 50%. Thus, with the assumption that FCCP does not directly influence fluorescence levels unrelated to NAD(P)H, reported NAD(P)H changes are expected to represent B50% of actual changes.
In some experiments, flavoprotein autofluorescence was monitored using excitation at 488 nm and emission detected using a 535 nm (50 nm BW) interference filter. Photobleaching was assessed for both NAD(P)H and flavoprotein measurements. Photobleaching was minimal for NAD(P)H measurements when measured using identical exposure times as used for assessment of responses to brief stimulus trains (0.39% ± 0.08% decrease over 50 secs, n = 6) or extended stimulus trains (0.51% ± 0.02% over 100 secs, n = 6). For flavoprotein measurements, photobleaching was more significant and nonlinear (4.93% ± 1.43% decrease over 50secs; 6.47% ± 0.90% over 100secs, n = 6 each). Therefore, equivalent nonstimulus ‘bleach’ trials were conducted in each slice and used to correct evoked flavoprotein responses in each preparation by subtracting these values from the raw data. Differences between groups were determined using either Student's t-tests for single time point or single-drug application comparisons, or one-way analysis of variance (ANOVA) for multiple comparisons. Bonferroni's multiple-comparison test was used for post hoc analysis, where the effects of multiple drug treatments were compared against each other. Dunnett's multiple-comparison test was used for post hoc comparisons of multiple time points of single-drug treatment, when compared with responses immediately before drug treatment. P < 0.05 was considered significant in all cases. Response amplitudes varied between preparations and therefore for all tests of drug effects, comparisons were made between control and test responses determined within the same slices.
Synaptic Stimulation and Recording of Postsynaptic Potentials
Schaffer collateral inputs to the CA1 region were stimulated using a bipolar electrode (25 μm tip) placed on the surface of stratum radiatum and connected to a constantcurrent stimulus isolation unit (Isoflex, AMPI, Israel). Brief stimulation consisted of 2 to 10 pulses delivered at 50 Hz in the presence of bicuculline (Shuttleworth et al, 2003). Intermediate stimulation was 100 pulses at 100 Hz. Extended stimulation was 640 pulses delivered at 32 Hz (Kasischke et al, 2004). Bicuculline was not used in experiments testing longer-duration stimuli (1 to 20 secs, 50 to 100 Hz), as these stimuli often triggered spreading depression-type events in disinhibited slices and aminobutyric acid (GABA)A antagonists were not used in the recent work of Kasischke et al (2004). Imaging was begun 6.67 secs before the onset of the stimulus train and continued for a total of 33 or 66 secs . Throughout all experiments, 4-min intervals were maintained between successive brief stimulation trials (each up to 200 ms duration) and 10-min intervals were maintained between successive intermediate or extended stimulation trials (each of 1 to 20 secs duration). Before beginning any pharmacological manipulations, at least three control trials were performed in each slice to ensure that baseline-evoked responses were stable. Extracellular field recordings of excitatory postsynaptic potentials (fEPSPs) were made using glass microelectrodes (4 to 6 MΩ) filled with ACSF. Electrodes were placed in the stratum radiatum of CA1 approximately 150 μm from the pyramidal cell body layer. Signals were amplified (Neurodata IR-283 (Neurodata IR-283, Neurodata Instruments Corp. New York, NY)), digitized (Digidata 1322A; Axon Instruments, Union City, CA, USA), and then acquired using Axoscope software (v 8.1; Axon Instruments).
Solutions
ACSF contained (in mmol/l): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2, and 10 glucose, equilibrated with 95% O2/5% CO2. Cutting solution contained (in mmol/l): 3 KCl, 1.25 NaH2PO4, 6 MgSO4, 26 NaHCO3, 0.2 CaCl2, 10 glucose, 220 sucrose, and 0.43 ketamine. Except where noted, all drugs and salts were obtained from Sigma Chemical Co. (St Louis, MO, USA). TBOA was from Tocris Cookson (Ellisville, MO, USA). In some experiments, extracellular Ca2+ was removed by perfusion with modified ACSF lacking added CaCl2 and supplemented with 0.5 mmol/l EGTA. In all studies involving pharmacological inhibition of glycolysis, slices were pre-exposed to pyruvate (1 to 10 mmol/l, as described below) to prevent rundown of mitochondrial responses simply as a downstream consequence of glycolytic inhibition. Glycolytic inhibitor concentrations were chosen based on previous work in hippocampal preparations (Guo et al, 2001; Izumi et al, 2003; Newman et al, 1990; Zhao et al, 1997). In studies using radiolabeled 2DG in hippocampal slices, approximately 5 mins exposure has been shown to be sufficient for equilibration throughout 500-μm-thick slices (see Newman et al, 1990) and in the present study exposures of at least 17.5 mins were tested with 350 mm slices.
Results
Comparison of NADH Transients Evoked by Brief Versus Extended Stimuli
Figure 1 compares NADH transients evoked by brief and extended synaptic stimulation of Schaffer collateral inputs to the CA1 region. The recording conditions for brief stimuli (Figure 1A; 200 ms, 50 Hz, in the presence of the GABAA receptor antagonist bicuculline 30 μmol/L) were the same as used for our previous studies. Disinhibition was employed with this brief protocol to increase the size of the signal so that changes, or lack thereof, could be more accurately tracked (Shuttleworth et al, 2003). The extended stimuli (20 secs, 32 Hz, no GABAA block) were matched to those recently reported by Kasischke et al (2004). With both types of stimuli, there was an initial decrease in NAD(P)H fluorescence, followed by longer-lasting overshoot. The initial NAD(P)H decrease peaked within ~1 sec after termination of brief stimuli. With extended stimuli, the initial fluorescence decrease still peaked within ~1.5secs and then progressively reversed before termination of the stimulus train (Figure 1B). Except for these obvious time-course differences, the responses were very similar in appearance (compare Figures 1A and 1B, left panels).
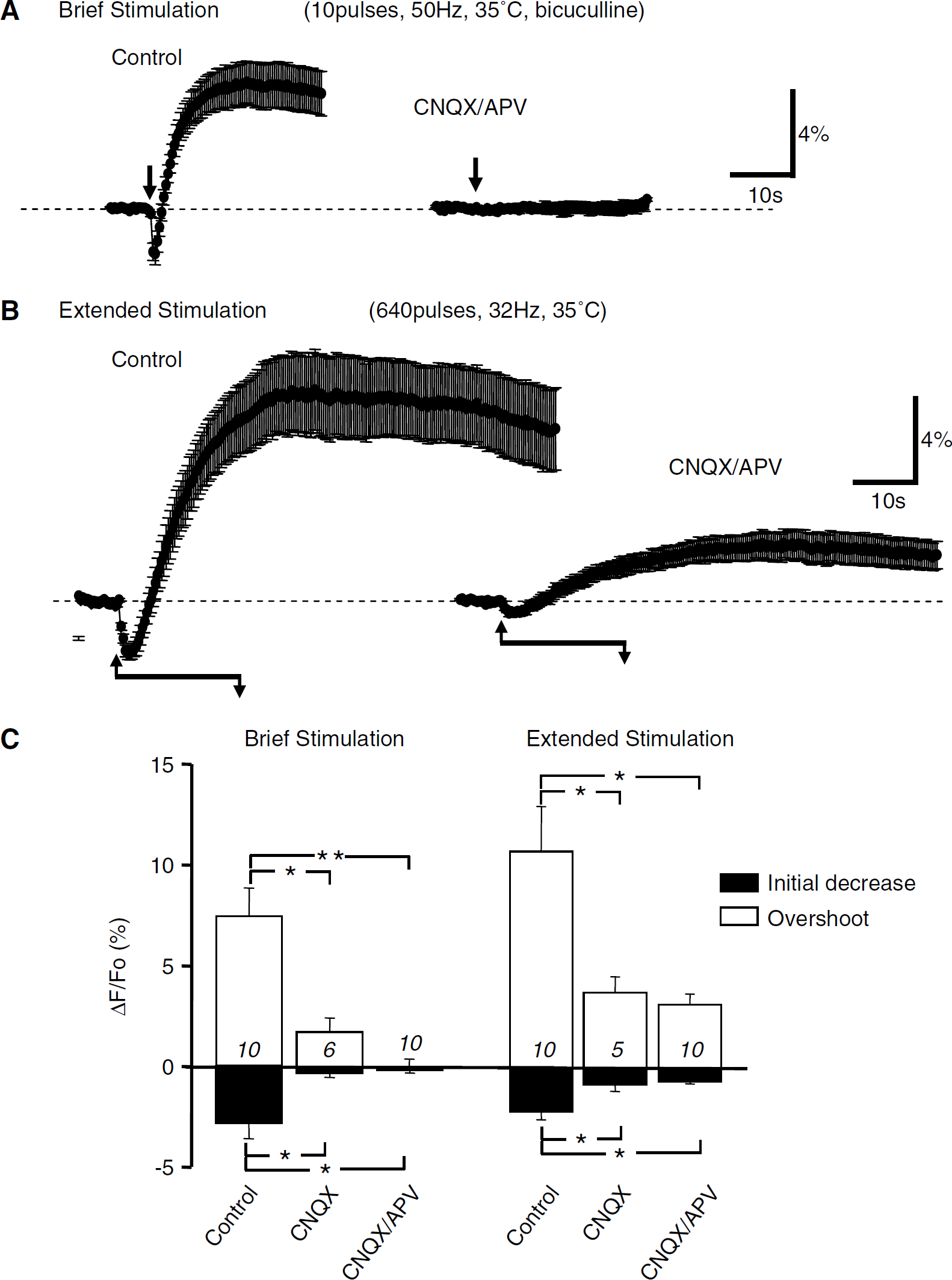
Comparison of NAD(P)H fluorescence transients evoked by either brief or extended periods of repetitive synaptic activity. (
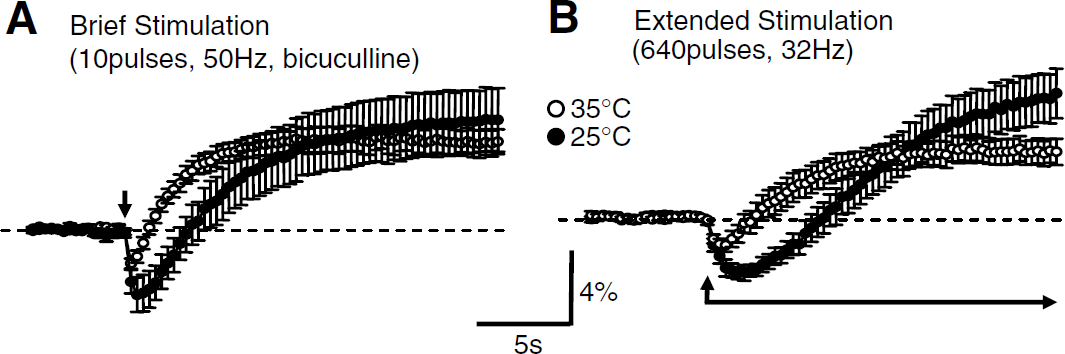
Temperature reductions increased the initial component of NAD(P)H responses. Responses to brief stimulation (
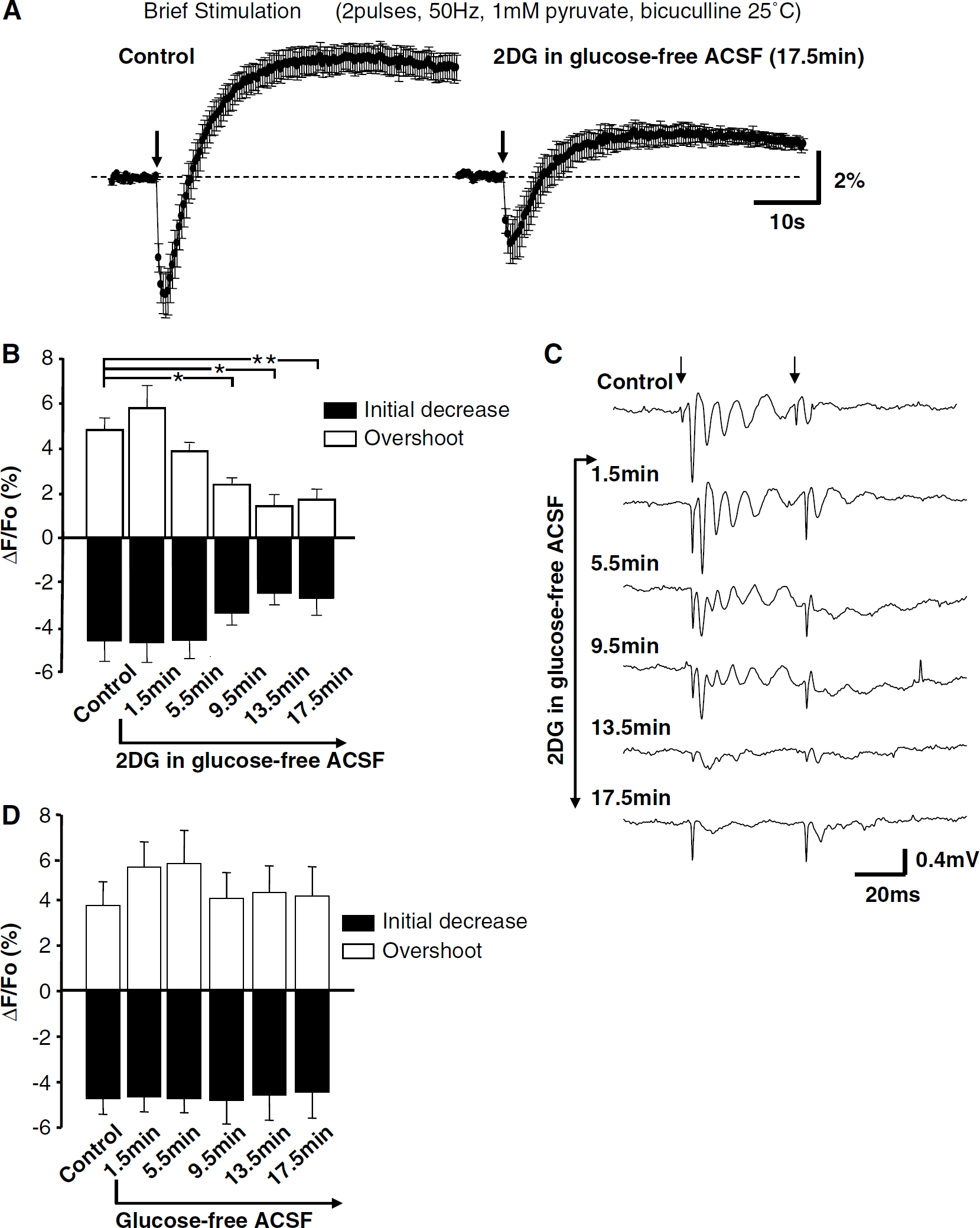
Exposure to 2-deoxyglucose (2DG) decreased synaptic efficacy and reduced NAD(P)H transients evoked by brief stimulation. (
It was previously concluded that NAD(P)H transients evoked by brief stimulus trains were predominantly because of postsynaptic activation (rather than perisynaptic glutamate uptake), as both components of responses were abolished by a combination of glutamate receptor antagonists CNQX and APV (Figure 1A, > 98% inhibition; see also Shuttleworth et al, 2003). Figure 1C (left panel) shows that both antagonists were required to block responses to brief stimulation, as CNQX alone reduced NAD(P)H overshoot responses by ~ 75% and together CNQX/APV abolished responses. Figure 1C (right panel) confirms that both phases of NAD(P)H transients evoked by extended stimulus trains were reduced by CNQX (consistent with Kasischke et al, 2004), and also shows that addition of APV did not result in further decreases.
Responses shown in Figure 1 were studied at 35 °C. Figure 2 shows that, at a lower recording temperature, the initial oxidation components of NAD(P)H responses were enhanced and overshoot components were delayed. The effects of iGluR blockers described above were not quantitatively changed by temperature reduction, as responses to brief stimulus trains were still abolished by CNQX/APV (data not shown) and after extended stimuli (20 secs, 32 Hz) a significant NAD(P)H overshoot was still observed in CNQX/APV at 25°C (see Figure 5 below).
Effects of Glycolytic Inhibition on Responses to Brief Stimulus Trains
To test the involvement of glycolysis in responses to brief stimulus trains, ACSF was modified by simultaneous removal of glucose with the addition of either 2DG (10 mmol/l) or IAA (1 mmol/l). Pyruvate (1 mmol/l) was supplied as an alternative substrate for mitochondrial metabolism. In preliminary studies, it was found that, under these conditions, synaptic stimulation at 35°C often triggered a propagating wave of NAD(P)H fluorescence increase with properties similar to that described for spreading depression (Foster et al, 2005; Obeidat et al, 2000; Rex et al, 1999). As this effect was not observed when the recording temperature was reduced to 25°C, studies using glycolytic inhibitors were performed at 25°C.
Figures 3A-3C summarize the effects of 2DG (with concomitant glucose removal) when tested on responses to brief stimulation (two pulses, 50 Hz, 1 mmol/l pyruvate, bicuculline). 2DG produced a rapid decrease in the amplitude of EPSPs (Figure 3C; see Bachelard et al (1984)). As would be expected from the loss of electrical activity, there was a concomitant reduction in synaptically evoked NAD(P)H transients (Figures 3A and 3B). The alternative inhibitor IAA also decreased synaptic potentials and, by 9.5 mins IAA exposure, NAD(P)H overshoots were significantly reduced (7.08% ± 1.48% versus 1.48% ± 1.02%, control and IAA, respectively; P < 0.001, ANOVA with Dunnett's multiple comparison test, n = 6). Figure 3D shows that removal of glucose alone (without the addition of 2DG or IAA) had no significant effect on NAD(P)H transients, implying that coadministration of glycolytic inhibitors was required for effects illustrated in Figures 3A and 3B.
Depression of synaptic transmission during glycolytic inhibition is thought to be mediated by adenosine receptor activation (Zhao et al, 1997). Figure 4 shows that the decrease in synaptic efficacy and reduction in NAD(P)H transients seen after 2DG exposure (illustrated in Figure 3) was prevented by the adenosine A1 receptor antagonist DPCPX (100 nmol/L). DPCPX also prevented depression of NAD(P)H transients produced by IAA exposure (overshoot amplitude 5.20% ± 1.35% versus 5.53% ± 0.73%, control and IAA + DPCPX, respectively, 20 mins IAA exposure, P = 0.84, ANOVA, Dunnett's multiple-comparison test, n = 6). Exogenous application of an A1 receptor agonist (CPA, 30 nmol/l) closely mimicked the actions of 2DG and IAA exposure. CPA reduced field EPSP amplitude and concomitantly reduced the amplitude of synaptically evoked NAD(P)H transients (n = 6, data not shown).
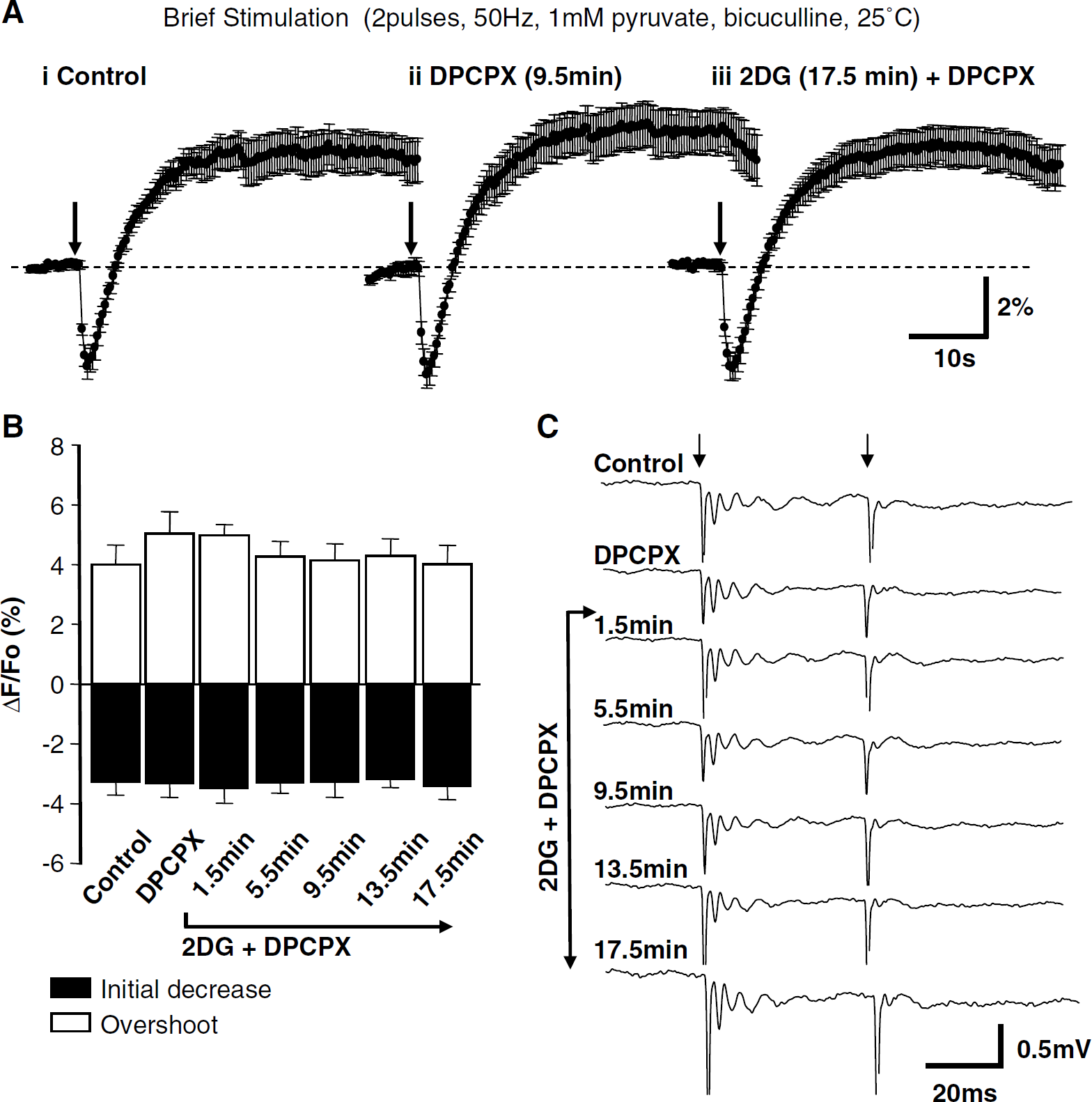
Responses to brief stimulation were not influenced by 2-2-deoxyglucose (2DG) when adenosine A1 receptors were blocked. (
We addressed the possibility that DPCPX could alone increase the amplitude of NADH transients and thereby mask a decrease in event amplitude that may be because of glycolytic inhibition. Exposure to DPCPX (100 nmol/L, without glycolytic inhibitors or glucose removal) resulted in no change in the amplitude of transients evoked by brief stimulation (two pulses, 50 Hz, bicuculline, 1 mmol/l pyruvate, 25°C). DPCPX did not influence initial NAD(P)H fluorescence decreases (P = 0.92) or overshoots (P = 0.91) at any time point when tested at 4-min intervals throughout 21.5 mins exposure to DPCPX (ANOVA with Dunnett's multiple-comparison test, n = 7).
In an additional set of studies, the effects of IAA were examined under identical conditions, but without the simultaneous removal of glucose. There was no significant effect of IAA on NAD(P)H overshoot amplitudes evoked by brief stimulation (25°C, DPCPX, bicuculline, 10 mmol/l glucose, and 1 mmol/l pyruvate throughout). Responses were 5.12% ± 1.45% under control conditions and 6.08% ± 2.67% in IAA (P = 0.50, n = 5).
Effects of Inhibition of Glycolysis on Responses to Extended Stimulus Trains
Like the experiments described above, the effects of glycolytic inhibitors on responses to extended stimulus trials (Figure 5) were tested at 25°C. DPCPX (100 nmol/L) was included to maintain synaptic efficacy and ACSF was supplemented with pyruvate (1 or 10 mmol/l, see below) to provide substrate for mitochondrial metabolism. Exposure to DPCPX alone (100 nmol/L, without glycolytic inhibitors or glucose removal) did not influence NAD(P)H transients evoked by extended stimulation (640 pulses, 32 Hz, 25°C, overshoot amplitudes were 7.28% ± 1.78% versus 8.51% ± 1.70%, control and DPCPX, respectively, P = 0.36, n = 4). CNQX and APV were also included in this set of experiments before the addition of glycolytic inhibitors. The iGluR antagonists reduced the incidence of spreading depression-like events with extended stimulation in the presence of glycolytic inhibitors. CNQX/APV-resistant ‘residual’ NAD(P)H overshoots triggered by extended stimulus trials are responses recently proposed to be because of glycolysis triggered by glial glutamate uptake (see Introduction).
When slices were supplemented with 1 mmol/l pyruvate, CNQX/APV produced significant decreases in both the initial NAD(P)H decrease and NAD(P)H overshoot (Figure 5Ai). It can be seen that after exposure to 2DG (with simultaneous glucose removal, in the continued presence of CNQX/APV) overshoot responses appeared smaller, but after correction for multiple comparisons was made, there was no significant effect of 2DG when compared with effects of CNQX/APV alone. Pre-exposure to a higher pyruvate concentration (10 mmol/l) alone increased prestimulus NAD(P)H fluorescence levels and decreased the amplitude of NAD(P)H overshoots; these effects of pyruvate are considered below (section ‘Use of elevated pyruvate concentrations to test the contribution of cytosolic NADH production'). Figures 5Aii and 5Aiii show effects of 2DG applied in the presence of 10 mmol/l pyruvate. When slices were pre-exposed to 10 mmol/l pyruvate, a significant effect of CNQX/APV was observed on the initial NAD(P)H decrease component, but not on overshoot responses. It can be seen that subsequent exposure to 2DG had no effect on overshoot amplitudes and that there was no significant effect of 2DG on the amplitude of initial NAD(P)H decreases.
IAA (1 mmol/l, with simultaneous glucose removal, 1 mmol/l pyruvate pre-exposure) did not significantly decrease the peak amplitude of CNQX/ APV-resistant NADH overshoots (Figure 5B). There was some trend toward a decrease in overshoot amplitude towards the end of the trial, but the mean Glucose-free ACSF amplitude measured at the termination of the trail (56 secs after the onset of stimulation) was not significantly different from responses in CNQX/APV (P = 0.21, n = 6, Student's t-test). Initial NAD(P)H decreases were significantly reduced by CNQX/APV and appeared further decreased after IAA in the continued presence of CNQX/APV (Figure 5Bi), but, after correction for multiple comparisons was made, there was no significant effect of IAA on initial NAD(P)H decreases when compared with effects of CNQX/APV alone.
In an additional set of studies, the effects of IAA were examined under identical conditions, but without the simultaneous removal of glucose. There was no significant effect of IAA on NAD(P)H overshoot amplitudes evoked by extended stimulation (640 pulses, 32 Hz, 25°C, DPCPX, 10 mmol/l glucose, and 1 mmol/l pyruvate throughout). Responses were 2.97% ± 1.0% in CNQX/APV and 3.08% ± 0.95% in IAA (in the continued presence of CNQX/APV, P = 0.87, n = 4).
Inhibition of glucose transport with the use of 3-O-methoxy glucose (3MG, 3 mmol/l, in the presence of 0 glucose ACSF, with 1 mmol/l pyruvate) resulted in no significant inhibition of NAD(P)H overshoot amplitudes. NAD(P)H overshoot amplitudes were 2.27% ± 0.24% and 1.62% ± 0.38%, control and 20 mins 3MG, respectively (P = 0.21, n = 7). A representative example is shown in Figure 5C, and in this case, 3MG exposure was extended to 60 mins without apparent decline in the NAD(P)H overshoot.
Effects of Glycolytic Inhibitors on Responses to an Intermediate Stimulus
Owing to its widespread use in hippocampal neurophysiologic studies, we also tested the effects of glycolytic inhibition on NAD(P)H transients produced by a standard LTP induction protocol, 1-sec stimulation at 100 Hz (1 mmol/l pyruvate, DPCPX). Bicuculline was not included in these experiments and we found that tests of glycolytic inhibition could be performed with this stimulus at 35°C, when CNQX and APV were present. Figure 6Ai illustrates the signal generated in control solution. The addition of CNQX/APV reduced the response to the same presynaptic stimulation, but there was still a significant remainder (Figure 6Aii). Subsequent addition of 2DG (in the presence of 1 mmol/l pyruvate, in the continued presence of CNQX/APV) did not reduce the NAD(P)H fluorescence overshoot (Figure 6Aiii).
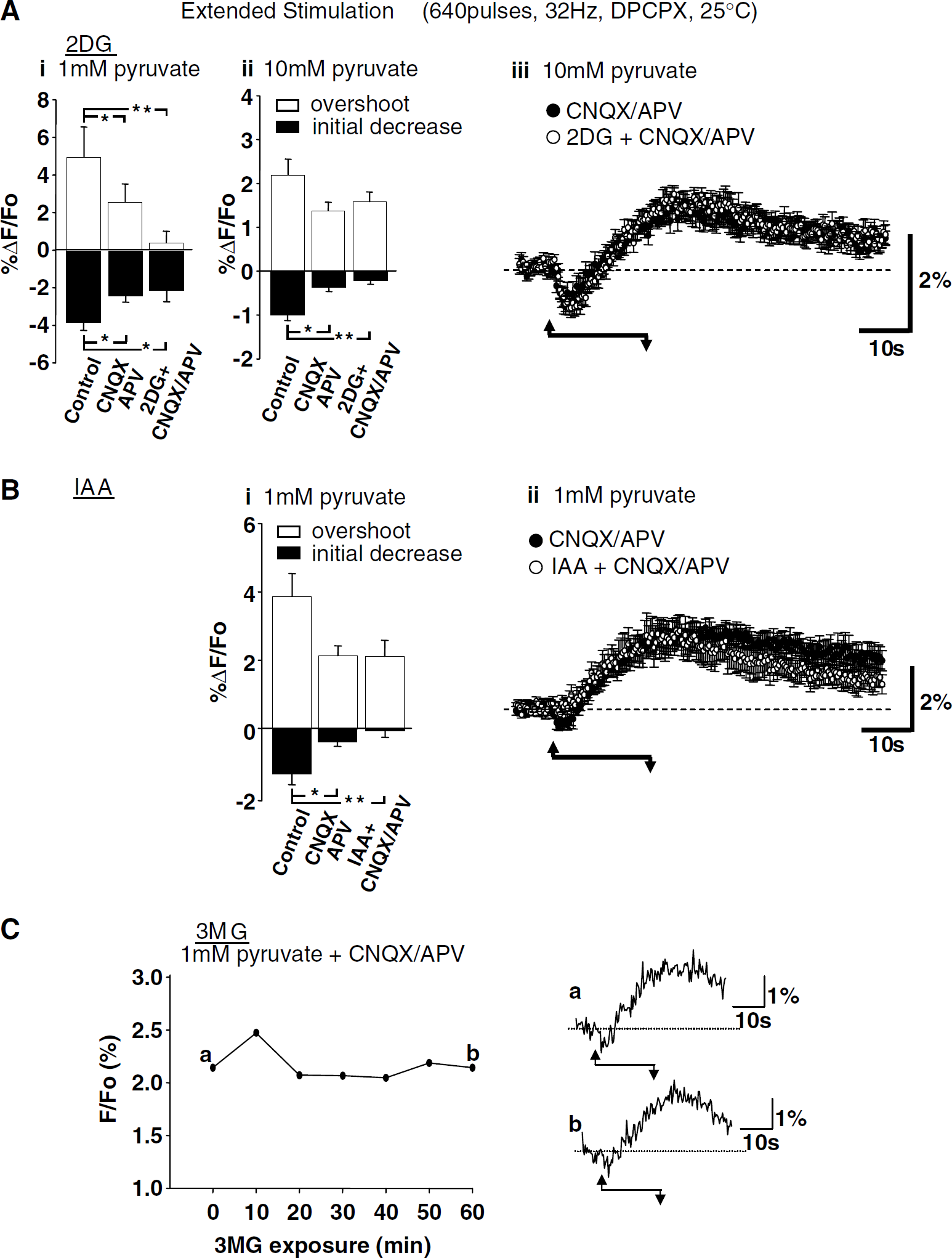
Effects of glycolytic inhibition on responses to extended stimulation (640 pulses, 32 Hz, DPCPX, 25°C). (A) Effects of 2-deoxyglucose (2DG). (i) Summary of effects in slices pre-exposed to 1 mmol/l pyruvate (mean ± s.e.m., n = 6). Control responses were reduced by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX)/2-amino-5-phosphonopentanoic acid (APV). Subsequent exposure to 2DG (in the continued presence of CNQX/APV) did not significantly reduce responses compared with CNQX/APV alone. * P < 0.05; **P < 0.01; analysis of variance (ANOVA) with Bonferroni's multiple comparison test. (ii) Summary of effects in slices pre-exposed to 10 mmol/l pyruvate (mean ± s.e.m., n = 6). Stimulation conditions were otherwise identical as those in (i). Initial NAD(P)H components were significantly decreased by CNQX/APV. Subsequent exposure to 2DG (in the continued presence of CNQX/APV) did not significantly reduce responses compared with CNQX/APV alone. *P < 0.05; **P < 0.01; ANOVA with Bonferroni's multiple comparison test. (iii) Time course of responses in CNQX/APV (closed circles) and after subsequent addition of 2DG (open circles). Data are from the same experiments summarized in (ii). (
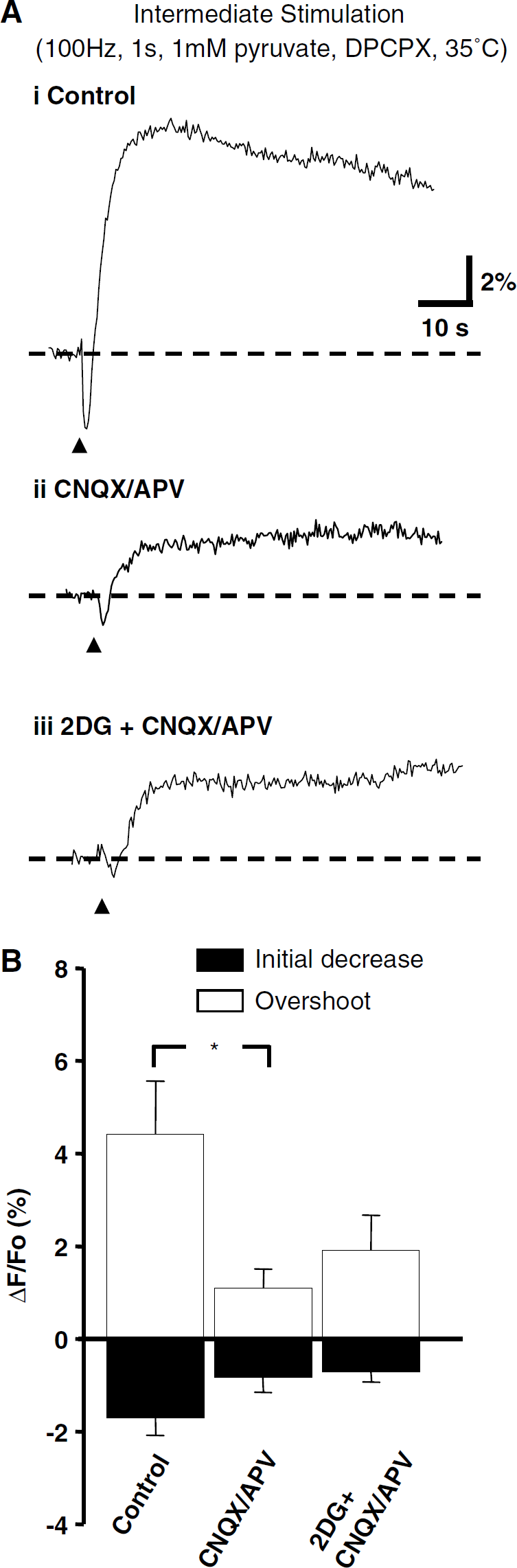
Lack of effect of glycolytic inhibition on NAD(P)H transients evoked by intermediate stimulation. (
Use of Elevated Pyruvate Concentrations to Test the Contribution of Cytosolic NADH Production
Exogenous pyruvate alone can lead to stimulation of cytosolic lactate dehydrogenase activity, resulting in consumption of cytosolic NADH (Scholz et al, 1969). High pyruvate concentrations greatly reduced NAD(P)H overshoots produced by 5 mins continuous electrical stimulation in brain slices (Lipton, 1973). We tested the hypothesis that if glycolysis is responsible for NAD(P)H overshoots, then brief pyruvate exposures should cause a larger NAD(P)H fluorescence decrease when applied at the peak of overshoots when compared with the effects of pyruvate exposures applied during ‘resting’ conditions. As this approach decreases cytosolic NADH levels without directly blocking glycolysis, these experiments could be performed at 35°C without the addition of CNQX/APV or DPCPX.
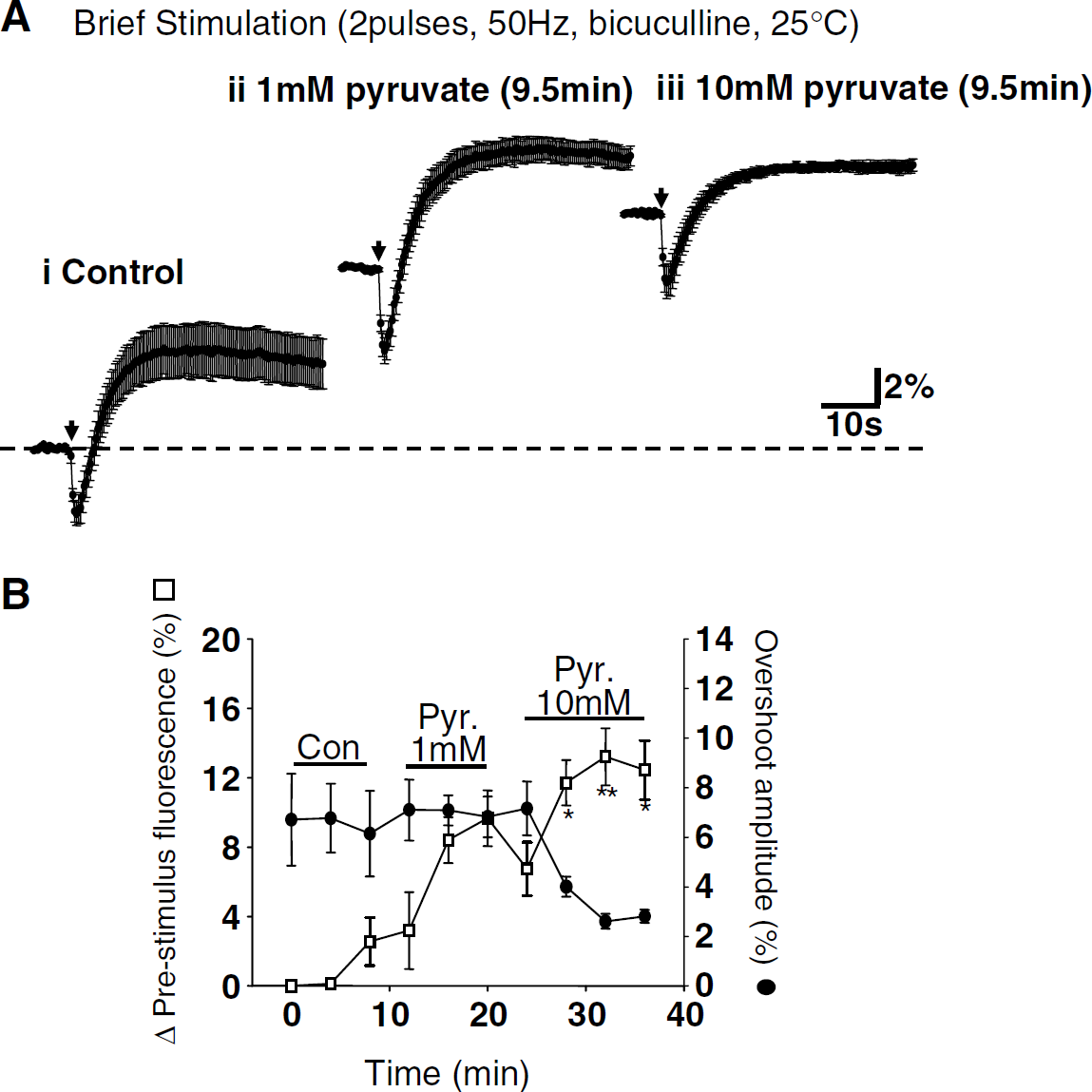
Responses to pyruvate alone (in the absence of stimulation) were established with brief applications (10 mmol/l, 2 mins). This procedure generated reproducible decreases in NAD(P)H resting fluorescence in 6/6 preparations, followed by a return to resting levels and a subsequent fluorescence increase (Figure 7). To ensure that decreases in resting fluorescence resulting from pyruvate applications were reproducible, we compared the mean decrease in three consecutive pyruvate trials, which were interleaved with three control synaptic stimulation trials. Intervals of 10mins were maintained between all synaptic stimulation and/or pyruvate application trials. There was no significant difference in the amplitude of the decrease of NAD(P)H resting fluorescence after application of 10 mmol/l pyruvate (first trial 3.27% ± 0.35%, second trial 3.77% ± 0.53%, third trial 3.83% ± 0.64%; ANOVA, P = 0.58, n = 6). Pyruvate (10 mmol/l, 2 mins) was then timed to arrive at the slice at the peak of overshoot responses evoked by extended stimulation (22 secs after onset of stimulation).
As would be expected because of the timing of the application, there was no significant difference between the peak overshoot amplitude generated by synaptic stimulation under control conditions and synaptic responses with pyruvate added at the peak of overshoots (Figure 7A, control amplitude 7.2% ± 0.81% versus synaptic stimulation + pyruvate application 7.4% ± 1.4%, P = 0.97, Student's t-test, n = 6). However, there was no clear decrease in NAD(P)H fluorescence at the plateau of synaptically evoked overshoots, coincident with pyruvate arrival. Comparison of Figures 7Ai and 7Aiii suggests that the mean rate of decline of the overshoot plateau might be slightly faster with pyruvate, but results from individual preparations showed no clear trend. Figure 7B illustrates effects in the individual preparations that make up the averaged data set in Figure 7A. Values are measurements of peak fluorescence changes that occurred during the time window indicated by the vertical dotted lines. This time window corresponds to the time at which responses to the pyruvate application are expected, based on the observed responses to pyruvate alone (Figure 7Aii). Negative values indicate a decline in the overshoot signal, determined relative to the highest value within the analysis window. Positive values indicate progressive fluorescence increases throughout the time interval. Lines in the figure connect responses of the same preparations and show that in most (5/6) preparations, changes in overshoot during this time window either increased or decreased < 2%.
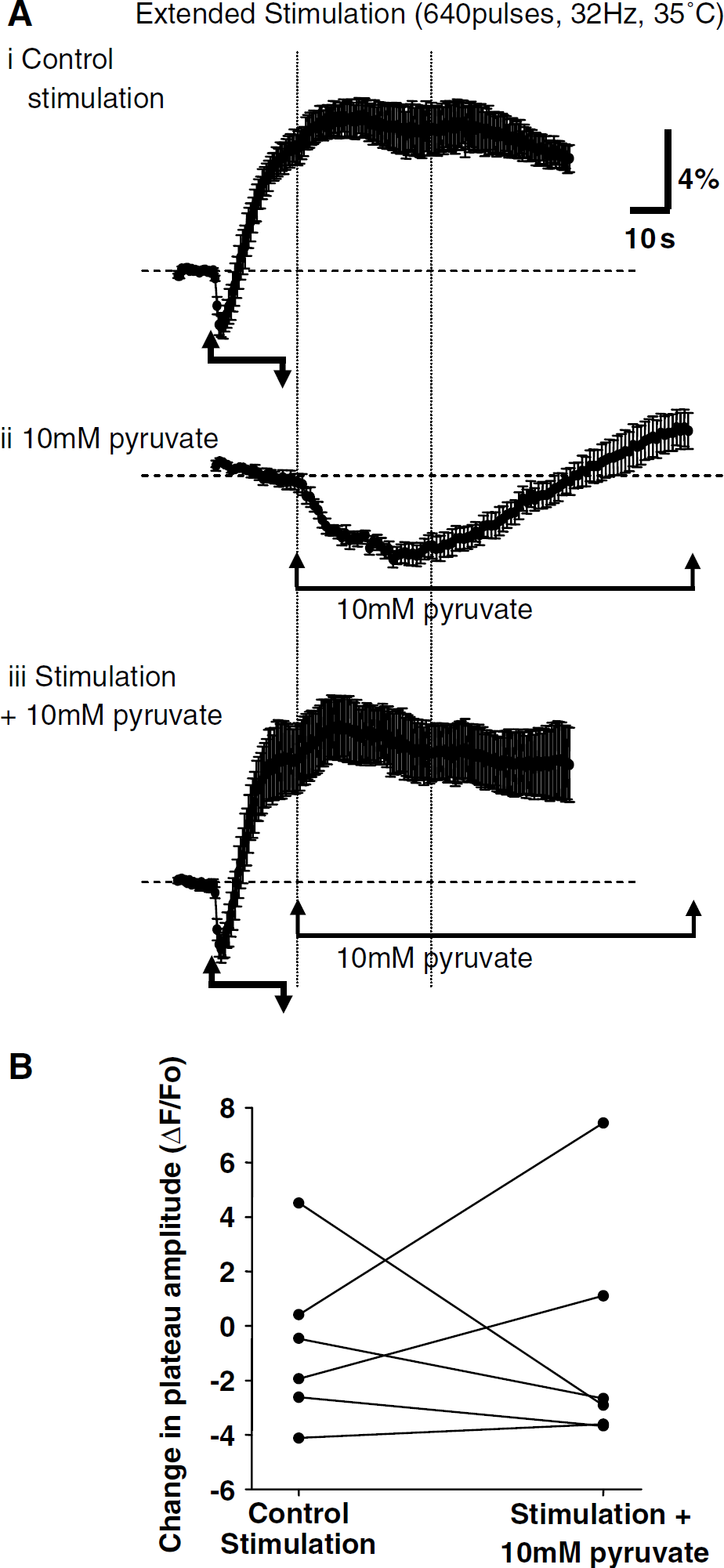
We also examined the consequences of sustained exposures to 10 mmol/l pyruvate on evoked NAD(P)H overshoots. Figure 5A suggests that responses to extended stimulation were smaller in slices pre-exposed to 10 mmol/l pyruvate, when compared with responses in different slices that were pre-exposed to 1 mmol/l pyruvate. In separate experiments to directly assess the effects of 10 mmol/l pyruvate on responses to extended stimulation, responses were established in CNQX/ APV and then the same slices were exposed to 10 mmol/l pyruvate for 10 mins (in the continued presence of CNQX/APV) before re-challenge with the synaptic stimulus (640 pulses, 32 Hz, DPCPX, 25 °C). There was a significant decrease in overshoot amplitude (2.48% ± 0.71% versus 0.49% ± 0.40%, P < 0.01, n = 4) and also a significant increase in pre-stimulus NAD(P)H fluorescence levels produced by pyruvate alone (1.04% ± 0.73% increase, P < 0.05, n = 4).
Figure 8 shows that a similar depression of NAD(P)H overshoots was also observed with responses to brief synaptic stimulus trains (two pulses, 50 Hz, bicuculline, 25°C). In this set of experiments, responses to brief stimulus trains were generated at 4-min intervals. After a series of three responses were obtained in control, ACSF slices were exposed to 1 and 10 mmol/l pyruvate (10 mins each). These long exposures to pyruvate significantly increased prestimulus resting fluorescence levels. Pyruvate (1 mmol/l) increased prestimulus NAD(P)H levels, but had no significant effect on either component of evoked NAD(P)H transients. Subsequent exposure to 10 mmol/l pyruvate resulted in a further increase in prestimulus fluorescence levels and a significant depression of the overshoot component (3.01% ± 0.26%, P < 0.03, n = 4).
Effects of brief exposures to high pyruvate. (
Exogenous pyruvate can decrease the amplitude of evoked NAD(P)H increases. (
Flavoprotein Fluorescence Transients Evoked by Extended Stimulation
Inverted flavoprotein autofluorescence signals have been used to indicate mitochondrial metabolism in neuronal preparations (Duchen, 1992; Mironov and Richter, 2001; Reinert et al, 2004), and we previously reported that (for brief stimulus durations) flavoprotein transients were of similar kinetics but inverted in sign when compared with NAD(P)H transients evoked by identical stimuli (Shuttleworth et al, 2003). Figure 9 compares NAD(P)H and flavoprotein transients elicited the same preparations by extended stimulus trains. These experiments were all performed at 25°C without supplementation of recording solutions with either pyruvate or DPCPX. Experiments were interleaved, and bleach runs were conducted (without stimulation) to identify and correct for photobleaching that occurred during flavoprotein measurements in each slice (see Materials and methods). As shown, NAD(P)H and flavoprotein transients were similar in onset time and amplitude and inverted with respect to each other. This was the case for responses generated without iGluR blockers and also for residual responses observed in CNQX/APV.
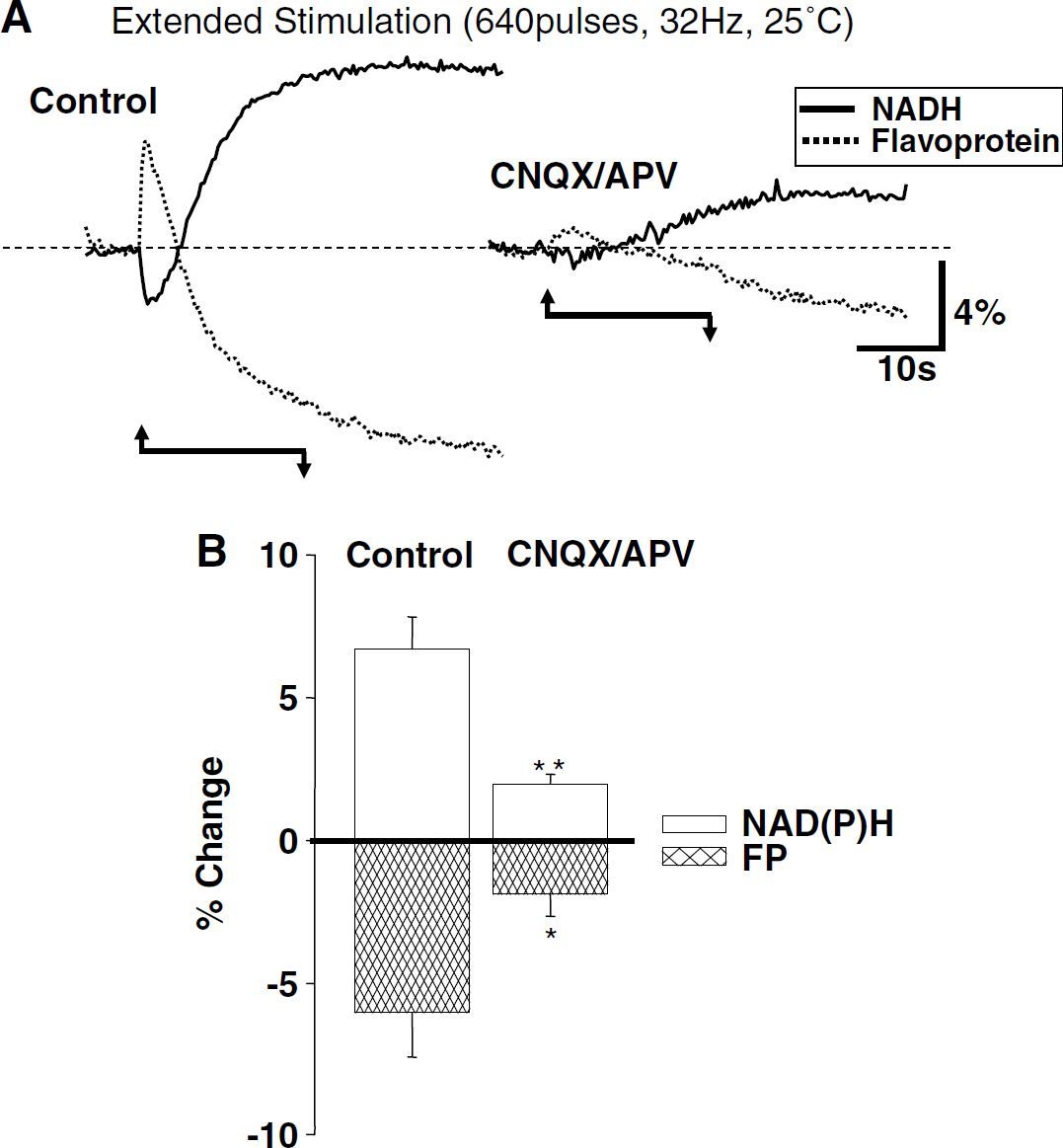
Correspondence between NAD(P)H and flavoprotein fluorescence transients generated by extended stimulation. (
Other Mechanisms Underlying the CNQX/APV-Resistant NAD(P)H Overshoots
The observation that responses to longer duration stimuli were not abolished by iGluR blockers (Kasischke et al (2004) and Figure 1) leaves open the question of the involvement of cell types other than postsynaptic neurons in responses to longer stimuli. Extracellular Ca2+ removal (see Materials and methods) virtually abolished the NAD(P)H overshoots produced by extended stimulation in CNQX/APV (640 pulses, 32 Hz, 25°C) (2.39% ± 0.67% versus 0.68% ± 0.16%, control and Ca2+ -free solution, respectively, P < 0.03, Student's t-test, n = 6). An inhibitor of nitric oxide synthase (NG-nitro-L-arginine methyl ester (L-NAME), 100 μmol/l) had no significant effect on NAD(P)H overshoots produced by extended stimulation in CNQX/APV (640 pulses, 32 Hz, 25°C; 1.97% ± 1.09% versus 2.17% ± 0.34% in L-NAME, P = 0.38, Student's t-test, n = 6). A combination of two metabotropic glutamate receptor (mGluRs) antagonists, 1-aminoindan-1,5-dicarboxylic acid (AIDA) (500 μmol/l) and LY341495 (10 μmol/l) was used to inhibit responses to both group I and group II mGluRs (Fitzjohn et al, 1998; Hermans et al, 1999; Johnson et al, 1999), but did not significantly modify NAD(P)H overshoots observed in CNQX/APV using either the LTP protocol (100 Hz, 1 sec; 2.07 ± 0.49 versus 4.10 ± 1.09, control and blockers, respectively, P = 0.09, n = 3) or extended stimuli (32 Hz, 20 secs; 5.55 ± 1.06 versus 4.43 ± 0.89, control and blockers, respectively, P = 0.33, n = 4). In contrast, there was a significant effect of these mGluR blockers on initial NAD(P)H decreases, observed during the intermediate stimulus protocol (100 pulses, 100 Hz) in the presence of CNQX/APV. As the decay time course of the early transient overlaps with the development of the delayed component, the initial slope of the early transient was analyzed to reduce contamination in the peak signal because of the delayed component. With the intermediate stimulus protocol (100 pulses, 100 Hz), initial slopes were significantly reduced (-12.68% ± 4.46% versus −6.79% ± 3.34% s−1, P < 0.04, n = 3), but no significant effect was observed with the more extended stimulus protocol (640 pulses, 32 Hz) (-3.69% ± 0.50% versus −1.52% ± 1.06% s−1, P = 0.16, n = 4).
Lack of Effect of Glutamate Uptake Inhibition
A final set of studies tested whether the NAD(P)H overshoot that persists in CNQX/APV is triggered by glial glutamate uptake. TBOA is a nontransportable, competitive inhibitor of glutamate transporters (Shimamoto et al, 1998) that increases extracellular glutamate concentration in hippocampal slice cultures (Jabaudon et al, 1999). Figure 10 shows that responses to extended stimulation (640pulses, 32 Hz, CNQX/APV, 25°C) were not reduced by TBOA. NAD(P)H overshoots showed an increasing trend that was not statistically significant. Initial fluorescence decreases were on average 1.04% ± 0.18% versus 0.98% ± 0.45%, control and TBOA, respectively (P =0.87, n = 6). Peak NAD(P)H overshoots were 1.03% ± 0.32% and 4.03% ± 1.34% (P = 0.06, n = 6), control and TBOA, respectively.
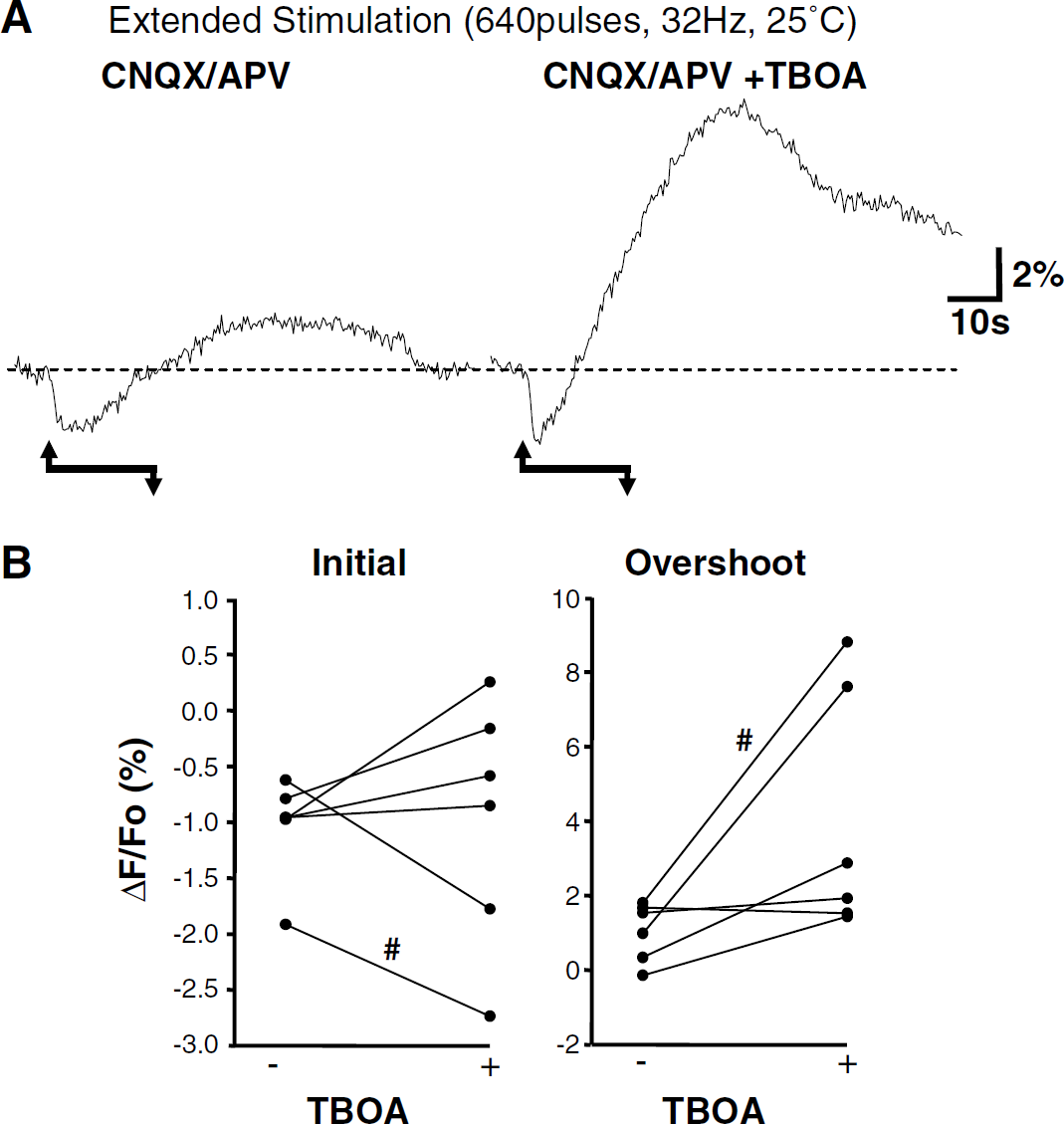
An inhibitor of glutamate transport did not antagonize responses to extended stimulus trains (640 pulses, 32 Hz, 25°C). The effects of TBOA (100 nmol/l) were tested on responses generated in CNQX/APV. (
Discussion
General
Experiments presented here were designed primarily to determine whether there was a difference in NAD(P)H-regenerating mechanisms recruited by what we have termed brief and extended stimulation protocols. Of particular interest was whether characteristic ‘overshooting’ NAD(P)H increases observed after synaptic glutamate release provide a means of indirect optical detection of transient increases in glycolysis (Kasischke et al, 2004). Glycolysis was clearly important for metabolic competence of slices, but if an alternative substrate was provided for oxidative phosphorylation, NAD(P)H increases were maintained during glycolytic inhibition over the wide range of stimuli tested. Comparison of flavoprotein and NAD(P)H transients is consistent with activation of mitochondrial metabolism by brief and extended stimulation, and attempts to reduce cytosolic NADH levels by brief exposures to high pyruvate concentrations did not abolish NAD(P)H overshoots generated by extended stimulation. Thus, the pharmacological approaches used in the present study do not support an immediate role for glycolysis in the NAD(P)H transient increases that occur after presynaptic stimulation.
Effects of Glycolytic Inhibitors
Inhibition of glycolysis using either 2DG or IAA (with concomitant removal of glucose) appeared to lead to rapid accumulation of extracellular adenosine, which in turn resulted in a substantial inhibition of synaptic transmission and inhibition of evoked NAD(P)H overshoots. Removal of glucose alone was without significant effect. We take this as evidence pointing to the effectiveness of glycolytic inhibition with the inhibitors used here, although whether glycolysis is completely blocked cannot be determined from these results. Synaptic transmission loss was prevented by block of A1 receptors using DPCPX (Coelho et al, 2000; Pearson et al, 2001; Zhao et al, 1997), allowing evaluation of the nature of NAD(P)H transients triggered by a constant synaptic stimulus. It is possible that A1 receptor block could complicate interpretation of results here if, for example, adenosine receptor activation was a crucial link between synaptic stimulation and activation of glycolysis. However, DPCPX alone (applied without glycolytic inhibitors) had no effect on either synaptic potentials or NAD(P)H transients evoked by either brief or extended stimulation. This suggests that A1 receptor activation was only significant during 2DG or IAA exposure and was not required for coupling of synaptic stimuli to NAD(P)H overshoots.
Responses to brief stimulus trains were not reduced by glycolytic inhibitors 2DG or IAA, suggesting, in line with a previous report, that both phases of responses to brief stimulus trains are a consequence of mitochondrial metabolism (Shuttleworth et al, 2003). With extended stimulation (20 secs), 2DG appeared to decrease the amplitude of NAD(P)H overshoots if the supporting pyruvate was only 1 mmol/l, although this effect was not significant after correction for multiple comparisons was made. If slices were pre-exposed to a higher pyruvate concentration (10 mmol/l), 2DG clearly had no effect on NAD(P)H overshoots (Figure 5A). It is possible that increasing pyruvate concentrations could compensate for other effects of 2DG. 2DG phosphorylation consumes ATP (Sols and Crane, 1954) and it is possible that this additional metabolic load may decrease effective intracellular pyruvate concentrations. During extended synaptic stimulation, this additional demand may be sufficient to limit the substrate available for generation of NAD(P)H overshoots. Alternatively, it is possible that glycolysis underlies NAD(P)H overshoots in both normal ACSF and with 1 mmol/l pyruvate supplementation, but addition of 10 mmol/l pyruvate could potentially relieve a reliance on glycolysis. Overshoots then generated by mitochondrial metabolism may appear more resistant to 2DG. The observations with IAA (an irreversible inhibitor of glycolysis that is not a substrate for hexokinase) support the first interpretation, as we found no effect of IAA on NAD(P)H overshoot amplitudes when preparations were supplemented with 1 mmol/l pyruvate. It remains possible that even 1 mmol/l pyruvate supplementation artificially increases mitochondrial metabolism, preventing the effects of glycolytic inhibitors being observed. This could occur as a consequence of activation of pyruvate dehydrogenase by pyruvate, mobilizing pyruvate into the TCA cycle. However, as discussed below, alternative experimental approaches do not provide evidence for glycolytic involvement in NAD(P)H overshoots, even when preparations were not supplied with pyruvate.
This study emphasized analysis of the amplitudes of NAD(P)H overshoots, but it was noted that there was a trend towards an increased rate of decay of NAD(P)H overshoot in preparations treated with IAA with extended stimulus trains. This was not significant and not observed with 2DG. However, this observation leaves open the possibility that with very much longer stimuli (perhaps approaching 5 mins continuous stimulation as reported previously; Lipton, 1973) glycolysis might contribute to NAD(P)H dynamics, but is not a significant contributor in the range of stimuli used in the present study (200 ms to 20 secs).
Flavoprotein and NAD(P)H Transients
Inverted flavoprotein and NAD(P)H signals in tissues can be used as a measure of mitochondrial redox state (Chance et al, 1979) and previous work has emphasized the usefulness of this approach for mitochondrial studies in a range of tissues and cells (Duchen et al, 2003; Kunz et al, 2002). Although glycolysis is not expected to contribute to flavoprotein fluorescence changes, it is possible that other cytosolic flavoproteins could modify slice fluorescence. However, recent studies showing disruption of mitochondrial function and consequences on flavoprotein transients produced by synaptic stimulation are consistent with the conclusion that signals are because of mitochondrial redox changes in intact brain (Reinert et al, 2004; Shibuki et al, 2003). We showed previously inverted flavoprotein and NAD(P)H signals for brief stimulus trains in hippocampal slices (Shuttleworth et al, 2003). In the present study, flavoprotein transients after extended stimulation also matched NAD(P)H transients with large ‘undershoots' after initial flavoprotein increases. These data provide evidence that mitochondrial electron transport carriers are reduced during NAD(P)H overshoots under both protocols. A similar correspondence was seen in the presence of iGluR antagonists, suggesting that mitochondrial redox state is similarly modified under these conditions. As these experiments could all be performed without supplementation with pyruvate, this provides evidence for mitochondrial redox changes after synaptic stimulation when additional substrate is not provided.
Glycolysis could potentially contribute directly to NAD(P)H signals, but is not expected to contribute directly to flavoprotein fluorescence changes. Although the amplitudes of NAD(P)H and flavoprotein transients are of similar amplitude (but inverted sign), it is difficult to make conclusions regarding the relative contribution of glycolysis to NAD(P)H signals from comparison with flavoprotein transients alone. It remains possible that cytosolic NADH production could still contribute to NAD(P)H overshoots and occur coincidently with mitochondrial NADH production (reported by flavoprotein transients). However, these data imply that mitochondrial NADH production contributes to NAD(P)H overshoots observed with extended and brief stimulus synaptic trains, including responses (observed after iGluR block) recently attributed to glial metabolism (Kasischke et al, 2004).
Effects of Pyruvate
Exogenous pyruvate alone can lead to the stimulation of cytosolic lactate dehydrogenase activity, resulting in consumption of cytosolic NADH (Scholz et al, 1969). Relatively high pyruvate concentrations reduce NAD(P)H overshoots in brain slices and this observation has been used as evidence to suggest that such responses are because of cytosolic NADH production (e.g., Foster et al, 2005; Lipton, 1973) and supports recent suggestions of glycolytic NADH production in responses to synaptic stimulation (Kasischke et al, 2004). We tested the effects of brief exposures to a high pyruvate concentration timed to arrive at the peak of NAD(P)H overshoots generated by extended stimulation. Reproducible (~3.5%) fluorescence decreases were observed when pyruvate (10 mmol/L, 2 mins) was applied under ‘resting’ conditions. However, when identical pyruvate applications were made at the peak of NAD(P)H overshoots, we did not observe the significant fluorescence decreases required if cytosolic NADH production were predominantly responsible for the overshoot. The fact that pyruvate produced little clear fluorescence decrease when applied at the peak of evoked overshoots may be due in part to increased use of mitochondrial substrate after stimulation.
Our results also suggest that sustained exposures to high pyruvate concentrations (as opposed to the transient exposures described above) reduce the amplitude of NAD(P)H overshoots (see Figure 8) by a mechanism unrelated to glycolysis. Sustained high pyruvate exposures resulted in elevated baseline NAD(P)H fluorescence levels (see Brauser et al, 1970). Pyruvate (1 mmol/l) increased baseline fluorescence levels, but did not reduce evoked NAD(P)H transients. Responses in 1 mmol/l pyruvate were not reduced by glycolytic inhibition. However, 10 mmol/l pyruvate produced a further increase in basal levels and coincident decrease in overshoot amplitude. From Figure 8A, it appears that the decrease in overshoot amplitude may be explicable in part because a ceiling level had been reached. Selective decreases in the amplitude of the overshoot component of NAD(P)H transients have been described previously, when repetitive stimuli are applied at the peak of overshoot responses in isolated neurons (Duchen, 1992) and in the hippocampal slice preparation (Kann et al, 2003).
Considerations from use of Multiple Pharmacological Inhibitors and Stimulation Conditions
With extended stimulus trains, testing of glycolytic inhibitors was limited to experiments where iGluR antagonists were present. These blockers reduced the onset of spreading-depression-type events that were otherwise commonly observed with extended stimulation after glycolytic inhibition. As described above (Introduction), glial glutamate uptake was recently suggested to be responsible for glycolytic NADH fluorescence increases (Kasischke et al, 2004; Pellerin and Magistretti, 2004). The iGluR antagonists themselves are not expected to interfere with glial glutamate uptake, and thus these inhibitors did not prevent tests of the main question of the study. However, it is possible that without the additional metabolic demands generated by iGluR receptor activation, glycolysis could be recruited. Previous work has provided evidence for glycolytic ATP production in the postsynaptic density (Wu et al, 1997). Two sets of experiments in the present study were performed without iGluR blockers: (1) comparison of flavoprotein and NAD(P)H transients and (2) the application of 10 mmol/l pyruvate applications on NAD(P)H overshoots. Results of neither support the hypothesis that neuronal glycolysis produces demonstrable NAD(P)H increases. However, it is important to emphasize that glycolysis may be significantly active under these conditions but not produce demonstrable cytosolic NADH accumulation, if, for example, pyruvate production is tightly coupled to lactate production.
A GABAA receptor antagonist (bicuculline) increased the amplitude of both components of evoked NAD(P)H transients evoked by brief stimulus trains and we concluded that this effect was likely because of increased postsynaptic depolarization (Shuttleworth et al, 2003). Bicuculline was present in all experiments with brief stimulus trains in the present study as it was helpful to generate robust signals to small numbers of stimuli. This antagonist was not used with extended stimulus trains in the present study, as (1) it was not required to generate large NAD(P)H transients, (2) it was not used in a recent study examining responses to identical stimulus durations (Kasischke et al, 2004), and (3) as spreading depression-type events were often triggered by extended stimulus trains in disinhibited slices.
Experiments were performed at both 25°C and 35°C. At the lower temperature, tests of glycolytic inhibition could be performed without triggering spreading depression-like events in the slices. At 25°C, initial NAD(P)H decreases were larger and the onset of the overshoot component was delayed; however, the peak amplitude of overshoots was not reduced when compared with responses at 35°C (Figure 2). Although it is expected that both mitochondrial and glycolytic metabolism are reduced at 25°C, it is difficult to estimate the overall temperature sensitivity of either glycolytic or mitochondrial NADH production in intact brain slice, especially under these conditions where substrate accumulation rates need to be taken into account. However, we note that reducing the recording temperature similarly affected responses to both brief and extended stimuli and the test of glycolysis that could be performed at 35°C (applications of brief high pyruvate concentrations) did not provide evidence for glycolytic NAD(P)H production at that temperature.
Glutamate Uptake
Previously, we showed that a combination of iGluR antagonists (CNQX/APV) virtually abolished NAD(P)H transients evoked by brief stimuli, a finding which suggested that postsynaptic neuronal activity was mainly responsible for NAD(P)H transients (Shuttleworth et al, 2003). This blockade (confirmed in Figure 1A here) does not rule out a role for other cell types (including astrocytes) in NAD(P)H responses to brief stimuli, as their activity may be a consequence of neuronal depolarization, for example, as a consequence of K+ efflux after neuronal depolarization (Brookes, 2000; Orkand et al, 1966). Alternatively, activation of iGluRs localized to glia (Matthias et al, 2003) could potentially contribute to these responses. However, it does argue against a major role for glial glutamate uptake contributing to NAD(P)H transients under these conditions, as the application of iGluR blockers should not substantially diminish extracellular glutamate concentrations during stimulation.
Regarding extended stimulus trails, the nontransportable inhibitor of glutamate uptake (TBOA; Jabaudon et al, 1999; Shimamoto et al, 1998) did not decrease the overshoot signal after extended stimulation (Figure 10). The concentration of TBOA used here (100 μmol/l) was the same as that used to block glial depolarizations evoked by synaptic stimulation in hippocampal slices (Kawamura et al, 2004). Therefore, although there must be significant glutamate uptake and astrocyte depolarization accompanying extended stimulation, it is possible that the glial metabolism mobilized in response to cytosolic Na+ accumulation does not cause measurable NAD(P)H fluorescence increases in these cells.
In this study, we only examined the effects of TBOA after pharmacological isolation of the CNQX/APV-resistant overshoot. Thus, it remains possible that TBOA may reduce transients in the absence of glutamate antagonists. It is technically difficult to assess this possibility, as increasing extracellular glutamate concentrations with TBOA greatly increases the incidence of spreading depression-type events in slices not previously exposed to iGluR blockers. Thus, these results suggest that, at least under the limited conditions tested here, measurements of synaptically evoked NAD(P)H transients do not provide an optical monitor of glycolytic metabolism triggered by glutamate uptake.
Possible Mechanisms Underlying CNQX/APV-Resistant NAD(P)H Increases
Unlike the responses to brief stimulation, which were almost totally eliminated by AMPA/NMDA receptor block, a significant component of overshoot responses to extended stimulation persisted after inotropic glutamate receptor blockade (Figure 1C; see also Kasischke et al, 2004). Thus, the differences in the effects of combined receptor blockers appears to be because of extended stimuli heavily recruiting mechanisms not activated by brief periods of activity. A significant NAD(P)H overshoot resistant to iGluR blockers was also observed with the LTP stimulus protocol, suggesting that the temporal threshold for iGluR-resistant NAD(P)H responses lies in the range of ~ 500 ms to 1 sec with high-frequency stimulation.
If CNQX/APV-resistant NAD(P)H increases are not generated by glutamate uptake, what other mechanisms may contribute? The block of responses observed after Ca2+ removal may be explained by inhibition of transmitter release, but the data also imply that strong depolarization and presumably changes in presynaptic metabolism because of Ca2+-independent mechanisms (e.g., Na+ influx) do not significantly contribute to the NADH overshoot phase that persists with extended stimulation in CNQX/APV. Ca2+-dependent metabolic changes in presynaptic terminals remains a possible mechanism, as do postsynaptic mechanisms (neuronal and/ or astrocytic) that are triggered by Ca2+-dependent release of presynaptic glutamate. Ca2+-dependent triggering of mitochondrial NADH production could also contribute to postsynaptic NADH increases (see Introduction), but the relative contribution of such a mechanism cannot be determined from these experiments using synaptic stimulation.
Such obvious things as reflectance changes that accompany neuronal depolarization appear unlikely to contribute to our responses (see Lipton, 1973; Reinert et al, 2004). This conclusion is supported by comparison of flavoprotein and NAD(P)H signals in the present study, which were inverted despite being generated using excitation wavelengths separated by < 100 nm. An additional possibility that we have given a preliminary examination to is the activation of mGluRss (Haak et al, 1997; Miller et al, 1995). Group I and II blockers did bring about a significant reduction in the initial decrease phase in the LTP protocol, but there was no change in the overshoot phase. A greater degree of glutamate spillover into extrasynaptic regions may be expected for the extended stimulus protocols, but the NAD(P)H overshoot was not reduced here either. Although this argues against a role of group I and II mGluRs in NAD(P)H overshoots, we note that these low-affinity antagonists might not result in complete block. The cost of going to higher concentrations using a slice preparation was prohibitive and further analysis may be facilitated by the development of more effective reagents. At this point then, the mechanisms responsible for triggering metabolic responses seen in CNQX/APV remain unknown.