
Abstract
Cardiocirculatory arrest is the most common clinical cause of global cerebral ischemia. We studied neuronal cell damage and neuronal stress response after cardiocirculatory arrest and subsequent cardiopulmonary resuscitation in rats. The temporospatial cellular reactions were assessed by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick endlabeling (TUNEL) staining of DNA fragments, in situ hybridization (heat shock protein hsp70; immediate early genes c-fos and c-jun), and immunocytochemical (HSP70; and myeloperoxidase, specific marker of polymorphonuclear leukocytes [PMNL]) techniques. Cardiac arrest of 10 minutes' duration was induced in mechanically ventilated male Sprague-Dawley rats anesthetized with nitrous oxide and halothane. After cardiopulmonary resuscitation, animals were allowed to reperfuse spontaneously for 6 hours, 24 hours, 3 days, and 7 days (n = 6 per group). Five sham-operated animals were controls. The TUNEL staining revealed an early onset degeneration in the thalamic reticular nucleus (TRN) at 6 hours that peaked at 3 days. In contrast, degeneration was delayed in the hippocampal CA1 sector, showing an onset at 3 days and a further increase in the number of TUNEL-positive cells at 7 days. A minor portion of TUNEL-positive nuclei in the CA1 sector showed condensed chromatin and apoptotic bodies, whereas all nuclei in the TRN revealed more diffuse staining. After 6 hours of reperfusion, levels of mRNA for hsp70 and c-jun were elevated in circumscribed areas of cortex, in all hippocampal areas, and in most nuclei of thalamus, but not in the TRN. After 24 hours, a strong expression of mRNA for hsp70 and c-jun could be observed in the second layer of the cortex and in hippocampal CA1 sector; hsp70 also was observed in hippocampal CA3 sector. Some animals showed expression of hsp70 and c-jun in the dentate gyrus. After 3 days, hsp70 and c-jun were detected mainly in the CA1 sector of hippocampus. At 7 days, mRNA for both returned to control values. Therefore, delayed cell degeneration in the CA1 sector corresponds to a prolonged expression of hsp70 and c-jun in this area. In situ hybridization studies for c-fos revealed a strong signal in CA3 and dentate gyrus and a less prominent signal in TRN at 6 hours. At 24 hours, CA4 and amygdalae were positive, whereas at 3 and 7 days, the signal reached control levels; no prolonged or secondary expression was observed in the CA1 sector. Immunohistochemical study confirmed translation of HSP70 in various areas corresponding to the detection of mRNA, including the CA1 sector. The number of PMNL increased significantly at 6 hours and 7 days after cardiac arrest; PMNL were distributed disseminately and were not regionally associated with neuronal cell damage. The current data support the view that CA1 neurons might undergo an apoptosis-associated death after cardiac arrest, but PMNL are not directly involved in this process. The marked differences in the time course and the characteristics of TUNEL staining and the neuronal stress response in CA1 sector and TRN point to different mechanisms of neuronal injury in the two selectively vulnerable areas.
Keywords
Brief periods of global cerebral ischemia result in delayed neuronal death in the vulnerable areas of the brain (Horn and Schlote, 1992; Kirino, 1982, Kirino and Sano 1984; Petito et al., 1987; Pulsinelli et al., 1982). In experimental models of isolated global cerebral ischemia, it was shown that the CA1 sector of hippocampus and the dorsolateral part of striate nucleus are particularly involved (Pulsinelli et al., 1982). Neurons in these regions are irreversibly damaged whereas glial and vascular cells survive (Pulsinelli et al., 1982). Selective neuronal death also has been shown to occur in humans after cardiocirculatory arrest and subsequent cardiopulmonary resuscitation, the most common clinical cause of global cerebral ischemia and reperfusion (Horn and Schlote, 1992; Petito et al., 1987). Therefore, the investigation of this phenomenon is of major interest.
Data obtained from experimental animal models of global cerebral ischemia, however, may not necessarily reflect the cardiac arrest situation (Böttiger et al., 1997). Most models using vascular occlusion techniques do not produce a complete standstill of cerebral circulation as occurs during cardiac arrest (Dietrich et al., 1987; Pulsinelli et al., 1982; Rehncrona et al., 1979). Moreover, reperfusion after cardiac arrest may be associated with additional injury from systemic extracerebral organ ischemia followed by activation of blood coagulation, complement system, platelets, polymorphonuclear leukocytes (PMNL), and various other systems, and is associated with strong lactacidosis (Böttiger et al., 1995, 1996; Caceres et al., 1995; Fischer et al., 1996; Safar, 1986). This may be the reason why, after cardiac arrest, thalamic reticular nucleus (TRN) neurons, and, in particular, somatosensory neurons of the middle segment of this nucleus, also exhibit early selective vulnerability (Blomqvist and Wieloch, 1985; Kawai et al., 1992, 1995; Ross and Duhaime, 1989). We, therefore, investigated neuronal stress response and neuronal death in global cerebral ischemia induced by cardiac arrest.
Delayed neuronal death and selective vulnerability have been correlated with characteristic patterns of neuronal stress response, as investigated by the transcription and translation of heat shock proteins (HSP) and immediate early genes (IEG) (Ikeda et al., 1990; Neumann-Haefelin et al., 1994; Nowak, 1990; Nowak et al., 1990; Onodera et al., 1989; Uemura et al., 1991a, 1991b; Wessel et al., 1991).
Transcription factors encoded by IEG are thought to play a crucial role in mediating alterations in gene expression in response to ischemia (Akins et al., 1996; Gass et al., 1992). The resulting alterations may be involved in active programmed or apoptotic neuronal cell death that differs fundamentally from necrosis, in which the initiating and promoting events are primarily associated with imbalances in ionic and metabolic homeostasis (Li et al., 1995; Schreiber and Baudry, 1995). Previous studies on HSP and IEG response after global cerebral ischemia have been restricted to artificial models of isolated cerebral ischemia (Neumann-Haefelin et al., 1994; Schreiber and Baudry, 1995; Uemura et al., 1991a, 1991b; Wessel et al., 1991). The current study determined special features of neuronal cell response to cardiocirculatory arrest using various histologic, in situ hybridization, and immunohistochemical techniques. In addition, the number and localization of PMNL were determined by immunohistochemical staining because recent data suggest a relation between PMNL and apoptotic neuronal cell death in experimental focal ischemia and reperfusion (Chopp et al., 1996).
MATERIALS AND METHODS
Animal preparation
After institutional approval by the Governmental Animal Care Committee was obtained, adult male Sprague-Dawley rats (body weight 320 to 450 g) were studied. All animals were handled according to the Guiding Principles published by the National Institutes of Health and the Council of the American Physiological Society (National Institutes of Health, 1985). The animals were anesthetized without premedication using halothane (0.8% to 1.5%) and 70% nitrous oxide in oxygen. Polyethylene catheters (PE 50) were placed into the tail artery and epigastric vein, and 0.9% saline was infused continuously (2 mL/kg/h) to maintain catheter patency. Heparin was not given during the entire study period. After endotracheal intubation (Braunüle-MT No. 3; B. Braun, Melsungen, Germany), the animals were paralyzed with pancuronium bromide (Pancuronium “Organon,” Organon Teknika, Eppelheim, Germany; 0.2 mg/kg/h) and mechanically ventilated at a rate of 30 breaths per minute (Harvard Rodent Ventilator, Harvard Apparatus, South Natick, MA, U.S.A.). The tidal volume was adjusted to ensure a PaCO2 within the physiologic range. The fraction of the inspired oxygen concentration (FIO2) was adjusted to maintain PaO2 above 100 mm Hg. The arterial catheter line was connected to a pressure transducer (Kombidyn Monitoring Set, B. Braun), which was leveled to midheart. The transducer output was displayed and recorded continuously (DASYLab Software SN #D4111444C, Datalog, Mönchengladbach, Germany).
Electrocardiographic recordings were performed using subcutaneous needle electrodes. All animals received an esophageal electrode for transesophageal induction of electrical ventricular fibrillation. The body temperature of the animals was kept at 37°C as long as the trachea was intubated before and after cardiac arrest with the use of a feedback-controlled heating pad.
Experimental protocol
Three minutes before cardiac arrest, halothane-but not nitrous oxide-was discontinued. Cardiac arrest was induced by electrical stimulation (alternating current: 12 V, 50 Hz) through the esophageal electrode (Böttiger et al., 1997) and confirmed by the abrupt decrease in mean arterial pressure to below 15 mm Hg. Ventilation and infusion were stopped, and the heating system was switched off. Cardiopulmonary resuscitation procedures, including mechanical ventilation (100% oxygen, respiratory rate 45/min, initiated 15 seconds before cardiac massage), closed-chest cardiac massage (200/min), and intravenous bolus administration of 0.02 mg/kg epinephrine and 0.5 mEq/kg sodium bicarbonate, were started 10 minutes after initiation of ventricular fibrillation. Two minutes later, external defibrillation (5 WS, DC-defibrillator DEFIPORT SCP912, Hellige, Freiburg, Germany) was carried out. If restoration of spontaneous circulation (ROSC) was not achieved immediately, DC countershocks were repeated after 30 to 60 seconds, accompanied by continuation of cardiopulmonary resuscitation procedures. The ROSC was confirmed by spontaneous cardiac rhythm accompanied by a rise of mean arterial pressure to above 50 mm Hg. The heating pad was switched on immediately after ROSC, and saline infusion was resumed. Blood gas analyses were performed 5 minutes, 30 minutes, and 1 hour after ROSC, and the ventilatory parameters were adjusted if needed. If base excess exceeded −5 mEq, additional sodium bicarbonate was administered.
Animals were allowed to recover for various time periods (6 hours, 24 hours, 3 days, 7 days; n = 6 per group). In animals surviving more than 6 hours, catheters were removed, and the wounds were infiltrated with 1% lidocaine (Xylocain, Astra, Wedel, Germany) and properly closed. The animals were weaned from the ventilator after stepwise reduction of FIO2 to 0.4 and allowed to breath spontaneously at 1 hour after ROSC. The trachea was extubated between 2 and 3 hours after ROSC, and the animals were placed in special cages with oxygen concentrations of about 50% for 1 more hour. Thereafter, animals were returned to their home cages with free access to food and water. They received 20 mL/day Ringer's solution subcutaneously until they started drinking. In animals subjected to 6 hours of reperfusion, anesthesia was maintained until the whole animal was frozen in liquid nitrogen. The other animals were reanesthetized and frozen in toto in liquid nitrogen. Five sham-operated animals, which received the same anesthesia and surgical treatment without cardiocirculatory arrest, were controls.
Brains were removed from the skull in a low-temperature cabinet at −20°C. At the hippocampal level (approximately at bregma −3.5 mm), coronal 10-μm cryostat brain sections were cut at −20°C and thaw-mounted on poly-L-lysine-coated glass slides.
TUNEL staining
For in situ staining of DNA fragmentation and apoptotic bodies, the histochemical TUNEL method (terminal deoxynucleotidyltransferase [TdT]-mediated d-uracil triphosphate[UTP]-biotin nick end-labeling [Gavrieli et al., 1992; Wijsman et al., 1993]) was used as described previously (Wiessner et al., 1996a). Cryostat sections were fixed for 15 minutes in ice-cold 4% paraformaldehyde. Subsequently, the sections were washed twice in 70% ethanol (1 minute), once in phosphate-buffered saline (PBS; 3 minutes), once in 2% hydrogen peroxide/PBS (5 minutes), and then again in PBS (5 minutes). Afterward, the sections were equilibrated for 15 minutes in buffer 1 (25 mmol/L Tris-HCl, pH 6.6; 200 mmol/L cacodylic acid; 200 mmol/L potassium chloride; 1 mmol/L cobalt chloride; 1.25 mg/mL bovine serum albumin [BSA]). The buffer was quantitatively removed, 10 μL of TdT mix (14 μmol/L biotin-16-dUTP [Boehringer Mannheim, Mannheim, Germany]; 60 U/mL TdT [Life Technologies, Eggenstein, Germany]; 500 mmol/L potassium cacodylate [pH 7.2]; 10 mmol/L cobalt chloride; 1 mmol/L dithiothreitol) per section were added, and the sections were covered with a coverslip. After incubation for 60 minutes at 37°C, the reaction was terminated by washing the sections for 15 minutes in buffer 2 (300 mmol/L sodium chloride; 30 mmol/L sodium citrate). Incorporated biotin was visualized using the avidin-biotin-peroxidase complex method (Vector Laboratories, Burlingame, CA, U.S.A.). Finally, the sections were dehydrated and embedded in Eukitt (Kindler, Freiburg, Germany).
The TUNEL method detects DNA fragmentation associated with apoptotic cell death (Chopp et al., 1996; Gavrieli et al., 1992; Li et al., 1995), although cells exhibiting morphologic features of necrotic cell death also contain stainable concentrations of DNA fragments. Staining of necrotic cells, however, is thought to be more diffuse than that of apoptotic cells, and characteristic apoptotic bodies are absent (Chopp et al., 1996; Li et al., 1995). Negative controls were performed using double-distilled water to replace TdT enzyme. Cells exhibiting DNA fragmentation (TUNEL positive) were counted in cortex, hippocampus, and TRN using light microscopic methods (×200 to 300 magnification). This was carried out by an investigator (B. S.) who was blinded to the experimental protocol. In addition, high-magnification light microscopic study (×1000) was performed in all animals to detect condensed chromatin and apoptotic bodies.
In situ hybridization
Antisense oligonucleotide probes specific for the mRNA of the inducible form of the HSP (hsp70) and of the IEG c-fos and c-jun have been described previously (Neumann-Haefelin et al., 1994; Wiessner et al., 1995). The specificity of all oligonucleotide probes was confirmed by Northern blots (Neumann-Haefelin et al., 1994; Wiessner et al., 1995). Each probe was 3′-OH end-labeled using TdT (GIBCO BRL, Eggenstein, Germany) and a 30:1 molar ratio of [35S]dATP (1200 Ci/mmol). In situ hybridization procedures were performed according to the methods described previously (Neumann-Haefelin et al., 1994; Wiessner et al., 1995). Brain sections were fixed for 15 minutes in ice-cold paraformaldehyde (4%) and PBS at pH 7.4. To reduce unspecific background signal, the sections were acetylated with acetic anhydride (0.25%) in triethanolamine for 10 minutes. After dehydration and air-drying, sections were covered with hybridization buffer (50% formamide; 10% dextran sulfate; 2× standard sodium citrate; 100 μg/mL poly-A; 120 μg/mL heparin; 5 mmol/L dithiothreitol; 1 mmol/L ethylenediamine tetraacetic acid; 1 mg/mL herring sperm DNA; 1 mg/mL BSA; 1 mg/mL polyvinylpyrrolidone; 1 mg/mL Ficoll 400; 0.5 mg/mL yeast tRNA; 10 mmol/L Tris-HCl, pH 8.0) containing 2 to 10 pg/μL 35S-labeled probe. The sections were covered with a coverslip and hybridized at 42°C for 16 hours. Thereafter, the coverslips were removed, and the sections washed twice (2 × standard sodium citrate; 50% formamide; 5 mmol/L dithiothreitol) at 42°C for 30 minutes. After dehydration and air-drying, hybridized radioactivity was visualized by film autoradiography (Hyperfilm-βmax, Amersham, Braunschweig, Germany) with exposure times of 2 to 3 weeks.
In control experiments performed with all oligonucleotide probes used, selected brain sections were pretreated with 20 μg/mL RNase A for 45 minutes.
Immunohistochemistry
Immunohistochemical staining was performed after fixation of cryostat sections in methanol at −20°C for 5 minutes and air-drying at room temperature. Thereafter, the sections were washed three times in PBS, followed by a block of endogenous peroxidase activity with 0.3% hydrogen peroxide in PBS for 20 minutes, and another rinse with PBS.
For immunostaining of HSP70, sections were incubated with normal rabbit serum (1:1,000) in PBS containing 1% BSA for 2 hours. The sections then were incubated for 48 hours at 4°C with a mouse monoclonal antibody specific for the inducible form of HSP (HSP70, ANTI-HSP70 monoclonal antibody SPA-810, StressGen, Victoria, Canada) diluted to a concentration of 2 μg/mL in PBS containing 1% BSA and 0.01% sodiumacide. Thereafter, the slides were washed in PBS, incubated for 2 hours with a rabbit anti-mouse monoclonal antibody (dilution 1:50; DAKO Z0109, DAKO Diagnostics, Glostrup, Denmark) in PBS containing 1% BSA, and immunoreactivity was visualized by the peroxidase-antiperoxidase method. For this purpose, sections were washed in PBS, incubated with a monoclonal mouse anti-horseradish peroxidase antibody (dilution 1:50; DAKO B0650, DAKO Diagnostics) in PBS and 1% BSA for 2 hours, and washed again in PBS. Staining was developed with 0.1% diaminobenzidine tetrahydrochloride in PBS with 0.01% hydrogen peroxide, and sections were dehydrated with increasing concentrations of ethanol and embedded.
For immunostaining of myeloperoxidase, which is a highly specific marker of PMNL (Barone et al., 1991), sections were incubated with normal goat serum (1:2,000) in PBS containing 1% BSA for 1 hour. The sections then were incubated for 16 hours at 4°C with the primary antibody (rabbit-anti-human myeloperoxidase, DAKO A0398, DAKO Diagnostics) diluted to a concentration of 1:100 in PBS containing 1% BSA. Thereafter, the slices were washed in PBS, incubated for 1 hour with a biotinylated secondary antibody (goat-anti-rabbit, dilution 1:50; Vector Laboratories) in PBS containing 1% BSA. After one more rinse in PBS, immunoreactivity was detected by streptavidin-Cy3 complex (dilution 1:100; Sigma S 6402, Sigma Chemical Company, St. Louis, MO, U.S.A.) in PBS with 1% BSA for 3 hours. After a final wash in PBS, sections were embedded in antifading fluorescent mounting medium (S3023, DAKO Diagnostics). The number of PMNL per tissue section was counted in a representative brain section from each animal using fluorescent microscopic methods (×200 to 300 magnification). This was carried out twice, and the investigator (B. W. B.) was again blinded to the experimental protocol.
In control experiments, sections were incubated with normal rabbit serum instead of each primary antibody.
RESULTS
Transesophageal electrical fibrillation led to circulatory arrest within 15 seconds in all experimental animals. No spontaneous defibrillations were observed during the period of circulatory arrest. All animals could be resuscitated successfully (ROSC) by the use of one to three countershocks (Fig. 1).
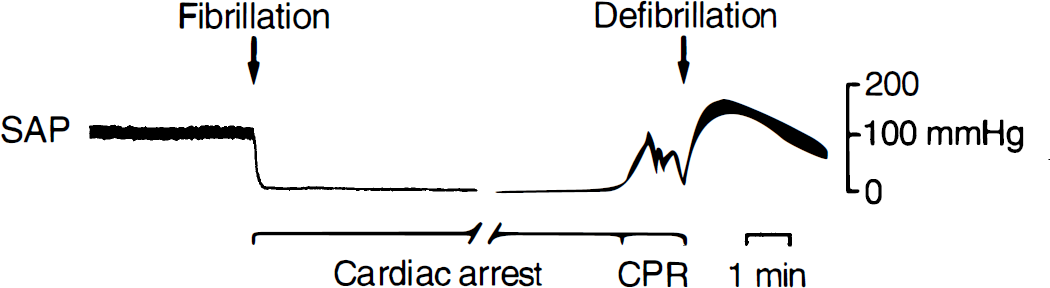
Recording of systemic arterial blood pressure (SAP) of a rat subjected to 10 minutes of cardiac arrest followed by cardiopulmonary resuscitation (CPR) using advanced cardiac life support procedures and external electrical defibrillation. Notice the immediate onset of complete circulatory arrest after fibrillation and rapid restoration of spontaneous circulation (ROSC).
Observations from TUNEL staining
The TUNEL staining detected DNA fragmentation in neurons of the hippocampal CA1 sector and in TRN. In the hippocampal CA1 sector, TUNEL-positive neurons were first detected at day 3 (4 ± 6 cells per section), and the number increased further at day 7 after cardiac arrest (91 ± 75 cells per section) (Figs. 2 and 3). At 7 days, about 30% of TUNEL-positive nuclei in the hippocampal CA1 sector showed condensed chromatin and apoptotic bodies (Fig. 2C). In TRN, TUNEL-positive neurons already could be detected at 6 hours (42 ± 37 cells per section), and the maximum of TUNEL-positive neurons was reached at day 3 after cardiac arrest (100 ± 56 cells per section; Figs. 2 and 3). The TUNEL staining in TRN nuclei was more diffuse and condensed chromatin and apoptotic bodies were not observed. No major differences in TUNEL staining were observed between different animals of the same group. The TUNEL-positive cells were neither observed in sham-operated control animals nor in the neocortex at any time after cardiac arrest. Negative controls did not reveal specific staining.
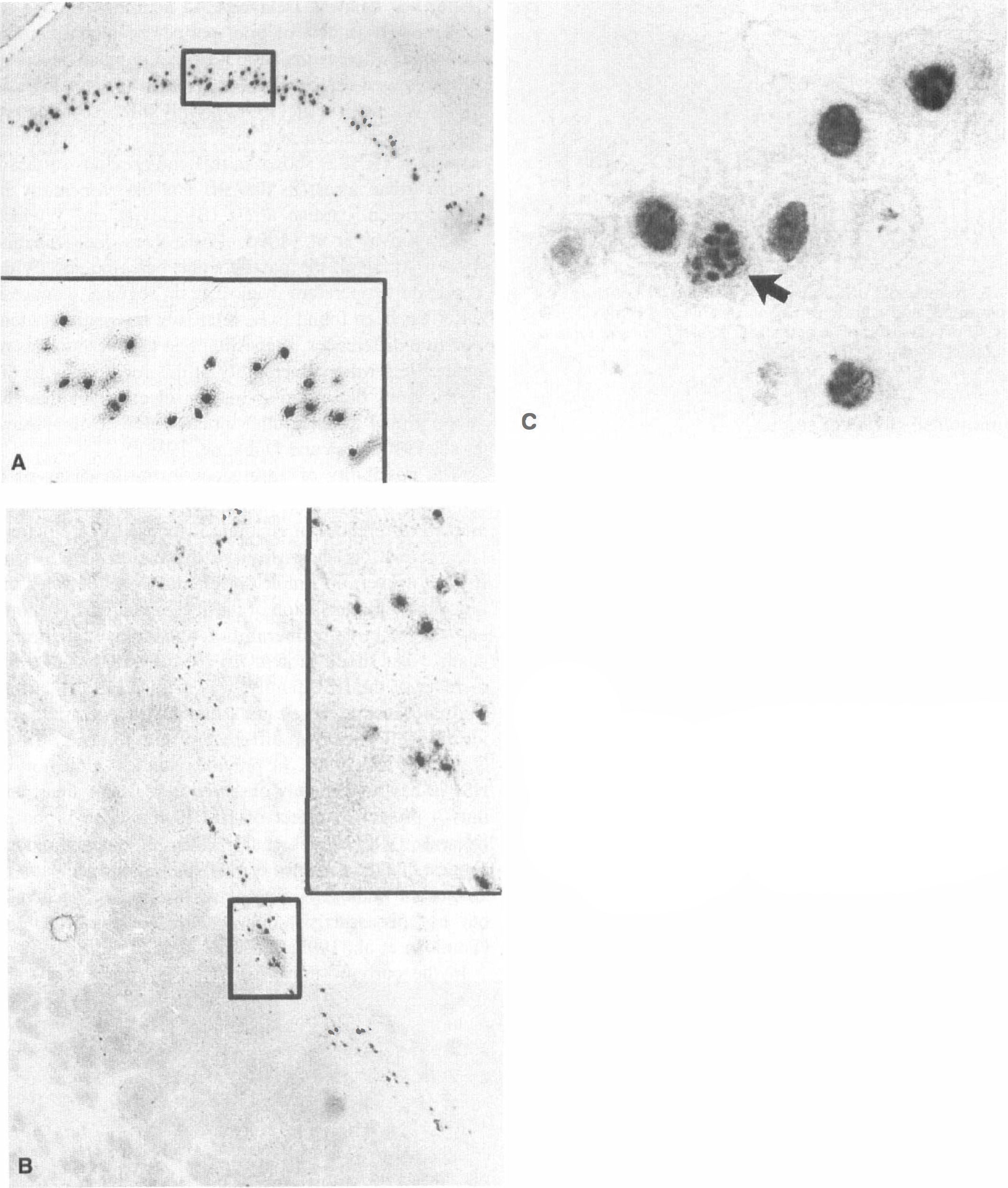
The TUNEL-positive neurons in the hippocampal CA1 sector at 7 days after 10 minutes of cardiac arrest
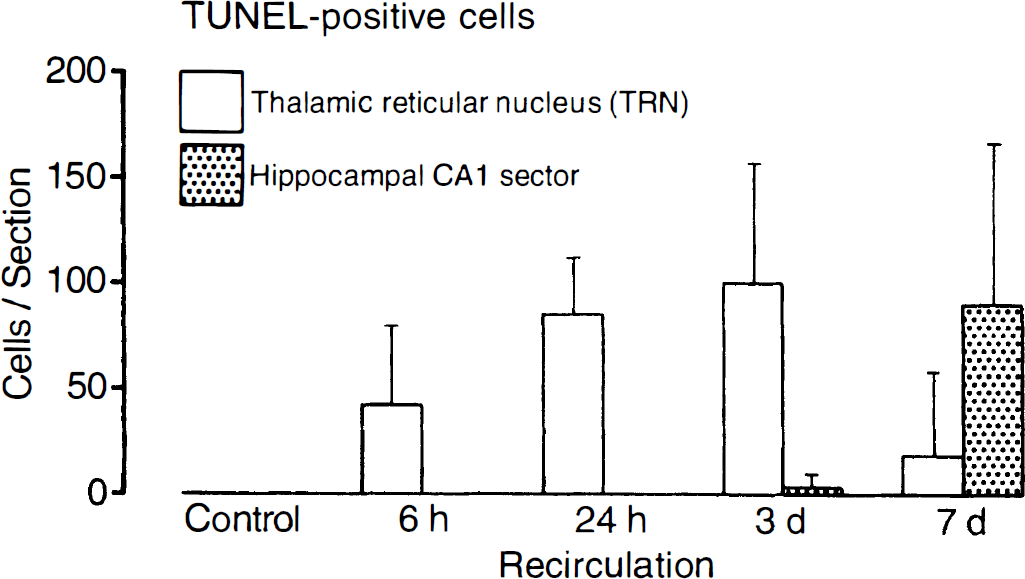
Number of TUNEL-positive cells in the CA1 sector of the hippocampus and in the thalamic reticular nucleus of the rat brain after 10 minutes of cardiac arrest and different times of reperfusion (numbers are means ± SD per tissue section).
In situ hybridization studies
Hybridization studies in sham-operated animals did not show any detectable signal for hsp70 mRNA. Six hours after cardiac arrest, hsp70 mRNA was detected throughout the neocortex (with a maximum in layers I and II) in the pyramidal layer of all hippocampal subfields, in the granule cells of the dentate gyrus, in most nuclei of thalamus, and in parts of the entorhinal cortex (Fig. 4). After 24 hours, hsp70 still was detectable in the superficial layers of neocortex and in hippocampal areas (preferentially CA1 and CA3 sectors) but not in the dentate gyrus. After 3 days, hsp70 was found mainly in the CA1 sector of hippocampus, but the signal intensity was less than at 24 hours. Some animals also demonstrated minor signal intensity in CA2 and CA3 sectors. After 7 days of reperfusion, hsp70 mRNA was no longer detectable. With the exception of one animal showing a faint expression of hsp70 mRNA in TRN at 24 hours, there was no hsp70 mRNA increase in TRN and striate nuclei at any time (Fig. 4).
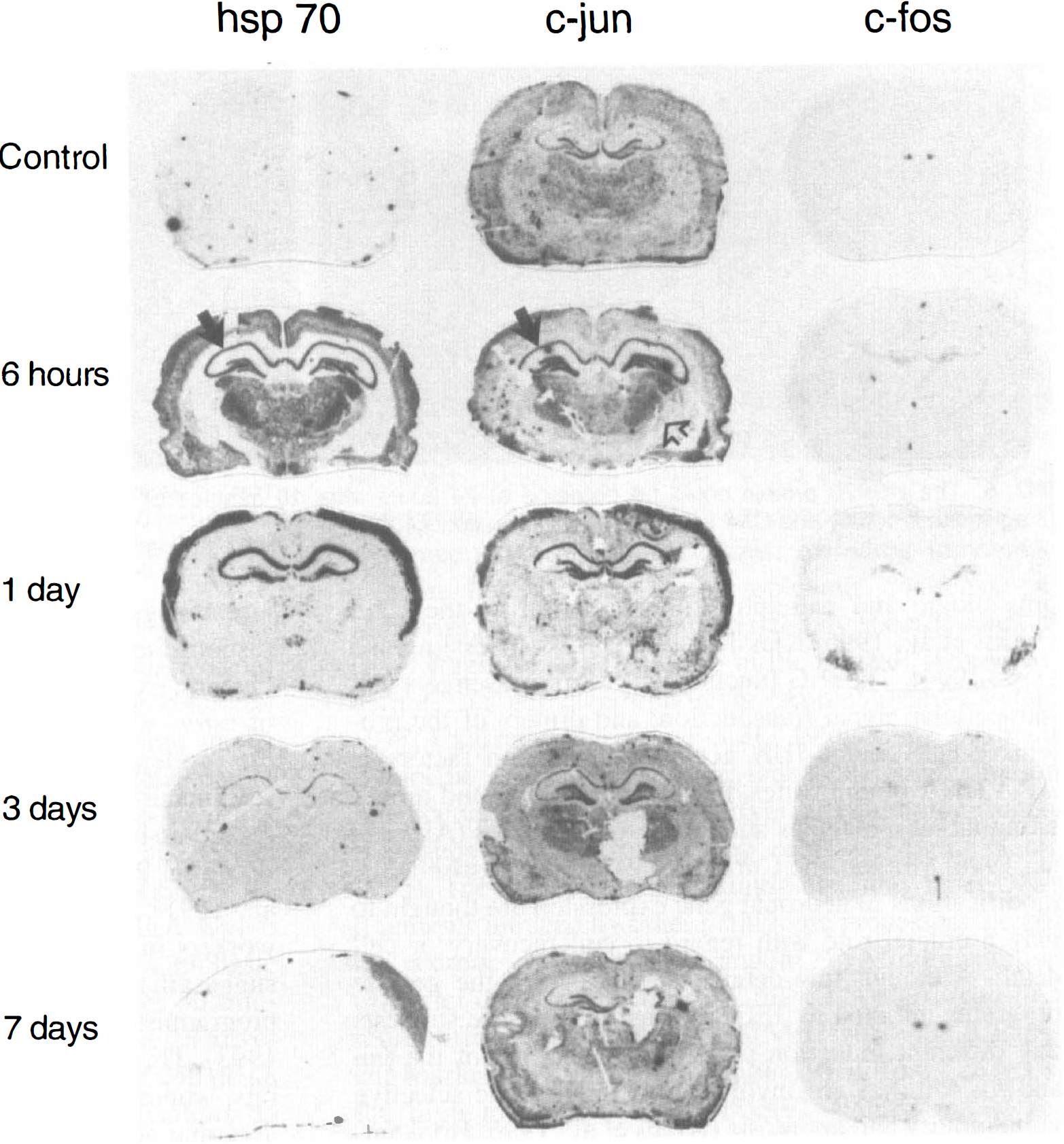
Genomic response after 10 minutes of cardiac arrest in rat. After 6 hours of reperfusion, hsp70 and c-jun mRNA were activated in circumscribed areas of cortex, in all hippocampal areas, and in most nuclei of thalamus but not in the thalamic reticular nucleus (TRN [open arrow]; black arrows, hippocampal CA1 sector). After 24 hours, a strong expression of hsp70 and c-jun mRNA could be observed in the first and second layer of the cortex and in the hippocampal CA1 sector; hsp70 also was observed in hippocampal CA3 sector. Some animals showed expression of hsp70 and c-jun in the dentate gyrus. After 3 days, hsp70 and c-jun were detected mainly in the CA1 sector of hippocampus. At 7 days, both mRNA returned to control values. In situ hybridization studies for c-fos revealed a signal in CA3 and dentate gyrus at 6 hours. At 24 hours, CA4 and amygdalae were positive, whereas at 3 and 7 days, the signal reached control levels (autoradiograms are from representative animals for each survival time).
Constitutive mRNA levels for c-jun were observed in dentate gyrus and, to a lesser extent, also in other hippocampal subunits (CA4 > CA3 > CA1; Fig. 4). After 6 hours of reperfusion, c-jun was increased in the cortex (layers I and II), all hippocampal areas, and in thalamic nuclei excluding TRN. After 24 hours, a strong expression of c-jun could be observed in layers I and II of neocortex and in hippocampal CA1 sector. Some animals also showed expression in the dentate gyrus. After 3 days, increased c-jun mRNA was detected almost exclusively in the CA1 sector of hippocampus and in the dentate gyrus. The intensity of the signal at this reperfusion time was less pronounced than in animals subjected to 24 hours of reperfusion. After 7 days, c-jun mRNA levels returned to control values (Fig. 4).
In sham-operated animals, in situ hybridization studies for c-fos revealed only weak signal intensity in dentate gyrus and in habenulae (Fig. 4). Six hours after cardiac arrest, c-fos mRNA was detected in the hippocampal CA3 sector and dentate gyrus and, to a lesser extent, in layer II of neocortex. In contrast to hsp70 and c-jun mRNA, c-fos was found to be increased in the TRN in five of six animals at this time point (Fig. 5). After 24 hours, c-fos mRNA was detected in the hippocampal CA4 sector and in amygdalae. Three of six animals revealed c-fos induction in the entorhinal cortex. Control levels of c-fos were reached 3 and 7 days after cardiac arrest (Fig. 4).
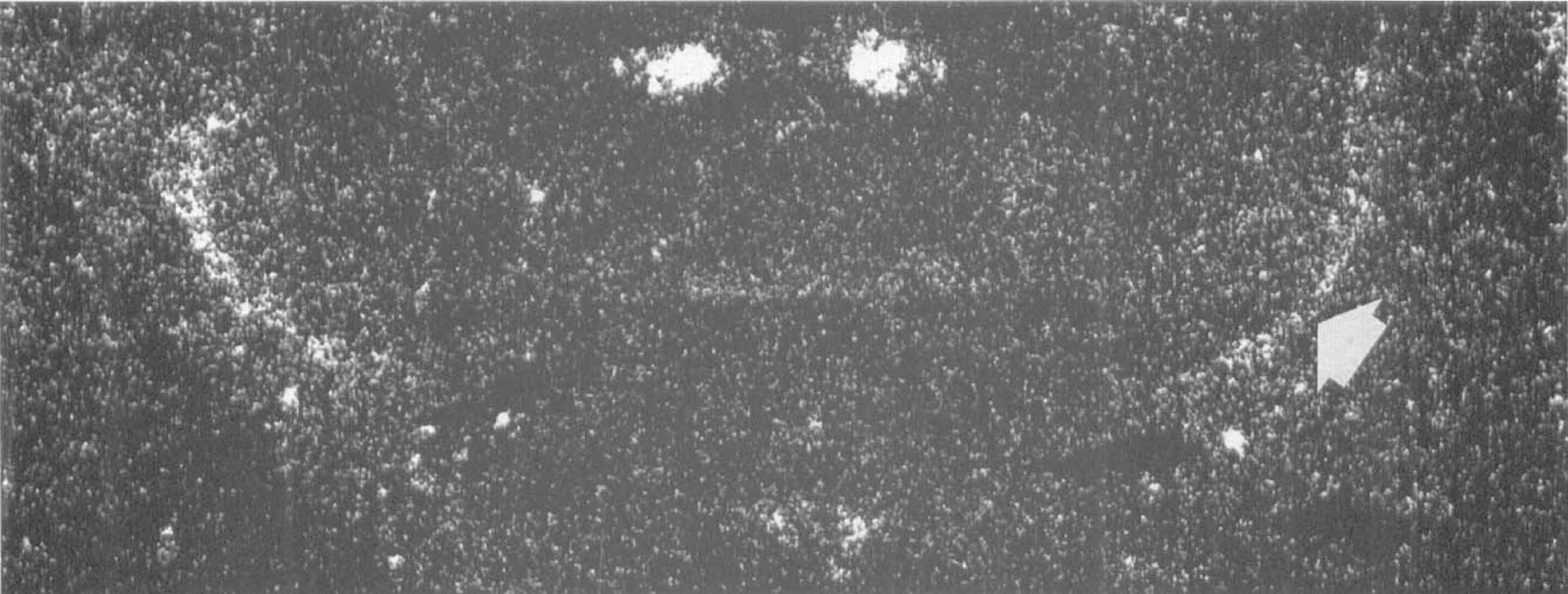
Upregulation of c-fos mRNA in the thalamic reticular nucleus after 10 minutes of cardiac arrest, detected by in situ hybridization at 6 hours of reperfusion (arrow).
There was only little variability with regard to gene expression between different animals per group. Control experiments with RNase A pretreatment did not result in any detectable signal.
Immunohistochemical studies
In sham-operated animals, HSP70 could not be detected. The spatial distribution of HSP70 after cardiac arrest was similar to the patterns obtained for hsp70 mRNA. After ischemia, no specific signal for HSP70 was observed at 6 hours of reperfusion. The HSP70 was first detected at 24 hours and reached a maximum at 1 to 3 days after cardiac arrest. At these times, a varying number of neocortical neurons and neurons of medial thalamic nuclei exhibited positive staining, whereas the hippocampal CA1 and CA4 sectors revealed a strong signal (Fig. 6). At 7 days, staining intensity was greatly reduced and could be detected only in the hippocampal CA1 sector. Interestingly, a varying number of hippocampal CA1 neurons exhibited strong immunoreactivity, which, however, persisted after omission of the primary antibody. These neurons probably were dead because they had a pyknotic morphologic makeup without discernible nucleus. The TRN did not stain for HSP70 at any time.
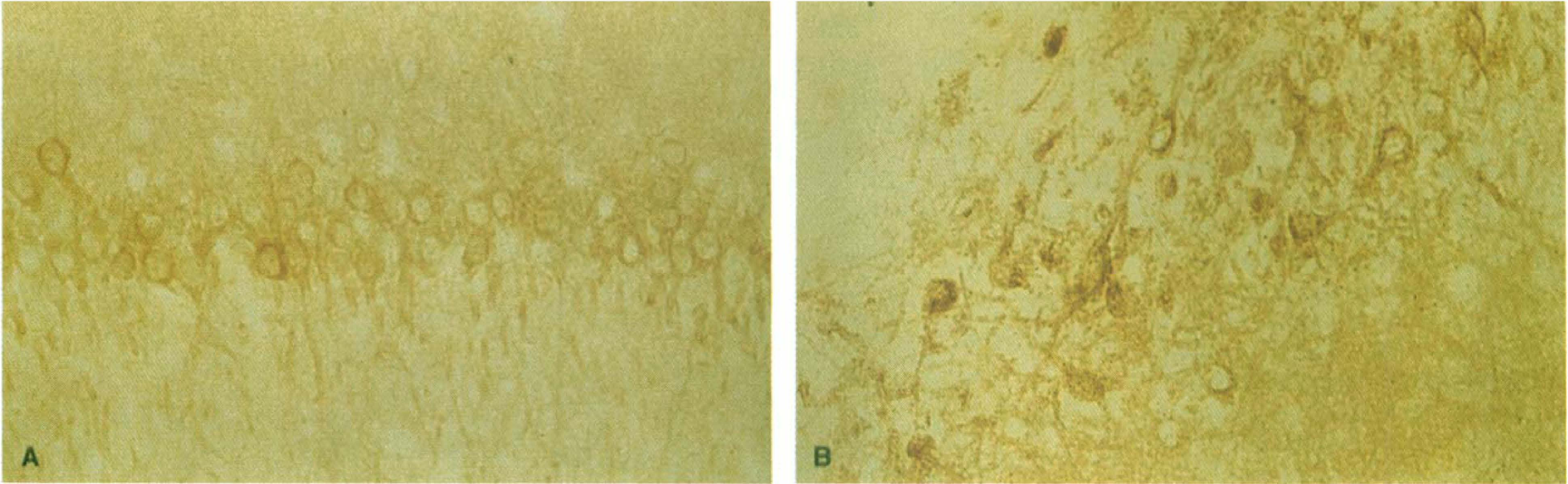
The HSP70 protein could be detected at 24 hours after 10 minutes of cardiac arrest in the hippocampal CA1 sector
Immunohistochemical staining for myeloperoxidase (Fig. 7) showed a significant increase in the number of PMNL at 6 hours and 7 days after cardiac arrest (Fig. 8). The PMNL were distributed disseminately throughout the brain and were localized both in microvessels and in the parenchyma (Fig. 7). They did not colocalize with areas of neuronal cell damage, as detected by the TUNEL technique.
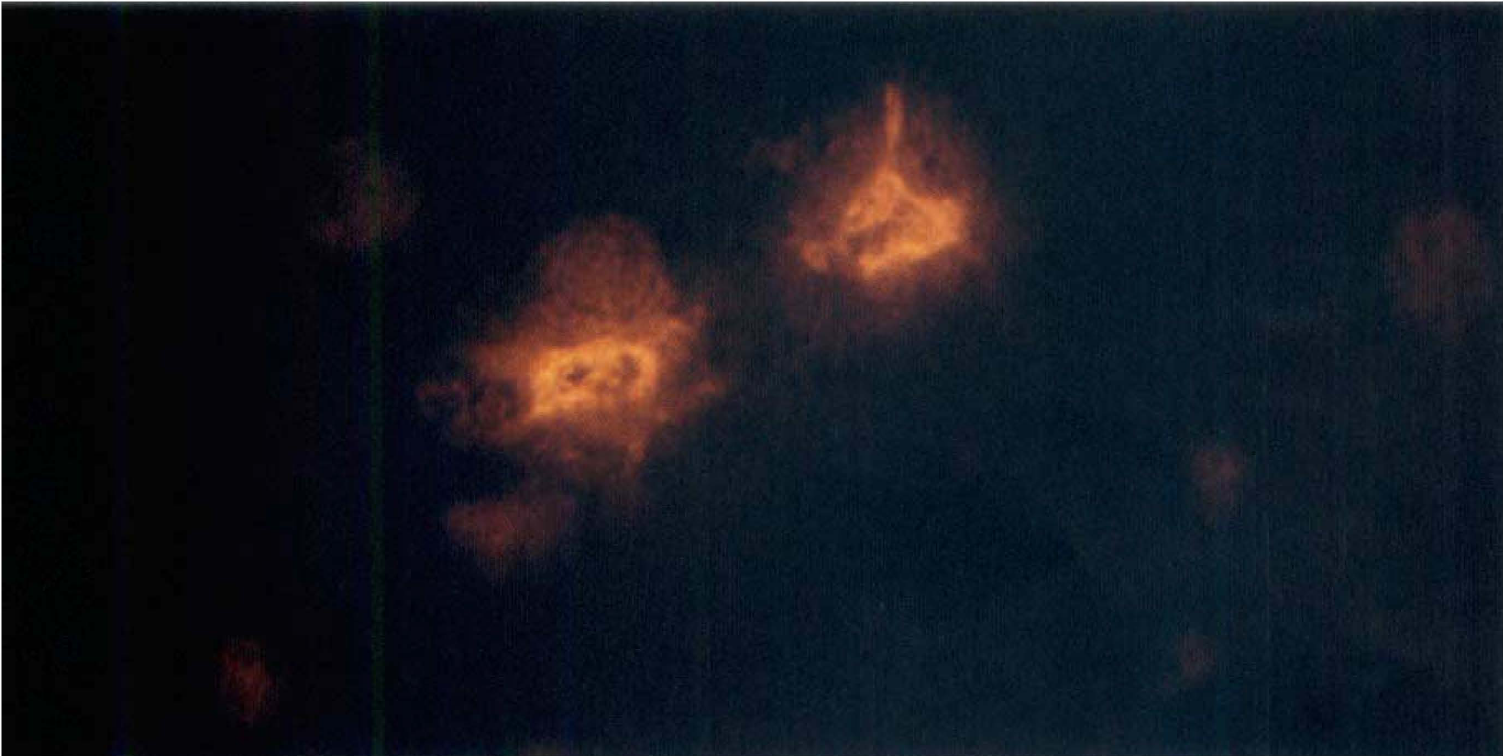
Polymorphonuclear leukocytes (PMNL) in brain tissue were stained with the use of a specific monoclonal antibody against myeloperoxidase. The figure shows two PMNL in an experimental animal subjected to 10 minutes of cardiac arrest and 6 hours of reperfusion (magnification ×1000).
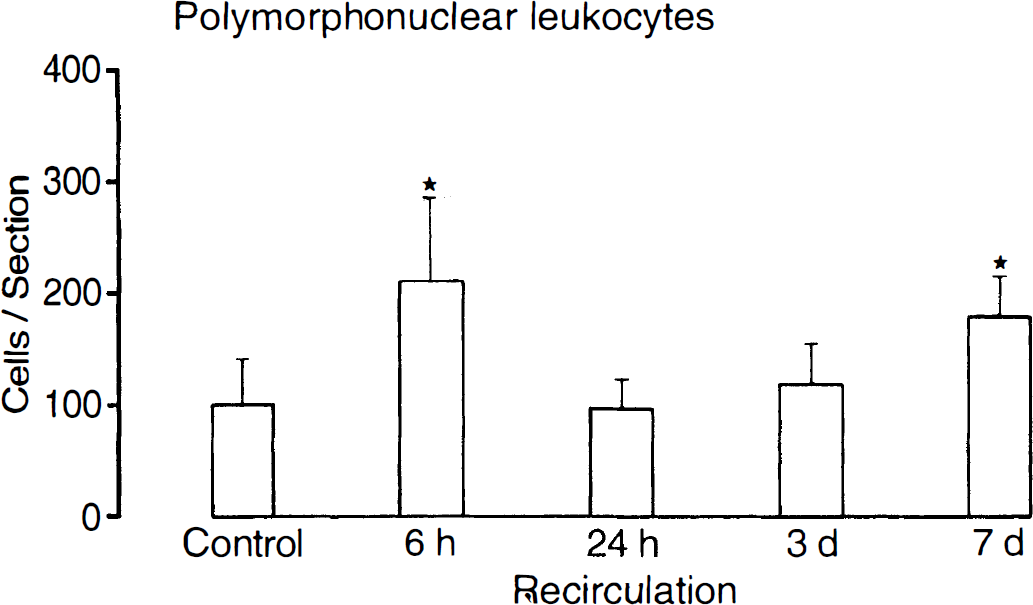
Immunohistochemical staining for myeloperoxidase revealed that the number of polymorphonuclear leukocytes in brain increased significantly at 6 hours and 7 days after 10 minutes of cardiac arrest (numbers are means ± SD per tissue section, *P < 0.01 versus control; t test).
After omission of the primary antibodies, no signal could be detected.
DISCUSSION
The current study demonstrates for the first time the precise topical relation between the neuronal stress response and neuronal damage after brief cardiac arrest. The TUNEL staining revealed early DNA fragmentation and thus neuronal death in γ-aminobutyric acid-ergic TRN, whereas neuronal damage in the hippocampal CA1 sector was delayed. Delayed CA1 neuronal damage is a well known sequela of brief periods of global cerebral ischemia, whereas thalamic neuronal damage occurs after prolonged cerebrocirculatory arrest (Pulsinelli et al., 1982; Wiessner et al., 1996a). Only after ischemia produced by cardiac arrest does an early neuronal injury appear in TRN, as shown in this and previous studies in both cardiac arrest in humans and experimentally induced rodent cardiac arrest (Blomqvist and Wieloch, 1985; Kawai et al., 1992, 1995; Ross and Duhaime, 1989). Although the postischemic outcome strictly depends on temperature in most brain regions, damage in TRN has been found to be relatively insensitive to temperature differences, suggesting that the mechanisms responsible for the vulnerability of this region may be different from those acting in the selectively vulnerable hippocampal areas or other brain regions (Minamisawa et al., 1990; Ross and Duhaime, 1989).
The possibility of differences in the mechanisms of cell death between CA1 sector and TRN is underlined by marked differences in genomic response to cardiocirculatory arrest, as shown by the expression of HSP and IEG. However, no simple correlation exists between the induction and translation of these genes and the phenomena of selective vulnerability and delayed neuronal death. Heat shock protein hsp70, the strictly inducible member of the HSP family, was included in the current study because it represents a marker for potentially injurious cell stress of different origin (Nowak, 1990; Nowak et al., 1990). In previous studies, induction of HSP70 has been mainly observed in resistant areas, and thus a protective effect of HSP70 has been proposed (Nowak, 1990; Nowak et al., 1990). More recent reports suggest that the presence of HSP70 is a good indicator of the general pathophysiologic stress response to ischemia, but not necessarily a marker for neuronal survival (Tomioka et al., 1993; Wiessner et al., 1996b).
In the current study, hsp70 mRNA increased transiently in different brain regions resistant to ischemia, including neocortex, hippocampal subunits, and most thalamic areas. Moreover, hsp70 was strongly induced in the selectively vulnerable CA1 sector of hippocampus between 6 hours and 3 days after cardiac arrest. However, most CA1 neurons survived at 7 days after cardiocirculatory arrest in the current model, and we could not establish whether neurons that failed to express hsp70 mRNA or HSP70 protein would die at a later time. Therefore, the observed pattern of hsp70 induction does not contradict the hypothesis of a possible protective role of hsp70 after cerebral ischemia. It has been hypothesized for transcription factor genes that prolonged mRNA expression in dying CA1 neurons may result from failure to synthesize the corresponding proteins that may feed back and normally suppress mRNA synthesis (Honkaniemi and Sharp, 1996). Therefore, the failure to see prolonged expression of hsp70 in the hippocampal CA1 sector may relate to CA1 cell survival in the current study. In contrast to the hippocampal CA1 sector, the selectively vulnerable TRN, which suffered severe injury, did not overexpress hsp70 mRNA or HSP70 protein at any time after cardiac arrest.
Comparable with heat shock response, the expression of IEG represents an early genomic response to various physiologic and pathophysiologic stimuli of the CNS (Akins et al., 1996; Kiessling et al., 1993; Kiessling and Gass, 1994). The IEG function as “third messengers” of intracellular signal transduction, and dimers of the proteins c-FOS and c-JUN act as transcription factors at AP-1 DNA binding sites, thereby controlling and modulating target molecules and late response genes (Akins et al., 1996; Morgan and Curran, 1989, 1991). Postischemic modifications in neuronal gene expression are thought to play a crucial role with regard to cell recovery or cell death. Although few details are known on the genetic programs initiated by IEG, increasing evidence suggests that different induction patterns of members of the jun and fos families are involved in postischemic selective vulnerability and apoptosis (Akins et al., 1996; Honkaniemi et al., 1996; Morgan and Curran, 1989, 1991; Neumann-Haefelin et al., 1994).
In the current study, c-jun mRNA was observed in various brain regions, including both resistant and selectively vulnerable areas. The expression almost paralleled the distribution of hsp70 and, similar to this mRNA, c-jun mRNA was not observed in TRN at any time. However, c-jun was expressed in hippocampal CA1 neurons between 6 hours and 3 days after cardiocirculatory arrest, suggesting that c-jun is involved in the neuronal response to ischemia in this selectively vulnerable area. Overall, expression of c-jun persisted far longer than that of c-fos, which is in line with previous observations in rats undergoing 30 minutes of four-vessel occlusion (Neumann-Haefelin et al., 1994). Others, however, observed a similar and parallel expression pattern for both IEG after 20 minutes of four-vessel occlusion (Wessel et al., 1991). Experimental studies by Dragunow and coworkers in newborn rats subjected to hypoxia-ischemia suggest that c-jun is involved in genetic events leading to programmed or apoptotic cell death (Dragunow et al., 1993). This is not confirmed by the current data in adult rats, where c-jun was expressed not only in regions undergoing neuronal cell death, but also in regions without neuronal cell damage. In the hippocampal CA1 sector, most neurons were still alive at 7 days despite marked c-jun expression. Since it is impossible to tell from an in situ autoradiograph whether neurons destined to die or neurons destined to survive express mRNA, it is possible that, also in the CA1 sector, c-jun mRNA is expressed in surviving neurons. This would be in line with the suggestion that c-jun expression is associated with attempts at neuronal regeneration (Herdegen et al., 1993).
The mRNA of c-fos also was induced in both resistant and selectively vulnerable areas. Interestingly, c-fos expression was found at 6 hours after reperfusion in the selectively vulnerable TRN but not in the hippocampal CA1 sector. It is well known, however, that c-fos peaks early after ischemia (Neumann-Haefelin et al., 1994), and it is possible that we missed the hippocampal response because the earliest time point studied was 6 hours after cardiac arrest. Others observed a biphasic pattern of c-fos expression in CA1 sector after 30 minutes of four-vessel occlusion and stressed the involvement of c-fos in delayed programmed neuronal cell death (Neumann-Haefelin et al., 1994). Our observations do not contrast with this conclusion, but it should be stressed that a causal role of c-fos for apoptotic cell death has not been established (Akins et al., 1996).
A characteristic feature of global cerebral ischemia is the persisting suppression of protein synthesis in the selectively vulnerable brain regions (Hossmann, 1993; Kiessling et al., 1993). This inhibition does not interfere with the expression of IEG, because transcriptional induction of IEG does not require de novo protein synthesis (Herschman, 1991). However, other genes may not be translated into their proteins. We, therefore, investigated the temporospatial expression of HSP70 by immunohistochemical study. The results indicate that the HSP70 pattern corresponds closely to the hsp70 mRNA pattern, with the exception of the 6 hours' survival time where no HSP70 could be detected. The HSP70 was observed both in selectively vulnerable and resistant brain areas. Thus, postischemic disturbances of protein synthesis do not seem to affect translation of early response and stress genes into their proteins, as supported by recent observations on FOS-like immunoreactivity and HSP70 in rats undergoing 30 minutes of four-vessel occlusion (Neumann-Haefelin et al., 1994; Wiessner et al., 1996a, 1996b). Therefore, the HSP70 and, possibly, other proteins as well, can act specifically in the postischemic brain. However, the observed translation of HSP70 in the hippocampal CA1 sector contrasts with previous studies in gerbils after 5 minutes of global cerebral ischemia, where the protein products of IEG did not increase despite elevated levels of mRNA (Kiessling et al., 1993).
Some authors refer to cells that exhibit TUNEL-positive staining as “apoptotic” cells, in particular, if the nuclei show condensed chromatin and apoptotic bodies (Choi, 1996; Chopp et al., 1996; Gavrieli et al., 1992; Li et al., 1995; Wijsman et al., 1993). In contrast to necrosis, apoptosis reflects an active process where different intracellular programs including expression of IEG are involved (Akins et al., 1996; Searle et al., 1982). Cells undergoing apoptotic cell death reveal a characteristic sequence of cytologic alterations, including membrane blebbing and nuclear and cytoplasmic condensation. Moreover, apoptosis is characterized by specific random DNA fragmentation resulting in the typical “DNA laddering” in gel electrophoresis (Appleby and Modak, 1977; Charriaut-Marlangue et al., 1996; Choi, 1996). It cannot be excluded, however, that DNA fragmentation also occurs in cells without the typical morphologic characteristics of apoptosis (Chopp et al., 1996; Li et al., 1995). Therefore, TUNEL staining also may label DNA fragments induced by other pathologic processes, including necrotic cell death. The random DNA fragmentation in nuclei damaged by necrosis may, however, lead to a more diffuse staining by the TUNEL technique (Charriaut-Marlangue et al., 1996; Choi, 1996). In all animals, the high magnification of TUNEL-stained nuclei revealed condensed chromatin and apoptotic bodies in about one third of the TUNEL-positive cells of hippocampal CA1 sector at 7 days. This histologic evidence supports the view that at least part of the CA1 neurons might undergo apoptosis after global cerebral ischemia from cardiac arrest. This is in line with previous results obtained from other models of global cerebral ischemia (Charriaut-Marlangue et al., 1996; Choi, 1996; Heron, 1993) and with data from our group demonstrating that CPP32 cysteine protease, an effector of the cell death program, is activated both at the transcriptional and the posttranscriptional level in the rat hippocampal CA1 sector after 10 minutes of cardiac arrest (Gillardon et al., 1997). In contrast to these observations in the CA1 sector, condensed chromatin and apoptotic bodies were not observed in the TRN in any animal at any time point.
In a previous investigation of focal ischemia, PMNL have been implicated for the induction of apoptosis (Chopp et al., 1996). In contrast to this study, we did not detect any regional associations of TUNEL-positive cells with PMNL, although the latter increased significantly at 6 hours and 7 days after cardiac arrest. However, it is known from various models of ischemia and reperfusion that PMNL may contribute to ischemic injury by promoting peroxidative changes during reperfusion (Chopp et al., 1996; Del Zoppo et al., 1991; Lucchesi et al., 1989). The current demonstration of PMNL influx may, therefore, be more relevant for reperfusion injury after brief periods of cardiac arrest.
Since the duration of global cerebral ischemia from cardiac arrest was similar in CA1 sector and TRN, the ischemic event and thus the severity of the initial injury was similar in both brain regions. However, it is possible that the neurons in the TRN are more sensitive to ischemia than those in the CA1 sector and, thus, the same ischemic event may lead to more pronounced ischemic injury in TRN. This could result in a different pattern of gene expression and a shorter delay for the appearance of TUNEL-stained neurons (Honkaniemi et al., 1996). The occurrence of apoptotic bodies only in CA1 neurons but not in TRN, however, supports the hypothesis of different mechanisms of neuronal death in these two selectively vulnerable areas.
In conclusion, after 10 minutes of reversible circulatory arrest, TUNEL-positive cells were detected early in TRN and later in the hippocampal CA1 sector. Some TUNEL-positive nuclei in the CA1 sector showed condensed chromatin and apoptotic bodies, whereas all nuclei in the TRN revealed more diffuse staining, suggesting that apoptosis-associated mechanisms may contribute to neuronal cell death only in the hippocampal CA1 sector. The number of PMNL increased in the brain during reperfusion but did not correlate with TUNEL staining, thereby excluding a causal relation. The marked differences in the time course and in the characteristics of TUNEL staining, as well as in the preceding neuronal stress response in the two selectively vulnerable brain regions, supports the view that after brief cardiac arrest, different mechanisms of neuronal injury may occur in different parts of the brain.
Footnotes
Acknowledgements
The authors thank P. Lorenz, U. Gerken, and M. Jagodnik for technical assistance. The authors also thank B. Huth and I. Mühlhöver for the artwork and the photographs, and M. Hahmann and D. Schewetzky for excellent secretarial help.