
Abstract

Several types of potassium (K+) channels are present in cerebral blood vessels. Opening or closure of these ion channels may have significant effects on membrane potential, which is a major determinant of entry of extracellular calcium and thus vascular tone. Because of the ionic properties of the cell membrane at rest, the change in activity of only a few K+ channels is sufficient to change membrane potential significantly and alter vascular tone (Nelson and Quayle, 1995; Quayle et al., 1997). This review will focus on recent findings regarding the characteristics and functional importance of K+ channels in the cerebral circulation.
THE MEMBRANE POTENTIAL OF CEREBRAL VASCULAR SMOOTH MUSCLE
Membrane potential of smooth muscle is a major determinant of cytosolic free calcium concentrations and thus vascular tone. Recent direct measurements of these variables in cerebral arteries have illustrated the close relationship between membrane potential and intracellular calcium, as well as membrane potential and vascular diameter (Knot and Nelson, 1998; Knot et al., 1998) (Fig. 1). Opening of K+ channels increases potassium conductance (K+ efflux) and causes the membrane potential to approach the equilibrium potential for potassium, typically about −85 mV (i.e., hyperpolarization). In contrast, closure of K+ channels will decrease the outward movement of K+ and depolarize the membrane. Modest changes in membrane potential can be associated with large changes in diameter of cerebral arteries. For example, Fig. 1 illustrates that from a resting membrane potential of about −45 mV, hyperpolarization is associated with an increase in vessel diameter, whereas depolarization is associated with vasoconstriction. The resulting change in diameter of cerebral arteries is significant with even small changes in membrane potential (Knot and Nelson, 1998). The concept illustrated in Fig. 1 is consistent with other studies that previously suggested a close relationship between membrane potential and cerebral vascular tone (Fujiwara et al., 1982; Harder and Waters, 1983; Knot et al., 1996; Nagao and Vanhoutte, 1993).
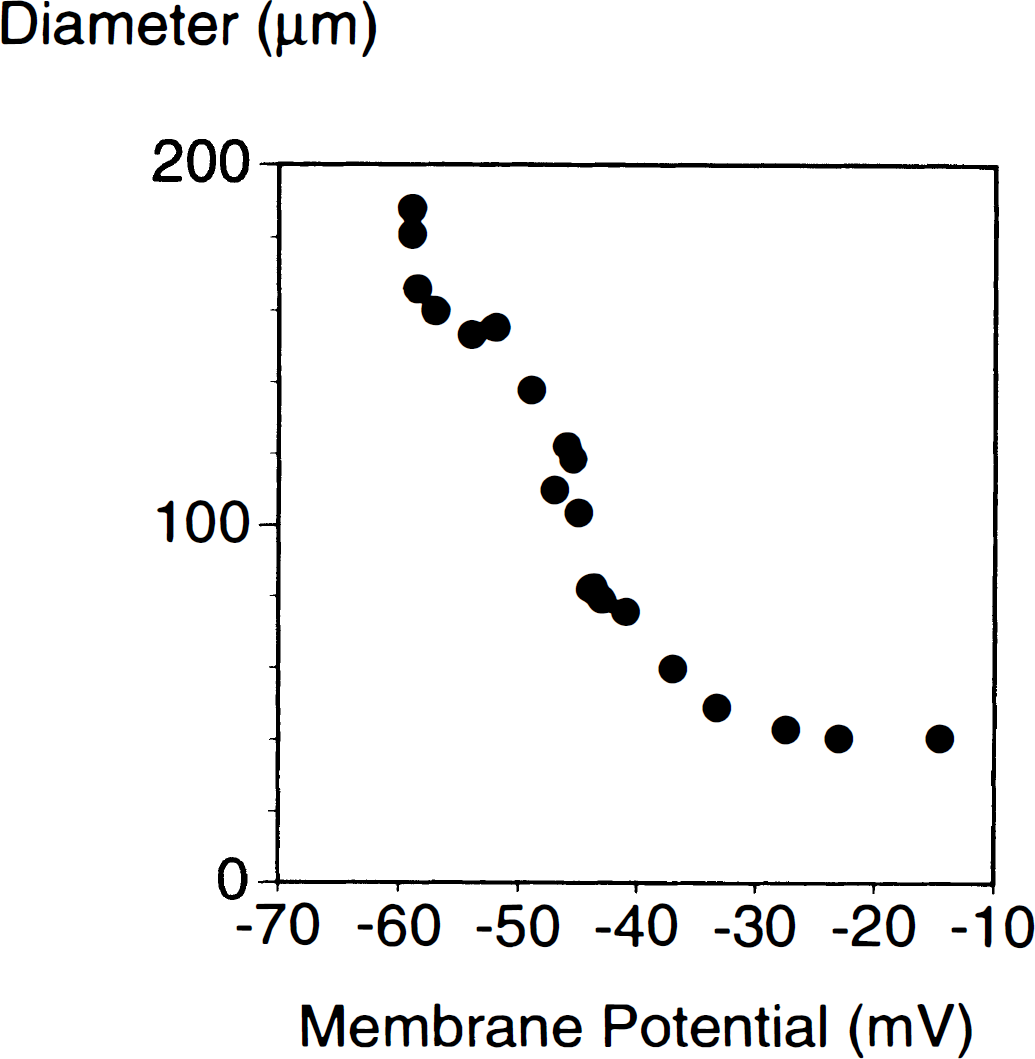
Relationship between membrane potential measured in vascular smooth muscle and diameter of cerebral arteries in vitro. Measurements were obtained in arteries pressurized to 60 mm Hg. Data were redrawn from Knot and Nelson (1998). Resting membrane potential in these arteries was approximately −45 mV. See Knot and Nelson (1998) for details of how these values were obtained.
What is the resting membrane potential of vascular smooth muscle under normal conditions? To our knowledge, membrane potential of cerebral vascular smooth muscle has not been measured in vivo. In other vascular beds (cremaster, mesenteric, and gracilis microcirculations), membrane potential averages approximately −40 mV in vivo (Stekiel et al., 1991). In the cheek pouch microcirculation, membrane potential of endothelium and vascular smooth muscle was found to be −36 and −35 mV, respectively (Welsh and Segal, 1998).
Many measurements of membrane potential have been made in cerebral vessels in vitro, and the values that have been obtained under baseline conditions have ranged from approximately −35 to −75 mV. This wide range of values was obtained under a variety of conditions (quiescent vessels, vessels with pharmacologically induced tone, pressurized arteries, etc.). We now know that intravascular pressure is an important determinant of resting membrane potential. For example, cerebral vascular smooth muscle depolarizes in response to increases in intravascular pressure in vitro (Brayden and Nelson, 1992; Harder, 1984; Harder et al., 1989; Knot and Nelson, 1995, 1998). Examination of data from cerebral arteries that have been cannulated and pressurized to physiologic levels in vitro reveals that the range of values for membrane potential under resting conditions is more narrow (−36 to −54 mV), and all but two studies report values between −36 and −46 mV (an average of −43 mV) (Brayden and Nelson, 1992; Brayden and Wellman, 1989; Dietrich and Dacey, 1994; Harder, 1984; Harder et al., 1987, 1989; Kauser et al., 1990; Knot and Nelson, 1995, 1998; Nelson et al., 1995, 1997; Zimmerman et al., 1997). Isolated parenchymal arterioles (resting diameter of approximately 44 μm) are the smallest vessels from the brain in which membrane potential has been measured in a pressurized preparation (Dietrich and Dacey, 1994). The value for membrane potential obtained in these arterioles (−38 mV) is similar to that measured in larger arteries under similar conditions. Thus, although measurements of membrane potential have not been made in cerebral blood vessels in vivo, the typical values obtained in pressurized cerebral arteries and arterioles studied in vitro are similar to those measured in other vascular beds in vivo (Stekiel et al., 1991; Welsh and Segal, 1998).
As discussed earlier, resting membrane potential in vascular smooth muscle under in vitro conditions can vary considerably. This is noteworthy as the level of resting membrane potential may be an important determinant of responses to vasoactive stimuli. For example, the magnitude of hyperpolarization of vascular smooth muscle in response to acetylcholine (the classic endothelium-dependent agonist) depends on the resting membrane potential (Chataigneau et al., 1998a). The level of resting membrane potential may also influence the effectiveness of some inhibitors of K+ channels (Frey et al., 1998).
A wide variety of vasoactive stimuli have been shown to alter membrane potential of cerebral blood vessels in vitro (Fig. 2). These stimuli include receptor-mediated agonists, second messengers, changes in pH, and synthetic openers of K+ channels (substances such as cromakalim). In some vessels, inhibitors of K+ channels depolarize vascular smooth muscle as well. A few substances (norepinephrine and morphine) produce hyperpolarization or depolarization depending on the preparation studied (Fallgren et al., 1990; Harder et al., 1981; Harder and Madden, 1984; Waters and Harder, 1983).
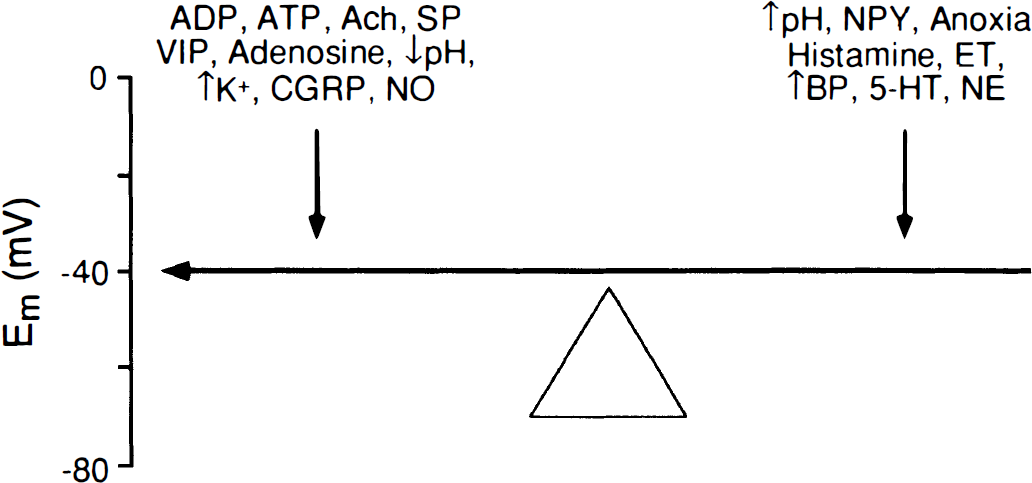
Effects of several vasoactive stimuli known to alter membrane potential in cerebral blood vessels. Hyperpolarization of vascular smooth muscle has been observed in response to modest elevations in extracellular K+ (Knot et al., 1996), adenosine diphosphate (ADP), adenosine triphosphate (ATP) (Chen and Suzuki, 1991; Brayden and Wellman, 1989), vasoactive intestinal peptide (VIP) (Standen et al., 1989), calcitonin gene-related peptide (CGRP) (Saito et al., 1989), adenosine (Nagao et al., 1996), substance P (Petersson et al., 1995), nitric oxide (NO) (McPherson and Stork, 1992; Rand and Garland, 1992; Zimmerman et al., 1997), acetylcholine (Brayden, 1990; Brayden and Wellman, 1989; Nishiye et al., 1989; Standen et al., 1989; Yamakawa et al., 1997), and acidosis (reduced pH) (Dietrich and Dacey, 1994). In contrast, increases in intravascular pressure (blood pressure, BP) (Brayden and Nelson, 1992; Harder, 1984; Harder et al., 1989; Knot and Nelson, 1995, 1998), serotonin (5-HT) (Brayden and Wellman, 1989; Clark and Garland, 1993; Fujiwara et al., 1982; Garland, 1987, 1989; Harder, 1980; Harder et al., 1981; Harder and Waters, 1983), anoxia (Nagao and Vanhoutte, 1993), endothelin (ET) (Jansen et al., 1990; Kauser et al., 1990; Salter and Kozlowski, 1998), alkalosis (increased pH) (Dietrich and Dacey, 1994; Harder and Madden, 1985), neuropeptide Y (NPY) (Abel and Han, 1989; Fallgren et al., 1990), histamine (Fallgren et al., 1990), and norepinephrine (NE) (Fallgren et al., 1990; Garland, 1989; Harder et al., 1981) have been reported to depolarize cerebral vessels.
Although membrane potential is an important determinant of vascular tone (Fig. 1), it should also be recognized that some vasoactive stimuli are capable of producing relaxation or contraction of cerebral blood vessels with no apparent change in membrane potential. For example, nitric oxide (NO) can produce marked relaxation of some blood vessels without changing membrane potential (Brayden, 1990; Plane and Garland, 1993, 1994; Yamakawa et al., 1997). In addition, adenosine diphosphate (Brayden, 1991), the calcium ionophore A23187 (Nishiye et al., 1989), papaverine (Fujioka, 1984), nicardipine (Fujiwara and Kuriyama, 1983), thromboxane A2 (Nishiye et al., 1989; Fujioka et al., 1986), neuropeptide Y (Brayden and Conway, 1988), and prostaglandin F2α (Fujiwara and Kuriyama, 1984) all have been reported to alter tone without altering membrane potential of cerebral arteries.
METHODS COMMONLY USED TO STUDY K+ CHANNELS
Several approaches are used routinely to study K+ channels in blood vessels. Each method has distinctive strengths and provides useful information that often cannot be obtained with any other method. Each method also has some potential limitations. One method involves the recording of current passing through individual ion channels using patch-clamp approaches. This is the most direct measurement of K+-channel activity. This method can be insightful in establishing the presence or absence of specific K+ channels, as well as defining variables such as channel density and electrophysiologic characteristics. However, the approach generally requires enzymatic digestion of vessels to obtain isolated myocytes. This procedure may injure cells, particularly extracellular and surface proteins. A recent extension of the patchclamp method has been applied to relatively intact cerebral arterioles (Quinn and Beech, 1998). With this approach, the extent of enzymatic digestion of tissue is minimized, perhaps resulting in a closer approximation of the physiologic situation.
The fact that patch-clamping is typically done in isolated myocytes may influence the outcome of some studies. An example of this is experiments that examine effects of NO on K+ channels. There is evidence that effects of NO and cyclic guanosine monophosphate (cGMP) (produced by soluble guanylate cyclase in response to NO) (Fig. 3) on activity of K+ channels are much greater when endogenous levels of NO and cGMP are low, such as in the absence of endothelium or in the presence of inhibitors of NO synthase (see discussion below). Because isolated myocytes have no associated endothelium, NO and cGMP levels in these cells are necessarily low, so measurements are made in a system that may exhibit enhanced responses to NO. Under such conditions, it may be easier to observe positive effects of NO on K+ channels. This limitation may explain, in part, why positive effects of NO on activity of K+ channels are seen fairly consistently with patch-clamping of isolated myocytes but less frequently in intact cerebral vessels.
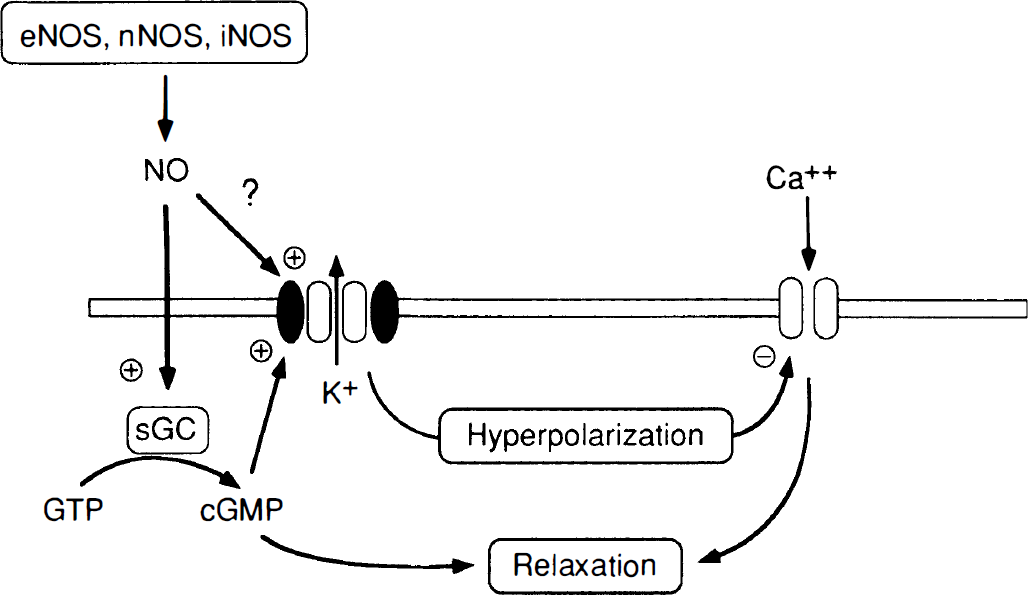
Mechanisms of relaxation of cerebral vascular smooth muscle in response to nitric oxide (NO). Nitric oxide is produced by three enzyme isoforms: endothelial (eNOS), neuronal (nNOS), and inducible (iNOS) NO synthase. Nitric oxide activates soluble guanylate cyclase (sGC), which converts guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). Activation of K+ channels by cGMP (or possibly by direct effects of NO) results in K+ efflux from the cell, hyperpolarization of the cell membrane, and closure of voltage-gated calcium (Ca2+) channels. Closure of Ca2+ channels reduces intracellular levels of Ca2+, producing relaxation of vascular smooth muscle.
A second electrophysiologic approach to study K+ channels is measurement of membrane potential in intact but isolated vascular rings or segments. This approach is also used in isolated and pressurized cerebral arteries. Studies using vascular segments allow some cell-cell interactions to occur as they would normally. A major strength of this method is that it allows simultaneous measurements of membrane potential and vascular diameter (or tone). Potential limitations of this approach include the fact that the majority of measurements have been made in nonpressurized vessels (which may be less physiologic). Even when arteries are studied under pressurized conditions, however, the vessels are almost never studied in the presence of blood flow (blood flow is probably an additional determinant of membrane potential), and no in vitro studies of cerebral vessels have included the use of pulsatile pressure and blood flow as occurs in vivo.
A third major approach in the study of K+ channels is to measure changes in tone or diameter of cerebral vessels in response to activators or inhibitors of K+ channels. Many of these studies are performed in vivo (some include measurements of cerebral blood flow rather than changes in diameter of blood vessels) and thus have the strength of being the most directly relevant to normal regulation of cerebral blood flow. This approach may provide insight into the functional importance of a particular K+ channel. A potential limitation exists, however, in that multiple cell types are present and other mechanisms that influence vascular tone, such as endothelium-dependent or neurohumoral mechanisms, are intact in vivo. Thus, activators and inhibitors of K+ channels may potentially affect cells other than vascular smooth muscle, resulting in indirect effects on vascular tone and complicating interpretation of findings. Membrane potential is not generally measured, but it is assumed to change in response to modulators of K+ channels in vivo. In addition, the use of high concentrations of KCl (used commonly to depolarize isolated vessels and implicate a hyperpolarization-dependent process) cannot be readily used in vivo because high concentrations of KCl cause cortical spreading depression. Despite these potential limitations, there are now many examples in which in vitro and in vivo approaches have produced results that support the same concept regarding K+ channels. Because each of these major approaches has unique strengths and limitations, we suggest that the most complete understanding of K+-channel function in cerebral blood vessels will be obtained by complementary in vitro and in vivo approaches.
The most recently developed approach for investigation of K+ channels involves studies at the molecular level (see discussion below). Very little data have been obtained regarding regulation of K+ channels at the gene and protein levels in the cerebral circulation (see Liu et al., 1998, for the single example). Such information will be essential to obtain a complete picture of mechanisms that control K+-channel expression, distribution, and regulation.
TYPES OF K+ CHANNELS PRESENT IN CEREBRAL BLOOD VESSELS
Four types of K+ channels have been described in cerebral blood vessels. These are (1) calcium-activated (Brayden and Nelson, 1992; Nelson and Quayle, 1995; Song and Simard, 1995), (2) ATP-sensitive (Kleppisch and Nelson, 1995; Standen et al., 1989), (3) voltage-dependent (also called delayed rectifier) (Knot and Nelson, 1995; Nelson and Quayle, 1995), and (4) inwardly rectifying K+ channels (Knot et al., 1996; Quayle et al., 1993, 1997). We will discuss each of these channels individually.
Calcium-activated K+ channels
As the name indicates, calcium-activated K+ channels are activated by increasing levels of intracellular calcium. This type of K+ channel may also be activated by membrane depolarization, although this mechanism also requires calcium at physiologic membrane potentials. Calcium-activated K+ channels are thought to be the most abundant in vascular smooth muscle, with up to 104 channels estimated to be present per cell (Nelson and Quayle, 1995).
Activity of calcium-activated K+ channels can be inhibited using tetraethylammonium ion (TEA). Concentrations of TEA of less than or equal to 1 mmol/L are thought to be selective for the calcium-activated K+ channel (Kitazono et al., 1995; Nelson and Quayle, 1995). Calcium-activated K+ channels are also inhibited by charybdotoxin or iberiotoxin, with the latter toxin thought to be highly selective (Kitazono et al., 1995; Nelson and Quayle, 1995) (Fig. 4). Activity of this ion channel is not inhibited by commonly used concentrations of glibenclamide or relatively low concentrations of barium ion (used to inhibit ATP-sensitive and inwardly rectifying K+ channels, respectively) (Nelson and Quayle, 1995). In contrast to pharmacologic inhibitors, discovery of selective openers of calcium-activated K+ channels has been slow. NS-1619 has been used in several studies as an activator of calcium-activated K+ channels. Although NS-1619 produces relaxation of cerebral arteries through a mechanism that may partially involve opening of calcium-activated K+ channels, this compound has other vasoactive effects, including inhibition of calcium channels (Holland et al., 1996a), that may complicate interpretation of experimental findings.
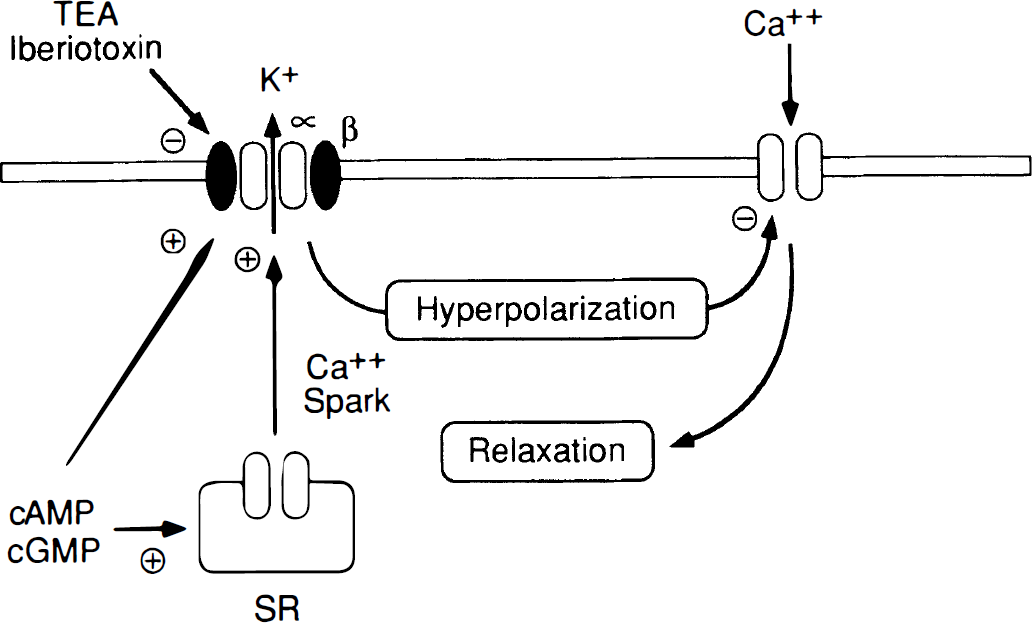
Summary of mechanisms by which calcium-activated K+ channels produce relaxation of vascular smooth muscle. Calcium-activated K+ channels are composed of α- and β-subunit proteins (see text). In addition to activation by intracellular calcium (Ca2+) and membrane depolarization (not shown), calcium-activated K+ channels may be activated by cyclic adenosine monophosphate (cAMP) or cGMP. Calcium sparks from sarcoplasmic reticulum (SR), produced in response to cAMP or cGMP, may also activate this ion channel. These K+ channels are inhibited by tetraethylammonium (TEA) or iberiotoxin. Activation of K+ channels results in K+ efflux from the cell, hyperpolarization of the cell membrane, and closure of voltage-gated Ca2+ channels. Closure of Ca2+ channels reduces intracellular levels of Ca2+, producing relaxation of vascular smooth muscle.
An important function of calcium-activated K+ channels may be to act as a buffering system in vascular smooth muscle to limit contraction. The best-studied example of this mechanism relates to the myogenic response. Elevations in arterial pressure produce membrane depolarization, increases in intracellular calcium levels, and vasoconstriction (Brayden and Nelson, 1992; Knot and Nelson, 1998; Knot et al., 1998). Calcium-activated K+ channels are activated in response to membrane depolarization and increases in intracellular calcium (including calcium sparks), resulting in hyperpolarization, which acts as a negative feedback mechanism to limit vasoconstriction (Brayden and Nelson, 1992; Knot and Nelson, 1998; Knot et al., 1998; Nelson et al., 1995).
In addition to modulating vasoconstrictor responses, calcium-activated K+ channels appear to play a major role as a mediator of vasodilation (see below) (Fig. 4). Activity of calcium-activated K+ channels also appears to influence resting tone of cerebral vessels, particularly in larger vessels. In cerebral arteries, inhibitors of calcium-activated K+ channels produce contraction in vitro (Brayden and Nelson, 1992; Geary et al., 1997; Gokina et al., 1996) and in vivo (Fujii et al., 1991; Sobey and Faraci, 1997b). In cerebral arterioles in vivo, these same inhibitors have no effect on vessel diameter or produce very modest vasoconstriction (reviewed in Faraci and Heistad, 1998). Thus, one interpretation of these findings is that the influence of calcium-activated K+ channels on resting tone varies along the vascular tree and may be more important in cerebral arteries than in cerebral microvessels. In addition, recent pharmacologic and patchclamp evidence suggest that calcium-activated K+ channels are silent in small arterioles outside the brain under basal conditions (Jackson and Blair, 1998). These data suggest that calcium-activated K+ channels in very small arterioles may have a relatively low calcium sensitivity. Because of this characteristic, these ion channels may not be active at rest in the smaller microvessels (Jackson and Blair, 1998).
A novel mechanism related to the activation of calcium-activated K+ channels in cerebral arteries involves calcium ‘sparks’ (Fay, 1995; Nelson et al., 1995). Calcium sparks are localized elevations in intracellular calcium caused by ryanodine-sensitive calcium release events (Nelson et al., 1995). These subcellular calcium sparks are triggered by activation of sarcoplasmic reticulum calcium-release channels that are in close proximity to calcium-activated K+ channels in the cell membrane (Fay, 1995) (Fig. 4). Thus, regulation of the frequency or amplitude of calcium sparks may be an important mechanism to control local activation of calcium-activated K+ channels and thus mediate vasodilation and modulate vasoconstriction (Nelson et al., 1995; Porter et al., 1998). For example, activators of protein kinase C produce contraction of cerebral arteries. At least part of the mechanism that produces this effect may involve a decrease in frequency of calcium sparks in cerebral vascular smooth muscle resulting in closure of calcium-activated K+ channels and depolarization (Bonev et al., 1997).
ATP-sensitive K+ channels
The characteristics and functional importance of ATP-sensitive K+ channels in blood vessels has been reviewed in detail recently (Quayle et al., 1997). One key characteristic of these ion channels is that their activity may reflect the metabolic state of the cell. As the name indicates, these K+ channels are sensitive to intracellular ATP, which inhibits channel activity (Nelson and Quayle, 1995). Dissociation of ATP from the channel results in channel opening and membrane hyperpolarization. Other metabolically related stimuli, including reductions in Po2 or pH, also open the channel and produce vasorelaxation (Quayle et al., 1997) (Fig. 5). It is estimated that a few hundred ATP-sensitive K+ channels are present per cell in arteries (Quayle et al., 1997). Although this number is much less than that for calcium-activated K+ channels, recall that opening of only a few K+ channels is sufficient to alter membrane potential (see above).
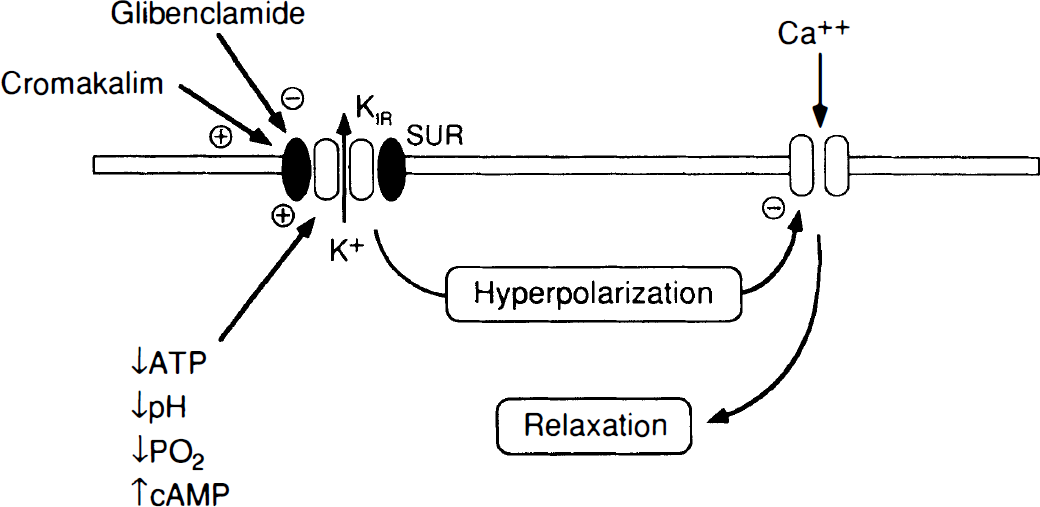
Summary of mechanisms by which ATP-sensitive K+ channels produce relaxation of vascular smooth muscle. ATP-sensitive K+ channels are composed of subunit proteins-an inwardly rectifying K+ channel (KIR) and a sulfonylurea receptor (SUR). These K+ channels may be activated by several stimuli including reductions in intracellular ATP, pH, or Po2, or increases in cAMP. Synthetic “K+-channel openers” such as cromakalim also activate this ion channel. ATP-sensitive K+ channels are inhibited by glibenclamide. Activation of K+ channels results in K+ efflux from the cell, hyperpolarization of the cell membrane, and closure of voltage-gated Ca2+ channels. Closure of Ca2+ channels reduces intracellular levels of Ca2+, producing relaxation of vascular smooth muscle.
Activity of ATP-sensitive K+ channels is inhibited by sulfonylureas, such as glibenclamide (Kitazono et al., 1995; Kleppisch and Nelson, 1995; Nelson and Quayle, 1995; Standen et al., 1989) (Fig. 5). Glibenclamide appears to be a selective inhibitor (in relation to effects on other K+ channels) of ATP-sensitive K+ channels at the most commonly used concentrations (⩽3 μmol/L), and has been used widely to examine the functional role of these ion channels in cerebral vessels. The selectivity and mechanism of action of glibenclamide has been reviewed in detail (Quayle et al., 1997).
Unlike the situation for other K+ channels, there are many pharmacologic activators of ATP-sensitive K+ channels (“K+-channel openers”) (Kitazono et al., 1995) (Fig. 5). These activators of K+ channels (levcromakalim, pinacidil, and nicorandil, for example) produce a hyperpolarization of cerebral arteries (Edwards et al., 1996; Harder et al., 1987; Kleppisch and Nelson, 1995; Nagao et al., 1996; Petersson et al., 1995; Plane and Garland, 1993) that is inhibited by glibenclamide (Edwards et al., 1996; Kleppisch and Nelson, 1995; Nagao et al., 1996; Plane and Garland, 1993). There is now a large body of data regarding functional effects of activators of ATP-sensitive K+ channels in cerebral blood vessels. Activators of ATP-sensitive K+ channels produce relaxation of cerebral arteries and arterioles in vitro (reviewed in Faraci and Heistad, 1998). This same response is present in human cerebral arteries (Hempelmann et al., 1995; Petersson et al., 1995; Thorin et al., 1998). Activators of ATP-sensitive K+ channels cause dilation of the basilar artery and cerebral arterioles in vivo (reviewed in Faraci and Heistad, 1998). Furthermore, intraparenchymal injection of cromakalim produces a glibenclamide-sensitive increase in local cerebral blood flow (Reid et al., 1995). Thus, there is strong evidence that ATP-sensitive K+ channels are present and functional in both large cerebral arteries and the cerebral microcirculation. Because glibenclamide does not produce depolarization or constriction of cerebral arteries in the vast majority of studies (reviewed in Faraci and Heistad, 1998), it seems unlikely that activity of ATP-sensitive K+ channels modulates resting tone.
Voltage-dependent K+ channels
Like calcium-activated K+ channels, voltage-dependent K+ channels are activated in response to membrane depolarization, but this process occurs independent of the intracellular calcium concentration. Activity of voltage-dependent K+ channels can be inhibited fairly selectively using low concentrations of 4-aminopyridine (<1 mmol/L) (Knot and Nelson, 1995; Nelson and Quayle, 1995. Because both voltage-dependent and calcium-activated K+ channels are activated by depolarization, 4-aminopyridine can be used to distinguish responses mediated by either channel. Tetraethylammonium is a poor inhibitor of voltage-dependent K+ channels unless very high concentrations are used (Nelson and Quayle, 1995). Voltage-dependent K+ channels can also be inhibited by dendrotoxin (Kitazono et al., 1995). The estimated number of voltage-dependent K+ channels per cell in arteries is about 103 (Nelson and Quayle, 1995).
Compared with the two K+ channels discussed previously, much less is known about the functional importance of voltage-dependent K+ channels. 4-Aminopyridine depolarizes cerebral vascular smooth muscle and constricts cerebral arteries (Knot and Nelson, 1995), suggesting that activity of voltage-dependent K+ channels influences resting cerebral vascular tone. These K+ channels are also activated by increases in arterial blood pressure (Knot and Nelson, 1995). Recent studies suggest that activation of voltage-dependent K+ channels may contribute to mechanisms that produce cerebral vasorelaxation in response to NO and endothelium-derived hyperpolarizing factor (EDHF) (Dong et al., 1998; Petersson et al., 1997a).
Inwardly rectifying K+ channels
Known properties of inwardly rectifying K+ channels in cerebral arteries are described in detail by Quayle et al. (1997). These K+ channels are inhibited by relatively low concentrations of external barium ion. Barium is considered to be the most effective and selective inhibitor of inwardly rectifying K+ channels (Quayle et al., 1997). 4-Aminopyridine and TEA are relatively weak inhibitors, whereas glibenclamide and charybdotoxin produce almost no inhibition (Nelson and Quayle, 1995).
Low concentrations of barium produce contraction of cerebral arteries in vitro suggesting that activity of inwardly rectifying K+ channels affects resting tone (Johnson et al., 1998; Knot et al., 1996). Although it is known that high concentrations of barium constrict cerebral arterioles in vivo (Rosenblum, 1985), we are not aware of in vivo studies that have used concentrations of barium thought to be selective for inwardly rectifying K+ channels. The estimated number of inwardly rectifying K+ channels per cell in arteries is estimated at a few hundred (Nelson and Quayle, 1995).
MOLECULAR BIOLOGY
Studies using molecular biology are revealing a large diversity in the subtypes of K+ channels that are expressed in vascular smooth muscle. Potassium channels are often composed of multiple gene products and complexes of proteins. For example, calcium-activated K+ channels consist of α- and β-subunit proteins (Kaczorowski et al., 1996; Tseng-Crank et al., 1996). The α subunit forms the ion-conduction pore of the channel and the β subunit is a membrane-spanning protein that acts as a regulatory component of the channel (Fig. 4). The β subunit is important in conferring calcium sensitivity to the channel (Jan and Jan, 1997) as well as sensitivity to inhibition by TEA and iberiotoxin (Kaczorowski et al., 1996; McCobb et al., 1995).
ATP-sensitive K+ channels are also a complex of proteins consisting of a pore-forming subunit (a member of the inwardly rectifying K+-channel family) (KIR) and the sulfonylurea receptor (SUR) (Quayle et al., 1997; Trapp and Ashcroft, 1997) (Fig. 5). The pore-forming subunit also serves as the sensor for ATP (Trapp and Ashcroft, 1997). As the name indicates, the sulfonylurea receptor is responsible for glibenclamide sensitivity of the channel and is probably the site that confers sensitivity of ATP-sensitive K+ channels to openers such as pinacidil and diazoxide (Quayle et al., 1997; Trapp and Ashcroft, 1997). In total, four sulfonylurea receptor subunits plus four pore-forming subunits are required to make a functional ATP-sensitive K+ channel (Trapp and Ashcroft, 1997).
More than one type of sulfonylurea receptor and more than one type of inwardly rectifying K+ channel are now known to exist (Babenko et al., 1998; Yokoshiki et al., 1998). There is also heterogeneity in the properties of ATP-sensitive K+ channels in different cell types, and this heterogeneity may be caused in part by differences in subunit proteins that make up the channel (Babenko et al., 1998). For example, ATP-sensitive K+ channels in vascular smooth muscle seem to be composed of SUR2B with either KIR6.1 or KIR6.2, whereas other tissues such as heart and pancreas may express other combinations of KIR and SUR subtypes (Babenko et al., 1998; Isomoto et al., 1996; Trapp and Ashcroft, 1997; Yokoshiki et al., 1998). It is noteworthy that the sulfonylurea receptor can couple to different types of inwardly rectifying K+ channels (Ammana et al., 1996). This characteristic may confer sensitivity to glibenclamide in K+ channels that are not ATP-sensitive. Thus, the presence of glibenclamide sensitivity implicates a K+ channel but does not necessarily indicate the presence of an ATP-sensitive K+ channel (Quast, 1996). In contrast to the calcium-activated and ATP-sensitive K+ channels, which are composed of protein complexes within the cell membrane, voltage-dependent K+ channels consist of a pore-forming α subunit and a cytoplasmic regulatory β subunit (Rettig et al., 1994). Multiple α and β subunits for voltage-dependent K+ channels may be expressed in vascular smooth muscle (Yuan et al., 1998b).
Expression of K+ channels may not be homogeneous throughout the circulation. For example, the Kv1.2 and Kv2.1 subtypes of voltage-dependent K+ channels are expressed to a greater extent in large arteries in lung, whereas Kv1.3 is present only in pulmonary resistance arteries (Patel et al., 1997). To our knowledge, there are no data at the molecular level regarding the regional or segmental distribution of K+ channels within the cerebral circulation.
Relatively little is known about factors that regulate expression of K+-channel proteins in vascular smooth muscle, or about the promoter regions for these genes. Recent evidence suggests that vascular expression of K+-channel proteins may change with hypertension (Liu et al., 1998; Yuan et al., 1998a) and hypoxia (Wang et al., 1997). For example, mRNA and protein for voltage-dependent K+ channel subunits (Kv1.2 and Kv 1.5) are decreased in response to hypoxia (Wang et al., 1997).
VASOACTIVE STIMULI
Nitric oxide
Nitric oxide plays a major role in regulation of cerebral vascular tone (reviewed in Faraci and Heistad, 1998). Nitric oxide is a potent dilator of cerebral blood vessels (Faraci and Heistad, 1998), and there are three sources of NO in brain (Fig. 3). Two of these sources, endothelial and neuronal isoforms of NO synthase (eNOS and nNOS, respectively), are expressed in a constitutive manner (Faraci and Brian, 1994). The third source is the inducible isoform of NO synthase (iNOS). Inducible NO synthase is not expressed under normal conditions in most cells but can be expressed in all major cell types (endothelium, vascular smooth muscle, neurons, and glia) in response to a variety of stimuli (primarily proinflammatory stimuli) (Faraci and Brian, 1994; Faraci and Heistad, 1998).
In general, there are two ways in which NO can produce relaxation of vascular smooth muscle-soluble guanylate cyclase-dependent or soluble guanylate cyclase-independent mechanisms. Both mechanisms can potentially involve activation of K+ channels (Fig. 3). Soluble guanylate cyclase, a heme-containing protein with a high affinity for NO, is responsible for the enzymatic conversion of guanosine-5′-triphosphate to cyclic guanosine-3′,5′-monophosphate (cGMP) (Hobbs, 1997) (Fig. 3). Submicromolar concentrations of NO evoke large increases in activity of soluble guanylate cyclase. For this concentration range of NO, the major cellular response is thought to be activation of soluble guanylate cyclase (Lincoln et al., 1996; Mayer and Hemmens, 1997). This mechanism occurs in cerebral blood vessels as well, where NO and NO donors produce marked increases in cGMP levels and vasorelaxation (Brian et al., 1994; Homayoun et al., 1989; Katusic et al., 1989; Kim et al., 1992; Onoue and Katusic, 1997, 1998a, 1998b; Pearce et al., 1994; Yang et al., 1991a). Several lines of evidence, including studies using inhibitors of soluble guanylate cyclase, suggest that relaxation of cerebral blood vessels (large arteries and microvessels) in response to NO is mediated predominantly by soluble guanylate cyclase (Dong et al., 1998; Jiang et al., 1998; Meng et al., 1998; Onoue and Katusic, 1998b; Sobey and Cocks, 1998; Sobey and Faraci, 1997a; Yang and Iadecola, 1998) (Fig. 3).
Although soluble guanylate cyclase appears to mediate the majority of cerebral vasorelaxation in response to NO, there is a component of the response that is not abolished by inhibitors of soluble guanylate and thus is presumably mediated by other mechanisms. These latter mechanisms may include direct activation of K+ channels by NO (Bolotina et al., 1994) (Fig. 3), inhibition of 20-hydroxyeicosatetraenoic acid formation (suggested to occur in kidney blood vessels) (Alonso-Galicia et al., 1997), and inhibition of calcium-stimulated chloride current resulting in hyperpolarization (Zhang et al., 1997).
The role of K+ channels in mediating relaxation of cerebral blood vessels to NO has been somewhat difficult to fully define. Under some conditions and in some blood vessels, activation of K+ channels appears to contribute to vasorelaxation in response to NO (Fig. 3). However, there are examples in which K+ channels do not appear to be involved in the vasodilator effects of NO.
In patch-clamp studies of isolated myocytes from cerebral arteries, NO, NO donors, and cGMP increase activity of calcium-activated K+ channels (Hoang and Mathers, 1998; Holland et al., 1996b, 1997a, 1997b, 1997c; Robertson et al., 1993). The study by Robertson et al. (1993) provided the first direct evidence that NO can activate calcium-activated K+ channels in cerebral vascular smooth muscle. This same response was not observed in isolated patch preparations (in which cytosolic enzymes are not present), providing some evidence against direct effects of NO on this ion channel (Holland et al., 1997b, 1997c). Although these studies provided evidence that NO is capable of activating K+ channels under some conditions, the technique used did not provide insight into whether this mechanism is functionally important in intact cerebral vessels.
Hyperpolarization in response to NO has been observed in cerebral blood vessels. For example, NO donors produce modest hyperpolarization in some preparations (Zimmerman et al., 1997), but the effect is sometimes only seen in response to very high concentrations of NO (McPherson and Stork, 1992; Rand and Garland, 1992). Nitric oxide may also activate calcium sparks, which open local calcium-activated K+ channels (Porter et al., 1998). Hyperpolarization in response to NO has also been observed in the carotid artery (Cohen et al., 1997; Corriu et al., 1996a). In addition, vasorelaxation in response to NO, NO donors, or cGMP can be attenuated by inhibitors of K+ channels (inhibitors of calcium-activated K+ channels in most studies) consistent with a role for K+ channels in NO-mediated hyperpolarization (Dong et al., 1998; Jiang et al., 1998; Onoue and Katusic, 1997, 1998a, 1998b; Paterno et al., 1996; Sobey and Faraci, 1997b). It should be noted however, that many of these findings were observed using vessels without endothelium (Dong et al., 1998; Jiang et al., 1998; Onoue and Katusic, 1997, 1998a, 1998b) or in intact vessels in the presence of inhibitors of NO synthase (Murphy and Brayden, 1995; Sobey and Faraci, 1997b) or soluble guanylate cyclase (Onoue and Katusic, 1998b). For example, charybdotoxin produced modest inhibition of vasorelaxation in response to DEA-NONOate (an NO donor), and the inhibitory effect was more pronounced after inhibition of soluble guanylate cyclase (Onoue and Katusic, 1998b). These findings provide some evidence that high concentrations of NO may activate K+ channels independent of activation of soluble guanylate cyclase and production of cGMP (Onoue and Katusic, 1998b) (Fig. 3).
There are also negative data in regards to effects of NO on K+ channels. In some studies, for example, NO has no effect on membrane potential of cerebral arteries but produces marked relaxation (Brayden, 1990; Murphy and Brayden, 1995; Plane and Garland, 1993, 1994; Yamakawa et al., 1997). There are also examples in which inhibitors of calcium-activated K+ channels do not affect vasorelaxation in response to NO (Armstead, 1997c; Sobey et al., 1996; Sobey and Faraci, 1997b; Taguchi et al., 1995b; Wellman and Bevan, 1995).
Basal production of NO exerts a tonic influence on resting vascular tone throughout the brain, but particularly in large cerebral arteries (Faraci, 1991; Faraci and Heistad, 1998). Part of this basal vasodilator effect may include an effect of NO on K+ channels. Recent measurements indicate that membrane depolarization occurs in the middle cerebral artery in response to inhibition of NO synthase, suggesting that basal NO produces tonic hyperpolarization (Zimmerman et al., 1997; Peng et al., 1998). Based on the majority of available data, we hypothesize that the following mechanism may be present. Nitric oxide can activate K+ channels under conditions in which NO production or cGMP levels are low. Thus, the basal dilator influence of NO on cerebral vessels may be mediated, in part, by activation of K+ channels. Activation of K+ channels may also contribute to vasorelaxation in response to additional NO (levels of NO greater than those produced under basal conditions) in some, but not all, preparations. Exogenous NO may therefore be more likely to activate K+ channels after endothelial removal, during inhibition of NO synthase, and perhaps during pathophysiologic conditions associated with impairment of NO signaling.
Endothelium-dependent hyperpolarizing factor
In addition to NO, the endothelium can produce other relaxing factors including dilator prostanoids and EDHF. Some investigators use the term “EDHF” when referring to an endothelium-derived relaxing factor that is not NO or a product of cyclooxygenase. Recent evidence suggests that this assumption has a potential limitation because it can be difficult to completely inhibit production of NO in endothelium (Cohen et al., 1997). An alternative definition is that any vasoactive factor produced by endothelium and which produces hyperpolarization of vascular smooth muscle (including NO) can be classified as an EDHF. Although there may be several such mediators in cerebral blood vessels, it is widely believed that the most likely mechanism of EDHF-mediated hyperpolarization of vascular smooth muscle is by activation of K+ channels (Cohen and Vanhoutte, 1995; Garland et al., 1995; Mombouli and Vanhoutte, 1997).
Several studies have described hyperpolarization of cerebral arteries in response to endothelium-dependent stimuli including acetylcholine (Brayden, 1990; Brayden and Wellman, 1989; Nishiye et al., 1989; Standen et al., 1989; Yamakawa et al., 1997) and substance P (Petersson et al., 1995). Not all endothelium-dependent agonists hyperpolarize cerebral vessels however, as A23187 and adenosine diphosphate can produce marked vasorelaxation but no hyperpolarization (Brayden, 1991; Nishiye et al., 1989).
To examine the functional importance of EDHF-mediated mechanisms of endothelium-dependent relaxation, effects of inhibitors of K+ channels on responses of cerebral blood vessels to endothelium-dependent agonists have been tested. Unfortunately, the findings obtained in these studies have been variable, and there is currently no consensus regarding the functional importance of EDHF in brain. When an EDHF has been implicated, there is also no consensus regarding which K+ channels mediate the vasoactive effect. For example, relaxation of cerebral vessels in response to acetylcholine has been reported to be inhibited by glibenclamide (Brayden, 1990; Faraci et al., 1994; Standen et al., 1989), apamin (an inhibitor of small conductance calcium-activated K+ channels (Yamakawa et al., 1997), or inhibitors of calcium-activated K+ channels (Dong et al., 1998; Petersson et al., 1997a; Wellman and Bevan, 1995). In some studies, the combination of charybdotoxin and apamin was much more effective in reducing responses to acetylcholine than either inhibitor alone (Corriu et al., 1996a; Petersson et al., 1997a). These differences may occur because of regional, segmental, or species differences in the models studied. Another inconsistency is that some, but not all, studies have been performed in the presence of inhibitors of cyclooxygenase and NO synthase.
A key point in the characterization of EDHF relates to the suggestion that EDHF may only be produced after inhibition of NO synthase. Recent evidence suggests that NO may inhibit formation or activity of EDHF (Bauersachs et al., 1996). Thus, EDHF may have a limited role under normal conditions, but may become functionally much more important after pharmacologic inhibition of NO synthase, or in disease states that are associated with impairment of the NO-cGMP signaling pathway.
To assess the functional importance of a non-NO EDHF, it is therefore necessary to determine whether a significant endothelium-dependent response remains in the presence of inhibition of NO synthase. In the brain, most studies have found inhibitors of NO synthase to be very effective in reducing responses to acetylcholine along the entire vascular tree. For example, relaxations in response to acetylcholine in the carotid and basilar arteries, as well as in pial and parenchymal arterioles, are greatly reduced by inhibitors of NO synthase (reviewed in Faraci and Heistad, 1998). Increases in parenchymal blood flow in response to acetylcholine are also markedly reduced by inhibitors of NO synthase (Iadecola and Zhang, 1996; Yang and Chang, 1998). Some studies suggest that a component of endothelium-dependent relaxation of the carotid artery is mediated by a non-NO EDHF (Lischke et al., 1995a, 1995b). In contrast, other data, including studies using mice that are deficient in expression of the gene for endothelial NO synthase, suggest the entire response to acetylcholine is mediated by NO (Cohen et al., 1997; Faraci et al., 1998). Relaxation of human pial arteries in response to substance P is also largely NO mediated, but an EDHF may contribute in arteries from some individuals (Petersson et al., 1995). These findings do not exclude the possibility that NO itself acts as an EDHF (Cohen et al., 1997). Despite NO being the major mediator of vasorelaxation in response to acetylcholine in most studies, portions of the response to acetylcholine appear to be mediated by other mechanisms. For example, in basilar arteries of the guinea pig, relaxation in response to acetylcholine is not attenuated by inhibitors of NO synthase or soluble guanylate cyclase (Petersson et al., 1997a; Zygmunt et al., 1997).
Some data from vessels other than cerebral arteries suggest that one EDHF is an epoxyeicosatrienoic acid (EET), a product of cytochrome P450 metabolism of arachidonic acid. This hypothesis is controversial, however, with evidence both for (Arima et al., 1997; Bauersachs et al., 1994; Campbell et al., 1996; Lischke et al., 1995a, 1995b; Chen and Cheung, 1996; Dong et al., 1997) and against (Cohen et al., 1997; Corriu et al., 1996a, 1996b; Urakami-Harasawa et al., 1997; Van de Voorde and Vanheel, 1997; Vanheel and Van de Voorde, 1997) a role for EET as a mediator of endothelium-dependent hyperpolarization (reviewed in Mombouli and Vanhoutte, 1997).
There is relatively little information regarding the vasoactive effects and functional importance of EET in the cerebral circulation. Studies examining vasoactive effects of EET have produced mixed results. Relaxation of the middle cerebral artery has been observed in response to 5,6-EET, 8,9-EET, and 11,12-EET in vitro, and responses to 11,12-EET were inhibited by a high concentration of TEA (30 mmol/L), suggesting activation of a K+ channel was involved (Gebremedhin et al., 1992). 14,15-Epoxyeicosatrienoic acid enhances outward K+ current in isolated myocytes from cerebral microvessels consistent with activation of a K+ channel (Alkayed et al., 1996). 5,6-Epoxyeicosatrienoic acid, 8,9-EET, 11,12-EET, and 14,15-EET all produce dilation of cerebral arterioles in newborn pigs with 5,6-EET being the most active (Leffler and Fedinec, 1997). In contrast, Ellis et al. (1990) reported that several EET (8,9-EET, 11,12-EET, and 14,15-EET) have little or no effect on diameter of cerebral arterioles in vivo, with the exception of 5,6-EET. In the carotid artery, EET (5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET) do not change membrane potential or affect vascular tone in vitro (Chataigneau et al., 1998a).
A potential limitation in attempting to define the functional role of EET relates to nonspecific effects of commonly used inhibitors of the cytochrome P450 pathway (the antifungal imidazoles) (Mombouli and Vanhoutte, 1997). These substances reportedly have several nonspecific effects unrelated to inhibition of the P450 pathway, including direct inhibition of K+ channels and NO synthase (Alvarez et al., 1992; Popp et al., 1996; Rittenhouse et al., 1997; Vanheel and Van de Voorde, 1997; Van de Voorde and Vanheel, 1997; Wolff et al., 1993; Yuan et al., 1995).
Very little is known regarding potential endothelial production of EET in the cerebral circulation. In cerebral arteries in vitro, relaxation in response to acetylcholine was not inhibited by 17-octadecynoic acid, ETYA (5,8,11,14-eicosatetraynoic acid, a nonselective inhibitor of arachidonic acid metabolism), or clotrimazole, suggesting no role for cytochrome P450 metabolism of arachidonic acid (Dong et al., 1998; Petersson et al., 1997a). Leffler et al. (1997) have provided some evidence that endothelium-derived EET may contribute to dilation of cerebral arterioles during hypoxia in piglets.
It has recently been suggested that anandamide, an endogenously produced cannabinoid (derived from arachidonic acid) may be an EDHF (Randall and Kendall, 1998). Although anandamide can produce dilation of cerebral arterioles (Ellis et al., 1995), whether it functions as an EDHF in the cerebral circulation is not known. Initial evidence suggests that anandamide does not alter membrane potential and that endothelium-dependent hyperpolarization of the carotid artery is not mediated by activation of cannabinoid receptors (Chataigneau et al., 1998b; Holland et al., 1997a). Furthermore, in the basilar artery of the guinea pig, relaxation in response to acetylcholine is not inhibited by an antagonist of cannabinoid receptors, suggesting EDHF is not an endogenously produced cannabinoid (Petersson et al., 1997b).
Cyclic adenosine monophosphate
In addition to cGMP, production of cyclic adenosine monophosphate (cAMP) in vascular smooth muscle is a major second messenger system that influences vascular tone. A wide variety of vasoactive stimuli are known to activate adenylate cyclase resulting in increased production of cAMP in blood vessels. These stimuli include receptor-mediated agonists (adenosine, vasoactive intestinal polypeptide, calcitonin gene-related peptide [CGRP], prostacyclin, isoproterenol, and adrenomedullin) and forskolin (a direct activator of adenylate cyclase) (reviewed in Faraci and Heistad, 1998). Activation of K+ channels appears to be a key mechanism that mediates cAMP-dependent vasorelaxation (Figs. 4 and 5).
The concept that relaxation of cerebral blood vessels in response to stimuli that increase cAMP is mediated by activation of K+ channels is based on several lines of evidence. First, studies using patch-clamping demonstrated that isoproterenol, forskolin, and cAMP increase current passing through calcium-activated K+ channels (Song and Simard, 1995). Adenosine and CGRP increase glibenclamide-sensitive current in cerebral arteries, consistent with activation of ATP-sensitive K+ channels (Kleppisch and Nelson, 1995). Second, forskolin and cAMP increase the frequency of calcium sparks, which activate calcium-activated K+ channels in cerebral arteries (Porter et al., 1998). Relaxation of cerebral arteries in response to forskolin is inhibited by ryanodine, an inhibitor of calcium sparks (Porter et al., 1998) (Fig. 4). Third, adenosine (Nagao et al., 1996), CGRP (Saito et al., 1989), prostacyclin (Corriu et al., 1996a; Dong et al., 1998), vasoactive intestinal peptide (Standen et al., 1989), and cAMP (Chen and Suzuki, 1991) hyperpolarize cerebral arteries. Fourth, relaxation of cerebral vessels in response to forskolin (Bari et al., 1997; Dong et al., 1998; Kitazono et al., 1993a; Taguchi et al., 1995b), cAMP (Armstead, 1997b; Paterno et al., 1996; Taguchi et al., 1995b), CGRP (Armstead, 1997a; Hong et al., 1996; Kitazono et al., 1993b; Louis et al., 1996), adenosine (Armstead, 1997e; Bari et al., 1998; Paterno et al., 1996), prostacyclin (Bari et al., 1996; Dong et al., 1998; Fredricks et al., 1994), norepinephrine (Kitazono et al., 1993a), adrenomedullin (Lang et al., 1997), and pituitary adenylate cyclase-activating peptide (Armstead, 1997d) are attenuated by inhibitors of K+ channels. Fifth, relaxation of cerebral arteries in response to forskolin is inhibited by precontracting arteries with high concentrations of KCl, which prevents hyperpolarization in response to activation of K+ channels (Dong et al., 1998; Porter et al., 1998). Taking all these findings into account, there is strong evidence that relaxation of cerebral vessels to cAMP is mediated by activation of K+ channels (Figs. 4 and 5).
Potassium ion
Normal concentrations of K+ in cerebrospinal fluid or brain extracellular fluid are approximately 3 mmol/L (Davson et al., 1987). In contrast to the depolarization and contraction of vascular smooth muscle that is commonly produced by high concentrations of K+, small to moderate increases in the concentration of extracellular K+ produce membrane hyperpolarization (Fig. 2) and relaxation of cerebral arteries in vitro (Johnson et al., 1998; Knot et al., 1996) and dilation of cerebral arteries and arterioles in vivo (Fujii et al., 1990; Kuschinsky et al., 1972). Because K+ is released during neuronal activation, this mechanism may play a role in the coupling of cerebral metabolism and local blood flow.
Potassium-induced vasodilation is generally thought to be endothelium-independent and not attenuated by inhibitors of ATP-sensitive, voltage-dependent, or calcium-activated K+ channels (Johnson et al., 1998; Knot et al., 1996; McCarron and Halpern, 1990). There appears to be at least two components of the K+-induced cerebral vasodilator response (Edwards et al., 1988; McCarron and Halpern, 1990). A transient vasodilation is produced by very low concentrations of K+ (1 to 5 mmol/L above control) and is blocked by ouabain, consistent with hyperpolarization produced by activation of Na+-K+-ATPase (McCarron and Halpern, 1990). In addition, a sustained vasodilator effect occurs in response to slightly higher concentrations of K+ (7 to 16 mmol/L) and this component is blocked by barium (Edwards et al., 1988; Johnson et al., 1998; Knot et al., 1996; McCarron and Halpern, 1990). The concentration of barium needed to produce this effect (<50 μmol/L) appears to selectively block inwardly rectifying K+ channels (Edwards et al., 1988; Quayle et al., 1993), suggesting that the sustained component of vasodilation in response to K+ is mediated by activation of inwardly rectifying K+ channels (Johnson et al., 1998; Knot et al., 1996). It is believed that this latter mechanism occurs because of unusual gating properties of this ion channel, which are affected by the concentration of extracellular K+ as well as the membrane potential. Thus, a small increase in extracellular K+ leads to an increase in resting outward current through inwardly rectifying K+ channels, and hence hyperpolarization and vascular relaxation.
Hypoxia and hypercapnia
Hypoxia and hypercapnia are potent dilators in the cerebral circulation. Although it has been known for many years that these stimuli increase cerebral blood flow, mechanisms that mediate these responses have been difficult to define completely. Recent evidence suggests that activation of K+ channels may contribute to cerebral vasodilation during hypoxia and hypercapnia.
Relaxation of the carotid artery (Taguchi et al., 1995a), large cerebral arteries (Fredricks et al., 1994), and cerebral arterioles (Shankar and Armstead, 1995; Taguchi et al., 1994) in response to hypoxia is inhibited by glibenclamide. Increases in cerebral blood flow during hypoxia are similarly reduced by inhibitors of ATP-sensitive K+ channels (Reid et al., 1993, 1995). Thus, activation of ATP-sensitive K+ channels appears to be important in mediating responses to hypoxia. Relaxation of the middle cerebral artery in response to hypoxia in vitro is inhibited by TEA, suggesting that calcium-activated K+ channels (Gebremedhin et al., 1994) may also contribute to hypoxia-induced vasorelaxation.
Activation of K+ channels may contribute to dilation of cerebral vessels during hypercapnia, although findings related to this stimulus have been inconsistent. Some studies suggest hypercapnia (or acidosis) relaxes cerebral vessels, in part, by activation of K+ channels (Faraci et al., 1994; Kinoshita and Katusic, 1997; Kontos and Wei, 1996). For example, relaxation of the basilar artery in response to acidosis was inhibited by a high concentration of KCl, barium, or glibenclamide (Kinoshita and Katusic, 1997). Acidosis is known to hyperpolarize cerebral arterioles, consistent with activation of K+ channels (Dietrich and Dacey, 1994). In contrast, glibenclamide did not affect dilation of cerebral arterioles in response to low pH in other studies (Janigro et al., 1997; Wahl et al., 1994). Thus, the overall importance of K+ channels in mediating vasorelaxation in response to hypercapnia/acidosis is not clear.
Reactive oxygen species
Reactive oxygen species (either applied directly in the case of hydrogen peroxide or produced enzymatically using xanthine-xanthine oxidase) produce dilation of cerebral microvessels in vivo (Rosenblum, 1983; Sobey et al., 1997b; Wei et al., 1985, 1996; Yang et al., 1991a, 1991b). Both arachidonic acid and bradykinin produce dilation of cerebral arterioles that is mediated by endogenous formation of reactive oxygen species (Faraci et al., 1997; Kontos et al., 1984; Rosenblum, 1987; Sobey et al., 1997b). Because intracellular production and concentrations of reactive oxygen species have not been measured in cerebral endothelium or vascular smooth muscle in vivo, the precise contribution of each reactive oxygen species (superoxide anion, hydrogen peroxide, and hydroxyl radical) as a mediator of the vasodilation is difficult to define. However, the pharmacologic profile obtained using inhibitory agents such as superoxide dismutase, catalase, and deferoxamine suggests that the primary mediator of vasodilation in response to bradykinin and arachidonic acid is hydroxyl radical or hydrogen peroxide, depending on the species (Kontos et al., 1984, 1989, 1990; Rosenblum, 1987; Sobey et al., 1997b; Yang et al., 1991a).
Recent experiments performed both in vitro and in vivo provide functional evidence that exogenously applied and endogenously produced reactive oxygen species produce vasorelaxation that is mediated by activation of K+ channels (Faraci et al., 1997; Iida and Katusic, 1997; Sobey et al., 1997b; Wei et al., 1996) (Fig. 6) It is noteworthy that hydrogen peroxide also increases cAMP levels in the basilar artery (Iida and Katusic, 1997), suggesting that effects of hydrogen peroxide on K+ channels in large arteries may not be direct but may be mediated by this second messenger system.
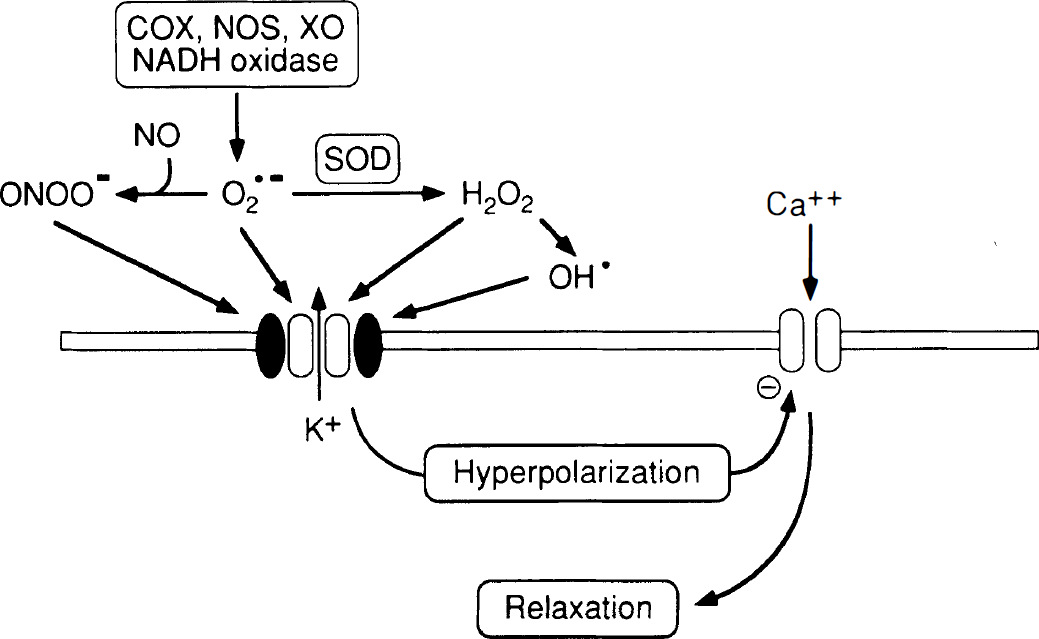
Summary of mechanisms by which reactive oxygen species (superoxide anion [O2−], hydrogen peroxide [H2O2], and hydroxyl radical [OH·]) and peroxynitrite (ONOO−) produce K+ channel-mediated relaxation of vascular smooth muscle. Potential sources of superoxide include cyclooxygenase (COX) (both COX-1 and COX-2), NO synthase (NOS) (NOS may produce superoxide in the absence of L-arginine or tetrahydrobiopterin), xanthine oxidase (XO), or reduced nicotinamide adenine dinucleotide (NADH) oxidase. Peroxynitrite is formed by the reaction of superoxide with NO. Hydrogen peroxide is produced from superoxide by superoxide dismutases (SOD). Hydrogen peroxide is degraded by catalase but can also be catalyzed to OH· radical by the Haber-Weiss reaction. Activation of K+ channels results in K+ efflux from the cell, hyperpolarization of the cell membrane, and closure of voltage-gated Ca2+ channels. Closure of Ca2+ channels reduces intracellular levels of Ca2+, producing relaxation of vascular smooth muscle.
Peroxynitrite, produced by the interaction of superoxide anion and NO (Fig. 6), is a reactive nitrogen species that also produces dilation of cerebral arterioles (Wei et al., 1996). Studies in feline pial vessels suggest that dilation in response to peroxynitrite is also mediated by activation of K+ channels (Wei et al., 1996) (Fig. 6).
PATHOPHYSIOLOGY
K+ channel-mediated effects on blood vessels are altered during pathophysiology
Several lines of evidence suggest that the function of K+ channels in cerebral blood vessels is altered in disease states. For example, relaxation of cerebral vessels in response to activators of ATP-sensitive K+ channels is impaired during chronic hypertension (Kitazono et al., 1993c) and diabetes (Mayhan, 1994; Mayhan and Faraci, 1993; Zimmerman et al., 1997) and after ischemia (Bari et al., 1996; Louis et al., 1996) or brain injury (fluid percussion injury) (Armstead, 1997a). Such changes may not be unique to ATP-sensitive K+ channels because brain injury also inhibits responses to NS-1619 and pituitary adenylate cyclase-activating peptide in the newborn pig (both produce iberiotoxin-sensitive vasodilation in this model suggesting responses are mediated by calcium-activated K+ channels) (Armstead, 1997d). In addition, function of inwardly rectifying K+ channels also appears to be impaired in cerebral arteries after ischemia with reperfusion (Marrelli et al., 1998). In contrast, responses to NS-1619 and agents thought to activate calcium-activated K+ channels (forskolin and cAMP) are not impaired by ischemia followed by reperfusion (Bari et al., 1997).
Functional effects of K+ channels in the cerebral circulation are not always impaired in disease states. For example, basal activity of calcium-activated K+ channels appears to be enhanced during chronic hypertension (Liu et al., 1998; Paterno et al., 1997). Data supporting this concept include the findings that inhibitors of calcium-activated K+ channels produce greater constriction of the basilar artery and cerebral arterioles in vivo in chronically hypertensive rats (Liu et al., 1998; Paterno et al., 1997).
The mechanisms that account for altered K+ channel-mediated vascular responses are poorly defined. One possibility is altered expression of K+-channel proteins, but very little is known regarding this potential mechanism. Recent studies have provided evidence for changes in expression of K+ channels at the mRNA and protein levels during chronic hypertension (Liu et al., 1998; Yuan et al., 1998a) and hypoxia (Wang et al., 1997). Other studies suggest that production of endothelin (Kasemsri and Armstead, 1997) and activation of protein kinase C can inhibit activity of K+ channels (Lange et al., 1997; Linde et al., 1997), including calcium-activated K+ channels driven by calcium sparks (Bonev et al., 1997). These latter findings are of interest as several pathophysiologic conditions are associated with increased production of endothelin and activation of protein kinase C.
K+ channels may be an important compensatory vasodilator mechanism
A concept that is emerging from recent studies is that K+ channels in the cerebral circulation may become functionally more important under some pathophysiologic conditions (especially conditions associated with impairment of the NO-cGMP signaling pathway). Several findings support this concept. First, NO produces hyperpolarization of the basilar artery in the presence, but not the absence, of an inhibitor of NO synthase (Murphy and Brayden, 1995). Hyperpolarization of the middle cerebral artery in response to NO is greater in vessels from diabetic compared with normal animals (Zimmerman et al., 1997). Second, inhibitors of K+ channels produce greater attenuation of relaxation of cerebral vessels in response to exogenous NO after inhibition of NO synthase or soluble guanylate cyclase than under control conditions (Onoue and Katusic, 1998b; Sobey and Faraci, 1997b). Third, the functional importance of K+ channels appears to increase in disease states associated with impairment of endothelium-dependent (NO-mediated) relaxation including subarachnoid hemorrhage (Harder et al., 1987; Sobey et al., 1996, 1997b; Zuccarello et al., 1996) and hypercholesterolemia (Najibi and Cohen, 1995). For example, inhibitory effects of charybdotoxin on vasorelaxation in response to NO are absent or modest in normal arteries but are enhanced after subarachnoid hemorrhage or in the presence of hypercholesterolemia (Najibi and Cohen, 1995; Onoue and Katusic, 1998a). Importantly, although several vasodilator mechanisms are impaired after subarachnoid hemorrhage, relaxation of the basilar artery in response to activators of potassium channels is preserved or enhanced (Ahmad et al., 1996; Harder et al., 1987; Hongo et al., 1989; Imaizumi et al., 1996; Inoue et al., 1996; Sobey et al., 1996, 1997a; Yanagisawa et al., 1998; Zuccarello et al., 1996). These findings suggest that K+ channels are potentially important therapeutic targets in disease states, including cerebral vasospasm.
In summary, several K+ channels are present in cerebral blood vessels. Multiple lines of evidence suggest these ion channels are functionally important, contributing to regulation of basal tone and as key mediators of vasodilation. The expression and functional importance of K+ channels may change in disease states. Future studies should provide insight into the molecular biology of K+ channels in the cerebral circulation as well as their potential as therapeutic targets.