
Abstract
Recent studies strongly suggest that oxidative stresses participate in ischemia/reperfusion-induced neurode-generation. In addition, heme oxygenase (HO) and major histocompatibility complex (MHC) antigens serve as functional molecules against oxidative stress and as self-recognition markers in the immune system, respectively. In this study, we examined the induction of HO and MHC antigens in the rat hippocampus after transient forebrain ischemia. The protein level of HO-1 was significantly enhanced after an episode of ischemia. After ischemia, HO-1 expression was observed early but transiently in the CA1 pyramidal neurons and later but continuously in glial cells. Glial cells expressing HO-1 were predominantly ameboid microglia, but not astrocytes. Ameboid microglia expressing HO-1 were predominantly localized with MHC class II antigens. These results indicate that (1) HO-1 expression in CA1 pyramidal neurons may be harmful, and (2) ischemia induces HO-1 in ameboid microglia that express MHC class II antigens, indicating a very specific microglial stress protein response.
One function of heme oxygenase (HO) (heme, hydrogen-donor:oxygen oxidoreductase [α-methene-oxidizing, hydroxylating], EC 1.14.99.3) is oxidation of the heme molecule in concert with NADPH-cytochrome P-450 reductase followed by the specific cleavage of heme into biliverdin, carbon monoxide, and iron (Maines, 1988). Recent studies have indicated that there are at least two isozymes of HO, the inducible type (HO-1) and the constitutive type (HO-2) (Maines, 1988). In addition, the HO-1 protein, which has a molecular weight of 33 kDa, has been cloned (Shibahara et al., 1985) and identified as heat shock protein-32 (HSP-32) in intact rat brain (Ewing and Maines, 1991; Ewing et al., 1992) and in cultured cells (Shibahara et al., 1987; Keyse and Tyrrell, 1989; Sauders et al., 1991). It has also been reported that one metabolite of heme, biliverdin, is rapidly metabolized by biliverdin reductase (bilirubin-:NAD(P)+ oxidoreductase, EC 1.3.1.24) to bilirubin, which is a powerful antioxidant (Stocker et al., 1987 a, 1987b; Llesuy and Tomaro, 1994). In addition, HO is the rate-limiting enzyme in the formation of bilirubin (Maines, 1988). Induction of HO-1 may protect cells from oxidative stress by increasing the formation of bilirubin (Nath et al., 1992) and by fostering the sequestration of catalytic iron with ferritin (Stocker et al., 1987a). It has been proposed that HO-1 induction protects the kidney against oxidative damage after ischemia and reperfusion (Maines, 1993).
Major histocompatibility complex (MHC) antigens are surface glycoproteins that play a central role in self-recognition in the immune system. These proteins are required for presentation of foreign antigens to CD4-positive lymphocytes (Widera, 1986). It is known that the CNS is an immunologically naive site from the following observations: (1) expression of MHC class I and class II antigens, which are critical for antigen presentation, is extremely low in the CNS; (2) the CNS is nearly devoid of a lymphatic system, which captures potential antigens; and (3) the blood-brain barrier imposes a complex barrier to cell extraversion from circulating blood. However, this naive nature is not absolute, as has been shown by the inability of foreign tissue grafts to survive indefinitely in the CNS and that rejection of the grafted tissue is accompanied by the upregulation of MHC antigen expression. It has been reported that MHC antigens are induced in in vivo experimental animal models, such as by treatment with kainic acid (Akiyama et al., 1988), in optic nerve degeneration (Rao and Lund, 1993), injection with cytokines and lipopolysaccharide (Kitamura et al., 1996), and by experimental allergic encephalitis (Traugott and Lebon, 1988) and in human CNS diseases such as Alzheimer's disease (Tooyama et al., 1990) and multiple sclerosis (Hayashi et al., 1988). These observations suggest that MHC antigens play a critical role in neuroimmunologic functions in the CNS.
In the transient forebrain ischemia model, it was recently shown that terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) -positive pyramidal neurons stained weakly 3 days after treatment and strongly 7 days after treatment (Nitatori et al., 1995). Biochemically, it is well known that cells undergoing apoptosis are associated with cleavage of genomic DNA, and the fragmented genomic DNA can be histologically detected by the TUNEL-staining method (Gavrieli et al., 1992). Thus, the delayed neuronal death in the CA1 is possibly caused by the apoptotic process. On the other hand, it also is known that the synthesis of many proteins decreases after cerebral ischemia (Kiessling et al., 1986) and specific families of genes are induced, including the HSP genes and immediate early genes. In fact, mRNA and protein levels of HSP-70 were markedly increased after ischemia (Kinouchi et al., 1993). However, it is unknown whether endogenous antioxidant factors, such as HO, are expressed in the neurodegenerative process by transient ischemia. In addition, the relationship between neuronal degeneration and immunologic activation is still unclear. In the present study, we examined whether HO-1 and MHC antigens are induced by transient forebrain ischemia, and we also explored what type of glial cells express the HO-1 protein.
MATERIALS AND METHODS
Animals and materials
Male Wistar rats, weighing between 180 and 220 g, were purchased from Charles River (Atsugi, Japan). The animals were acclimated to an maintained at 23°C under a 12-hour light/dark cycle. All animals were housed in standard laboratory cages and had free access to food and water throughout the study period. Institutional guidelines for experimental animal care were strictly observed. The experimental protocol was approved by the Committee for Animal Research at the Kyoto Pharmaceutical University.
The reagents used in this study were rabbit anti-rat heat shock protein-32 (HO-1) and HO-2 purchased from Stress Gen (Victoria, Canada). Rabbit anti-HO-1 and mouse anti-glial fibrillary acidic protein (GFAP) antibodies were purchased from Chemicon International, Inc. (Temecula, CA, U.S.A.), and mouse monoclonal antibodies against rat CD11b (MRC OX-42) and rat MHC class I (MRC OX18) and class II (MRC OX6) were purchased from Serotec Ltd. (Oxford, U.K.). The Bradford protein assay kit was purchased from BioRad Laboratories (Hercules, CA, U.S.A.), the Vectastain ABC Elite kit was purchased from Vector Laboratories (Burlingname, CA, U.S.A.), and the enhanced chemiluminescence detection system kit was purchased from Amersham (Buckinghamshire, U.K.).
Animal model
After the rats were fasted overnight with free access to water, they were subjected to transient forebrain ischemia as previously reported (Smith et al., 1984) with slight modifications (Kaku et al., 1993). Rats were anesthetized briefly with 3.5% halothane, intubated, and connected to a Starling-type respirator. Anesthesia was maintained with 0.7% halothane and 30% oxygen in nitrous oxide. The femoral artery and vein were exposed and silicone catheters were inserted for monitoring arterial blood pressure, arterial blood gases, and blood glucose levels. After isolation of the bilateral common carotid arteries, halothane was discontinued and rats were allowed to remain in a steady state for a period of 20 minutes, maintaining the rectal temperature at 37° ± 0.2°C, arterial PaO2 at 100 to 150 mm Hg, and PaCO2 at 20 to 40 mm Hg. Transient forebrain ischemia was induced by clamping the bilateral carotid arteries and lowering the blood pressure to 50 mm Hg for 8 minutes while administering trimetaphan camphor sulfonate and withdrawing blood from the venous catheter. After the 8-minute period of ischemia, the blood pressure was restored to normal levels by rapid reinfusion of the shed blood, and the bilateral carotid clamps were removed.
Immunoblotting and quantitative analysis
On days 1, 3, and 7 after ischemia, the rats were sacrificed and the hippocampi were removed. The brains were homogenized in 50 mM Tris-HCl buffer (pH 7.4) containing 1 mM EGTA, 1 mM EDTA, 1 mM phenyl methyl sulfonyl fluoride, and 1 μg/mL leupeptin. The homogenates were centrifuged at 50,000g at 4°C for 20 minutes, and the supernatant was used as the cytosolic fraction. The protein concentration was quantified by the Bradford protein assay kit according to the manufacturer's protocol.
Ten micrograms of cytosolic protein was run on an SDS-polyacrylamide gel (12.5%) and then transferred to a polyvinylidene difluoride membrane. To block nonspecific protein binding, membranes were incubated with 5% dehydrated skim milk and 1% normal goat serum in Tris-buffered saline (pH 7.5) containing 0.3% Tween-20. The primary antibodies, anti-HO-1 (1:10,000) or anti-HO-2 (1:500), were incubated with the membranes for 1 hour at room temperature and the enhanced chemiluminescence detection system kit was used for antibody detection.
For quantitative analysis, the X-ray film was scanned with a CCD color scanner (DuoScan, Agfa-Gevaert, Düsseldorf, Germany) and analyzed using the NIH Image 1.56 program (written by Wayne Rasband, National Institutes of Health, Bethesda, MD, U.S.A., and available from the internet by anonymous ftp from zippy.nimh.nih.gov). Statistical analysis was performed using a one-way analysis of variance. The Bonferroni/Dunn posthoc test was used for group comparisons. Data are presented as the mean ± SD.
Tissue preparation, immunohistochemistry, and quantitative analysis
On days 1, 3, and 7 after ischemia, the animals were perfused through the left cardiac ventricle with 150 mL of 10 mM phosphate-buffered saline (pH 7.4) under deep anesthesia (sodium pentobarbital, 100 mg/kg, intraperitoneally), followed by 300 mL of cold fixative consisting of 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). After perfusion, the brain was quickly removed and treated for 2 days with fixative at 4°C. After cryoprotection with 15% sucrose in phosphate buffer, the sections were cut into 20-μm slices in a cryostat. Some of the sections were stained with hematoxylin-eosin (H-E) to examine the level of neuronal damage.
The free-floating sections were incubated with the anti-HO-1 antibody (1:10,000) for 4 days at 4°C. The antibody was detected with the ABC Elite Kit and labeling was revealed by incubation with 50 mM Tris-HCl buffer (pH 7.6) containing 0.02% 3,3′-diaminobenzidine and 0.0045% hydrogen peroxide with nickel enhancement using 0.6% nickel ammonium sulfate to obtain blue-stained positive products. For double immunostaining, anti-HO-1 immunostained sections were incubated with 0.3% hydrogen peroxide to inactivate peroxidase activity, and then incubated with antibody against CD11b (1:2,000), GFAP (1:4,000), MHC class I (1:200), or MHC class II (1:200) for 4 days at 4°C. The antibody was detected as described above, and labeling was revealed by incubation with diaminobenzidine-hydrogen peroxide solution without nickel enhancement to obtain brown-stained products. For quantitative analysis, immunostained sections were scanned using a high-resolution camera (ProgRes 3008, Carl Zeiss, Jena, Germany) and then analyzed (WinRoof, Mitani Corp., Fukui, Japan). Data are presented as the mean ± SD. Immunostained sections were scanned using a high-resolution camera (ProgRes 3008) and the photographs were printed on a color photo printer (Pictrography 3000, Fuji Film, Tokyo, Japan).
RESULTS
Histologic changes in the hippocampus after ischemia
Although cell death of the pyramidal neurons was not detected on day 1 after the 8-minute period of forebrain ischemia, changes in cell shape and eosinophilic somata with pyknotic nuclei were observed in the CA1 subfield. The loss of pyramidal neurons in the CA1 began to increase 3 days after the 8 minutes of ischemia, and widespread neuronal loss was observed on day 7. This observation confirms that delayed neural death occurred in this experimental cerebral ischemia model (data not shown).
Enhancement of HO-1 protein in rat hippocampus after ischemia
The 33-kDa HO-1 protein was detectable in the cytosolic fraction from the normal rat hippocampus (Fig. 1A). Although protein levels of HO-1 did not change with the sham operation, HO-1 expression levels were markedly enhanced on days 1, 3, and 7 after ischemia (Fig. 1A). Conversely, the level of the 36-kDa HO-2 protein was detected in higher levels than the HO-1 protein in the untreated hippocampus, and the expression level of HO-2 was not altered by the sham operation or by ischemia (Fig. 1B). Quantitatively, the level of the HO-1 protein increased significantly on day 1 (227%, P < 0.05), day 3 (277%, P < 0.05), and day 7 (330%, P < 0.01) after ischemia (Fig. 1A), whereas the level of the HO-2 protein was unchanged (Fig. 1B).
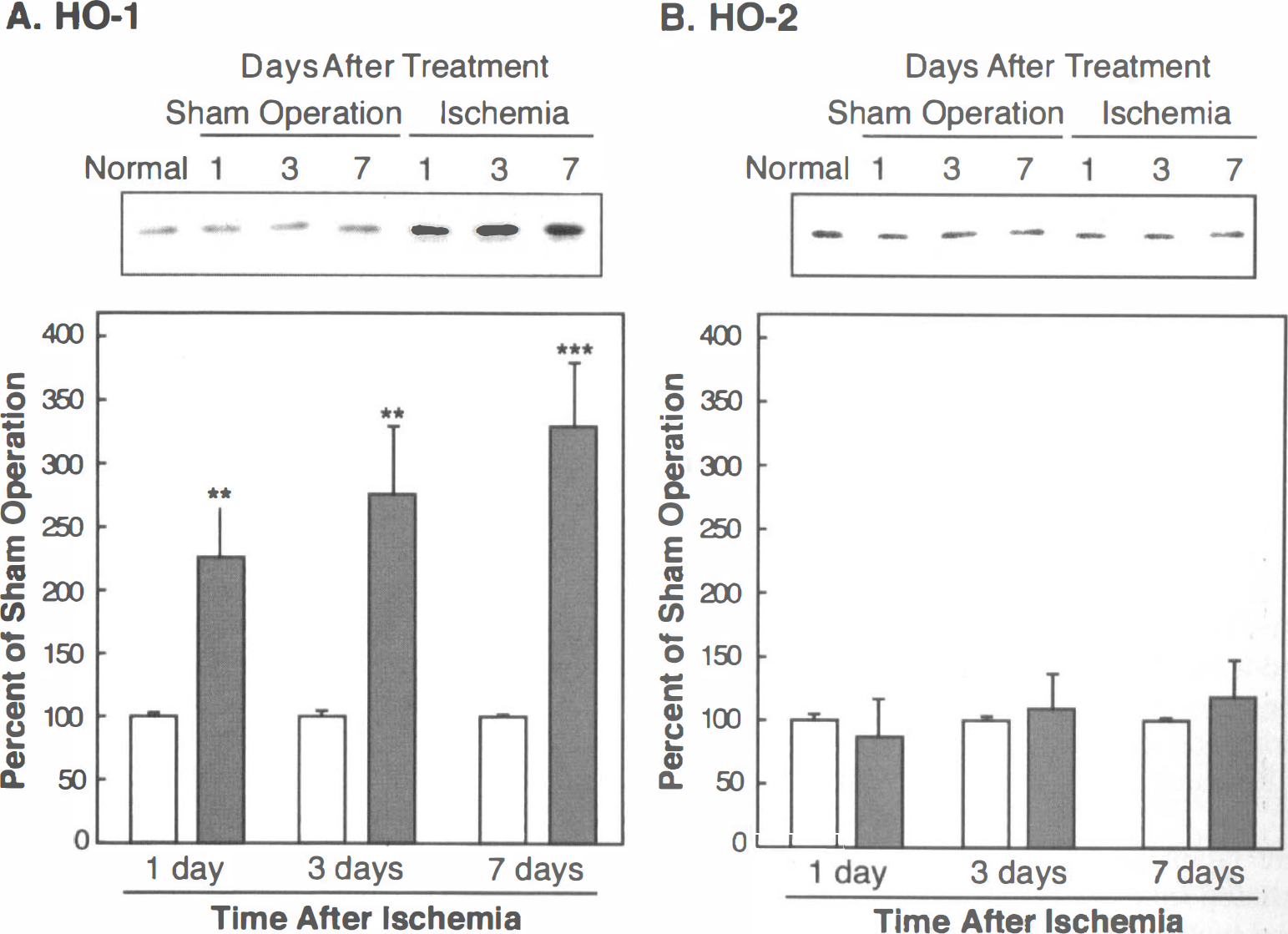
Alterations of proteins of inducible heme oxygenase (HO-1)
Changes of HO-1 immunoreactivity in rat hippocampus after ischemia
In the normal and sham-operated rat hippocampus, HO-1 immunoreactivity was distributed in a limited area, for example, in some of the neurons in the hilus of the dentate gyrus (Fig. 2A). Heme oxygenase-1 immunoreactivity was induced in the CA1 subfield, but was not induced in other subfields after ischemia (Fig. 2B and C). On day 1 after ischemia, HO-1 immunoreactivity was observed in the pyramidal neurons in the CA1 subfield (Figs. 2B, 3B and E). Subsequently, on days 3 and 7 after ischemia, HO-1 immunoreactivity was observed in the glial-like cells, but not in neurons in the CA1 subfield (Figs. 2C, 3C, 3D and 3F).
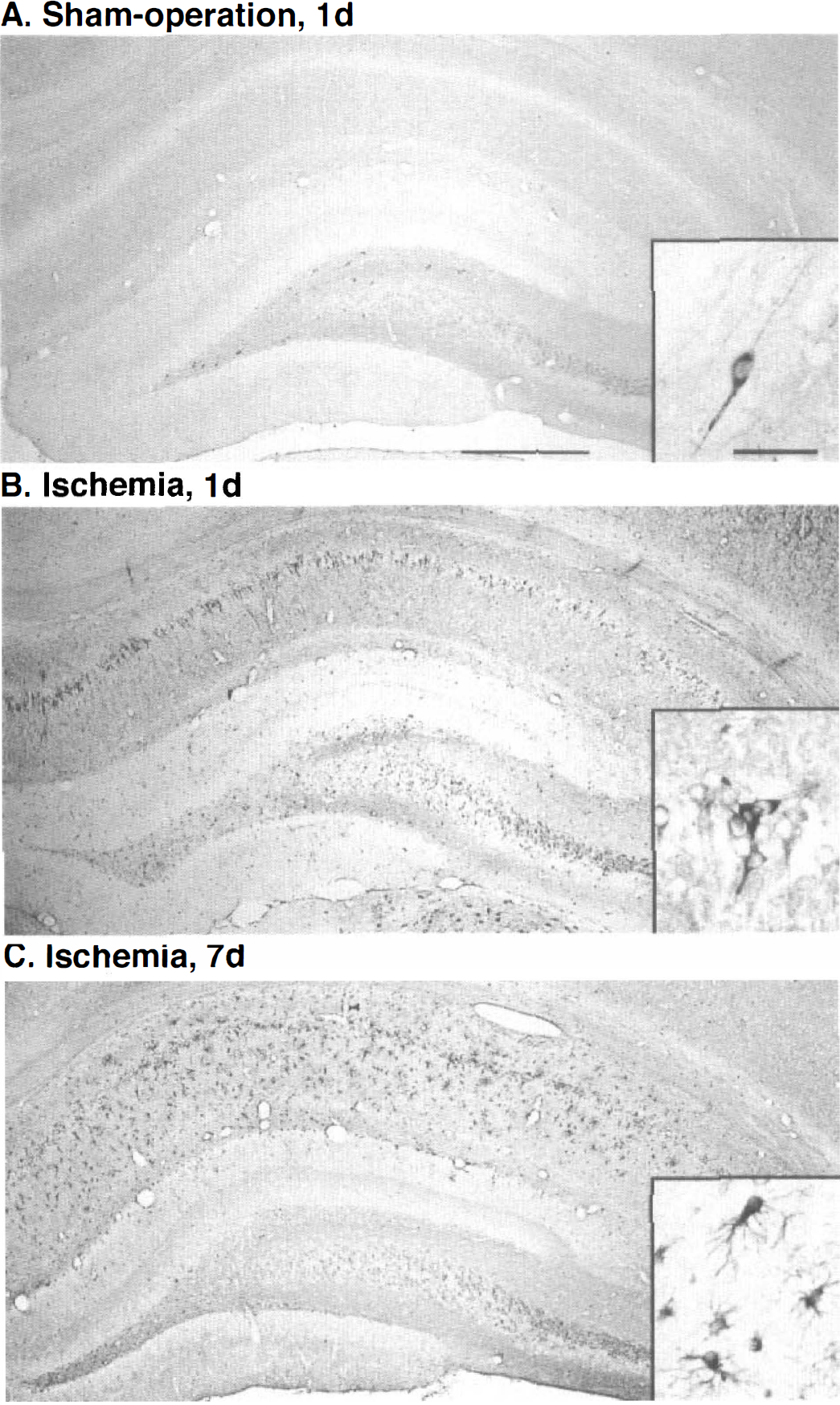
Heme oxygenase-1 immunoreactivity in rat hippocampal formation after sham operation or ischemia.
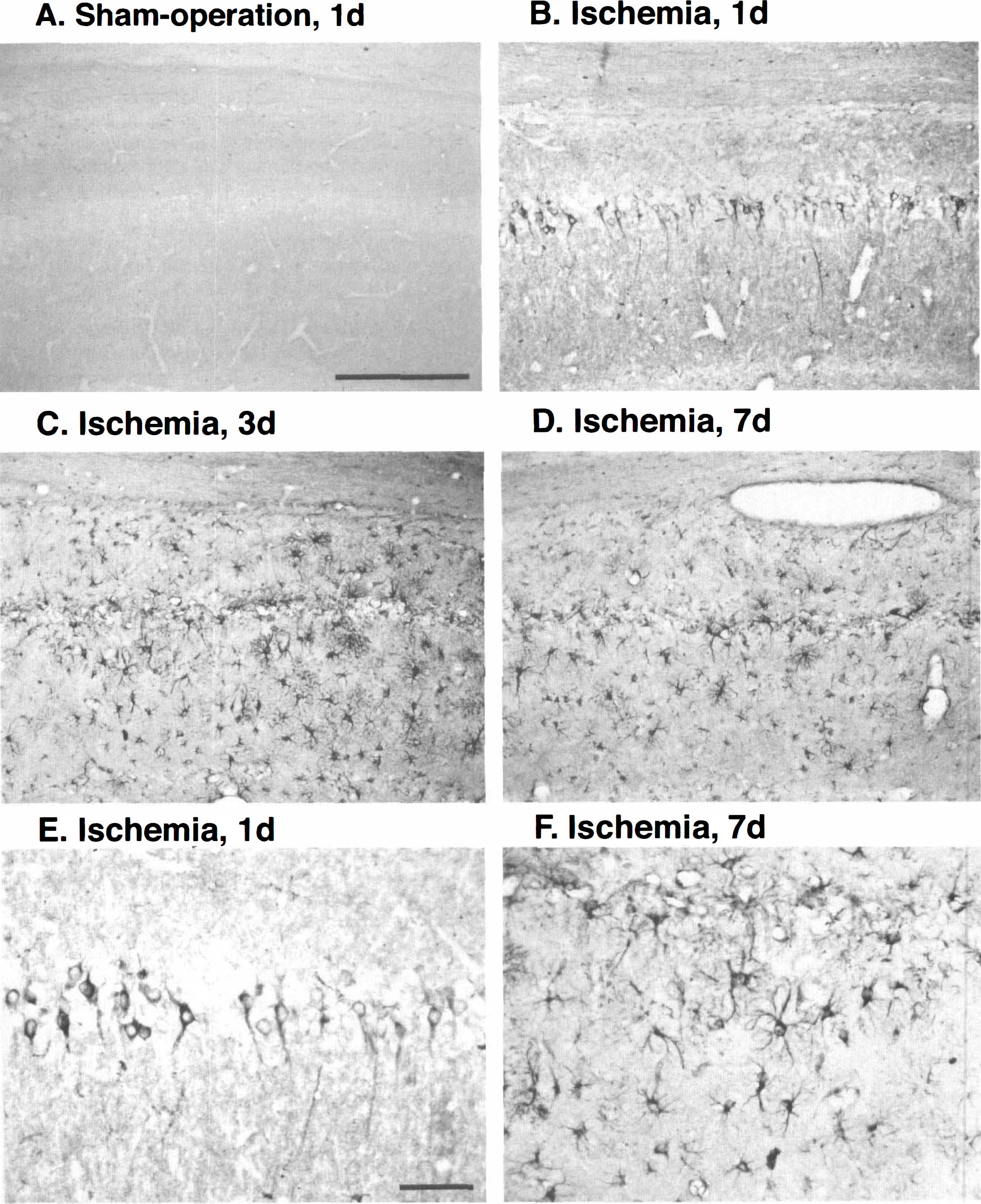
Heme oxygenase-1 immunoreactivity in the CA1 subfield after ischemia.
In the normal and sham-operated rat hippocampus, the CD11b-immunoreactive microglia have many long, thin processes like the ramified type. After ischemia, numerous CD11b-immunoreactive microglia, which have short, thick processes like the ameboid type, were observed in the neurodegenerative area of CA1 (data not shown). On the other hand, GFAP-immunoreactive astrocytes were slightly activated (data not shown). To clarify which cell type expresses HO-1, we further examined the double immunostaining with anti-HO-1 antibody and glial cell markers, for example, anti-CD11b antibody for microglia and anti-GFAP antibody for astrocytes. Heme oxygenase-1 immunoreactivity was colocalized predominantly with CD11b immunoreactivity in ameboid microglia (Fig. 4A and C). However, HO-1 immunoreactivity in GFAP-immunoreactive astrocytes was hard to detect (Fig. 4B and D).
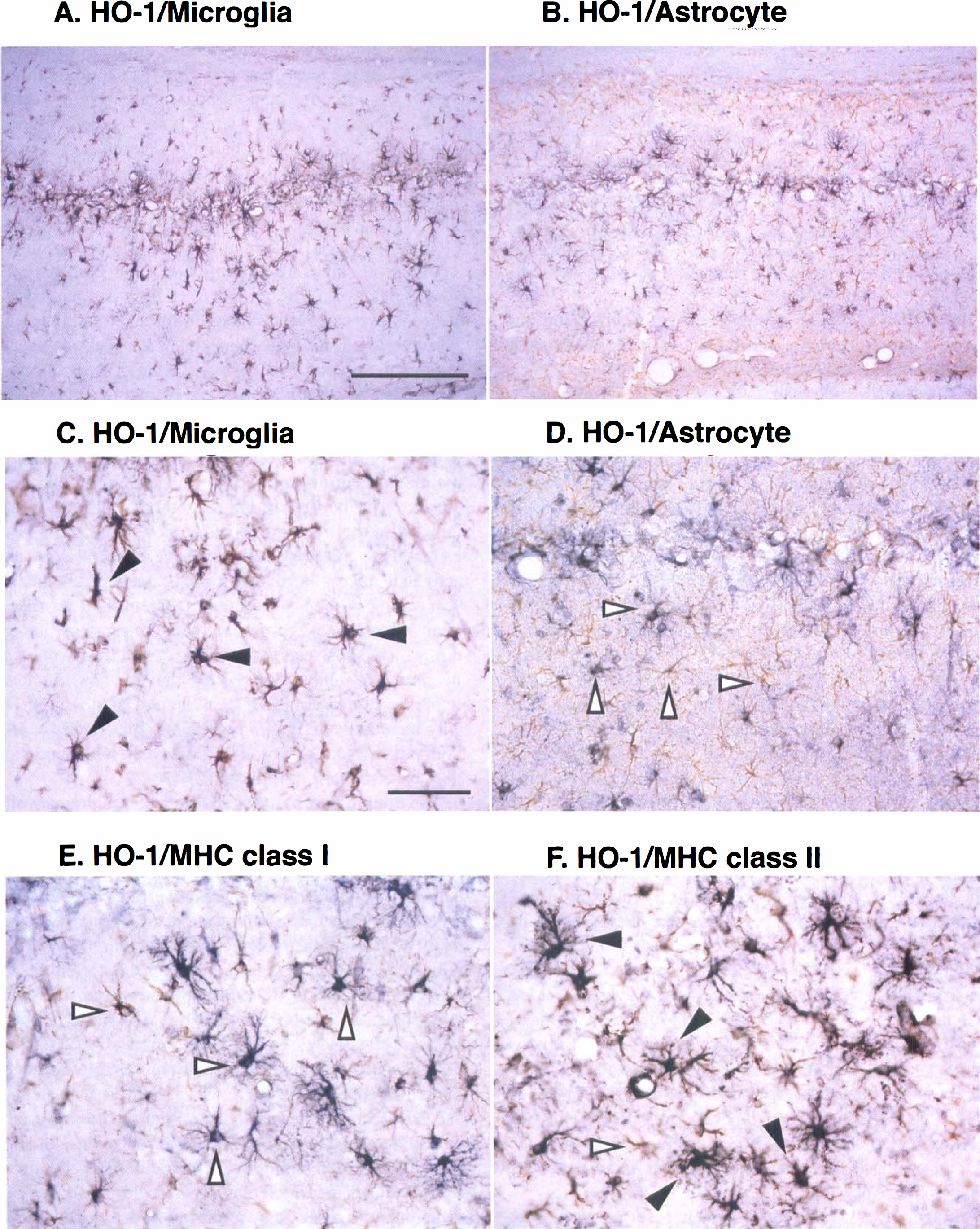
Double immunostaining with HO-1 and glial cell markers, CD11b for microglia
Colocalization of HO-1 with MHC antigens
In the normal and sham-operated rat hippocampus, neither MHC class I nor class II immunoreactivity was detectable. After ischemia, MHC immunoreactivities were induced in glial cells in the CA1 subfield. After 3 days, the immunoreactivities of MHC class I and II in ameboid microglia were observed in the CA1 subfield, but not in the other subfields in the hippocampal formation (data not shown). In addition, HO-1 immunoreactivity in ameboid microglia was mainly colocalized with MHC class II (Fig. 4F) and little with class I (Fig. 4E). Quantitative analysis shows almost all HO-1-immunopositive microglia were MHC class II immunoreactive (Table 1).
Quantitative analysis of colocalization of HO-1 with MHC class II antigen in the CA1 subfield 7 days after ischemia

The number of HO-1 immunoreactive microglia with or without MHC class II immunoreactivity (+ and −, respectively). Data are presented mean ± SD. HO-1, heme oxygenase-1; MHC, major histocompatibility complex.
DISCUSSION
In in vivo rat hippocampus, it is known that the level of expression of HO-1 mRNA is very low (Ewing et al., 1992), and HO-1 immunoreactivity is observed in limited numbers of neurons (Vincent et al., 1994). Conversely, the HO-2 protein is more widely expressed in neurons (Vincent et al., 1994). Recently, it was suggested that HO also functions as a defense system against oxidative stress, because biliverdin and bilirubin may act as physiologic antioxidants and potent scavengers of oxygen radicals (Stocker et al., 1987a, 1987b). Because neurons have low concentrations of glutathione and other antioxidants, bilirubin and biliverdin produced by HO may act as important antioxidants in stressed cells (Sauders et al., 1991). Ascorbic acid protects excitatory amino acid-induced neuronal degeneration in the rat hippocampus by serving as an antioxidant (MacGregor et al., 1996). In addition, recent studies strongly suggest that oxidative stresses participate in ischemia/reperfusion-induced neurodegeneration (Kondo et al., 1997). In fact, stress-related proteins such as HSP-70 (Kinouchi et al., 1993), nuclear factor-κB (Salminen et al., 1995), and c-Fos (Uemura et al., 1991) were expressed or activated by ischemia in animal models. Heat shock protein-70 (Sharp et al., 1993) and nuclear factor-κB (Terai et al., 1996) have also been expressed or activated in the human brain by ischemia. On the other hand, mRNA and protein of HO-1 are reported to be induced by heat shock (Keyse and Tyrrell, 1989; Okinaga and Shibahara, 1993), oxidative stress (Applegate et al., 1991), hypoxia (Murphy et al., 1991; Lavrosky et al., 1993), heme (Maines, 1988), and injection of blood into the subarachnoid space (Matz et al., 1996a, 1996b, 1997). These observations suggest the possibility that HO-1 is activated after ischemia. In fact, global and transient forebrain ischemia induced HO-1 mRNA (Paschen et al., 1994; Takeda et al., 1994; respectively), and focal ischemia induced HO-1 protein in neurons in the regions of focal ischemia, and also in microglia throughout the ischemic hemisphere (Nimura et al., 1996).
Although in the present study, HO-2 protein was constitutively expressed in the normal rat hippocampus, the protein level remained unchanged after ischemia. On the other hand, recent studies have shown that excessive production of nitric oxide by the inducible nitric oxide synthase in glial cells also participates in delayed neuronal death in ischemia (Endoh et al., 1994; Iadecola et al., 1995). It is also known that nitric oxide induces the S-nitrosylation and ADP-ribosylation of cysteine residues in several enzymes, which results in a reduction of enzyme activity (Brüne et al., 1994; Zhang et al., 1994). In contrast to rat and human HO-2 proteins, which contain three and two cysteine residues respectively, rat and human HO-1 proteins each lack a cysteine residue (McCoubrey et al., 1992; Rotenberg and Maines, 1990), suggesting that HO-1 proteins may not be influenced by nitric oxide. In addition, HO-1 and biliverdin reductase are resistant to heat shock whereas HO-2 is not (Ewing et al., 1993; Maines, 1988). The overexpression of HO-1 is associated with marked decreases in cell growth and DNA synthesis and increased survival in response to hyperoxic injury (Lee et al., 1996).
In the present study, HO-1 protein levels were significantly enhanced in the hippocampus on days 1, 3, and 7 after ischemia. On day 1, although neuronal death could not be detected, shape changes of the pyramidal neurons were observed in the CA1 subfield, as well as the transient induction of HO-1. Between 3 and 7 days after ischemia, however, HO-1 immunoreactivity in the neurons was eliminated in conjunction with neuronal loss in the CA1. It is known that HO-1 produces not only biliverdin but also iron and carbon monoxide. Iron could lead to increased oxidative stress if not handled properly, and carbon monoxide is harmful. Therefore, it is still unclear whether induction of HO-1 is helpful or harmful. Although protein synthesis in the CA1 subfield is markedly inhibited after ischemia (Kiessling et al., 1986), HO-1 was induced in the CA1 pyramidal neurons on day 1. However, the CA1 pyramidal neurons died between day 3 and day 7. With respect to HO-1 induction in CA1 neurons, we consider two possibilities. First, heme as a substrate for HO-1 in damaged CA1 neurons is not enough to produce protective biliverdin. The second possibility is that harmful iron or carbon monoxide produced by HO-1 causes delayed neuronal death. Thus, HO-1 induction may be harmful rather than helpful to CA1 neurons. Further studies of the function of HO-1 in CA1 pyramidal neurons are necessary before definitive conclusions can be drawn. On the other hand, HO-1 was induced in the microglia between day 3 and day 7. Because heme might be released from dead neurons and activated microglia phagocytoses damaged neurons by scavenging in the CA1 subfield (Jørgensen et al., 1993) after 3 days, there is sufficient heme to produce biliverdin, which is resistant to ischemia. In fact, many microglia were still richly observed even after 7 days. In addition, overexpression of HO-1 was associated with markedly increased survival in response to hyperoxic oxidant injury (Lee et al., 1996). Thus, delayed HO-1 induction may be of benefit in microglia.
The immunoreactivity of HO-1 increased dramatically in glial cells 3 days after ischemia. Double immunostaining using the anti-HO-1 antibody and glial cell markers clearly indicates that HO-1 immunoreactivity was colocalized with CD1 1b-immunoreactive microglia, but not with GFAP-immunoreactive astrocytes. It is known that CD11b-immunoreactive microglia contain many long, thin processes of the ramified type in the normal brain; however, numerous CD11b-immunoreactive microglia with short, thick processes like the ameboid type were induced after ischemia (Jøregensen et al., 1993). In the present study, it was observed that numerous ameboid microglia appeared in the CA1 subfield after ischemia, and HO-1 immunoreactivity was seen predominantly in ameboid microglia and not in ramified microglia. Recently, it has been reported that interleukin-1β and tumor necrosis factor-α, which play pivotal roles in a variety of inflammatory processes, were produced from microglia after focal cerebral ischemia (Buttini et al., 1996). In the present study, although MHC class I and II were also expressed 3 days after ischemia in microglia, HO-1 immunoreactivity was predominantly colocalized with MHC class II antigens in ameboid microglia. Recent papers have stated that kainic acid lesions induce MHC class II immunoreactive ameboid microglia (Akiyama et al., 1988) and that reactive microglia, which express MHC class II antigens, have been observed phagocytosing degenerated neuronal elements in Alzheimer's disease, Parkinson's disease, acquired immunodeficiency syndrome, and in other neuronal degenerative disorders (Dickson et al., 1993; McGeer et al., 1993). These observations suggest that numerous ameboid microglia, in which HO-1 protein was induced, may acquire resistance to oxidative stress and express MHC class II antigens, which can then have a neuroimmunologic function after ischemia.
In conclusion, we examined the induction of HO-1 and MHC antigens in rat hippocampus after transient forebrain ischemia. The protein level of HO-1 was significantly enhanced after ischemia, although the level of the HO-2 protein was unchanged. Heme oxygenase-1 was expressed early but transiently in pyramidal neurons and later but continuously in microglia in the CA1 subfield. In addition, most of the HO-1-positive cells at day 7 were ameboid microglia, but not astrocytes. After transient forebrain ischemia, MHC class I or II antigens were also expressed in microglia. Double immunostaining showed that HO-1 immunoreactivity in ameboid microglia was predominantly colocalized with MHC class II antigens.
Footnotes
Acknowledgements
The authors thank Ms. K. Iguchi for technical assistance.