
Abstract
The ability of diazepam, a benzodiazepine full agonist, and imidazenil, a benzodiazepine partial agonist, to protect hippocampal area CA1 neurons from death for at least 35 days after cerebral ischemia was investigated. Diazepam (10 mg/kg) administered to gerbils 30 and 90 minutes after forebrain ischemia produced significant protection of hippocampal area CA1 pyramidal neurons 7 days later. In gerbils surviving for 35 days, diazepam produced the same degree of neuroprotection (70% ± 30%) in the hippocampus compared with 7 days after ischemia. The therapeutic window for diazepam was short; there was no significant neuroprotection when the administration of diazepam was delayed to 4 hours after ischemia. The neuroprotective dose of diazepam also produced hypothermia (~32°C) for several hours after injection. To assess the role of hypothermia in neuroprotection by diazepam, hypothermia depth and duration was simulated using a cold-water spray in separate gerbils. Seven days after ischemia, neuroprotection by hypothermia was similar to that produced by diazepam. However, 35 days after ischemia, there was no significant protection by hypothermia, suggesting that hypothermia does not play a significant role in long-term diazepam neuroprotection. Imidazenil (3 mg/kg), which produced only minimal hypothermia, protected area CA1 of hippocampus to the same degree as that by diazepam 7 days after ischemia. At 35 days after ischemia, significant protection remained, but it was considerably reduced compared with 7 days. Like diazepam, the therapeutic window for imidazenil was short. Imidazenil neuroprotection was lost when the drug was administered as early as 2 hours after ischemia. The ability of ischemia to produce deficits in working memory and of benzodiazepines to prevent the deficits also was investigated. Gerbils trained on an eight-arm radial maze before ischemia demonstrated a significant increase in the number of working errors 1 month after ischemia. The ischemia-induced deficits in working memory were completely prevented by diazepam but not by imidazenil. There was a significant, but weak, negative correlation between the degree of CA1 pyramidal cell survival and the number of working errors in both the diazepam and imidazenil groups. Thus, if given early enough during reperfusion, both benzodiazepine full and partial agonists are neuroprotective for at least 35 days, but the lack of sedating side effects of imidazenil must be weighed against its reduced efficacy.
Transient cerebral ischemia, resulting from cardiac arrest or cardiopulmonary bypass surgery, leads to neuronal damage in specific areas of the brain of both rodents and humans (Ito et al., 1975; Kirino, 1982; Petito et al., 1987; Pulsinelli et al., 1982). The pyramidal cells in area CA1 of the hippocampus are particularly sensitive, but death of these principal cells is delayed at least 3 to 4 days after a transient ischemic event (Kirino, 1982; Pulsinelli et al., 1982; Crain et al., 1988). The cell loss in area CA1 of the hippocampus often is accompanied by anterograde memory loss in humans (Cummings et al., 1984; Zola-Morgan et al., 1986) and impairment of working memory in rodents (for review, see Davis and Volpe, 1990; Nunn and Hodges, 1994).
The delay in neuronal degeneration provides an opportunity for therapeutic intervention. Because ischemia-induced neuronal death involves a cascade of numerous cellular events, the neuroprotective efficacy of several different classes of drugs has been examined. Most neuroprotection studies focus on the excitatory amino acid receptor antagonists, which have produced varying degrees of neuroprotective efficacy, depending on the ischemia model, species, receptor subtype, dosing regimen, and presence of hypothermia (Boast et al., 1987; Gill et al., 1988; Sheardown et al., 1990; Buchan and Pulsinelli, 1990; Nellgard and Wieloch, 1992; Li and Buchan, 1993). In each of these studies, neuroprotection was assessed at short survival times (less than 7 days). Although excitatory amino acid receptor antagonists have good neuroprotective efficacy in animal models, their usefulness in humans may be limited because of adverse CNS side effects and peripheral toxicity (Lipton, 1993; Browne and McCulloch, 1994; Foutz et al., 1994; Xue et al., 1994; Grotta et al., 1995).
Other neuroprotective approaches are successful. One approach is to enhance neuronal inhibition after the ischemic event, thus limiting depolarization and subsequent increases in intracellular calcium. When administered before or after ischemia, drugs that enhance γ-aminobutyric acid (GABA) neurotransmission, such as benzodiazepines, barbiturates and GABA uptake inhibitors, produce neuroprotection for at least 1 week after ischemia (Levy and Brierley, 1979; Sternau et al., 1989; Voll and Auer, 1991; Schwartz et al., 1994; 1995; Inglefield et al., 1995). We have shown that diazepam, administered early during reperfusion, protects both hippocampus and striatum from cell death 4 to 7 days after ischemia (Schwartz et al., 1994; 1995). Another neuroprotective approach is to produce hypothermia, especially in humans (Swain et al., 1991). In rodents, the neuroprotective efficacy of postischemic hypothermia depends on the duration of the hypothermia. When hypothermia is maintained for 24 hours after ischemia in gerbils, neuroprotection of the hippocampus still is evident up to 6 months later (Colbourne and Corbett, 1994; 1995). However, in rats and gerbils, the neuroprotection produced by postischemic hypothermia of 3 to 12 hours' duration is either reduced (Colbourne and Corbett, 1994) or lost (Dietrich et al., 1993; Shuaib et al., 1995) when assessed 1 and 2 months after ischemia. At shorter survival times (1 week), even deep hypothermia of less than 3 hours' duration after ischemia fails to be neuroprotective in gerbils (Welsh and Harris, 1991).
The current study addresses issues that are important when assessing the neuroprotective efficacy of drugs. First, we have extended our original findings (Schwartz et al., 1994) and determined the therapeutic window for diazepam. Second, we determined if diazepam, given after ischemia, can produce long-term protection of pyramidal cells within area CA1 of the hippocampus. The neuroprotective efficacy of hypothermia (of similar degree and duration to that produced by diazepam) also was assessed long term. Third, we determined the neuroprotective efficacy of imidazenil, a benzodiazepine partial agonist that is nonsedating (Haefely et al., 1992; Giusti et al., 1993). Last, we also determined if diazepam and imidazenil could prevent ischemia-induced deficits in working memory long term. These results have been presented in preliminary form (Schwartz-Bloom et al., 1995; Chadwick et al., 1996).
METHODS
Subjects
Adult male Mongolian gerbils (50 to 70 g; Tumblebrook Farms, West Brookfield, MA, U.S.A.) were used in all studies. For behavioral studies, three to four gerbils were group-housed in cages in a room with a radial arm maze, on a 12/12-hour light-dark cycle. They had ad libitum access to drinking water but were maintained on a restricted diet to keep their body weight at ~85% of free feeding levels. Testing took place during the light phase of the cycle, and gerbils were fed daily after behavioral testing.
Transient forebrain ischemia
Forebrain ischemia was produced by bilateral carotid occlusion as described previously (Schwartz et al., 1994). Briefly, gerbils were anesthetized with 2.5% halothane in breathing air. A small midline incision was made in the ventral neck, and the carotid arteries were exposed and isolated. Each of the arteries was clamped with aneurysm clamps. Rectal temperature was monitored throughout the surgery and was maintained between 36.5° and 37.5°C by using a heating pad and an incandescent lamp placed over the head. Previously, we showed that these rectal temperatures corresponded to brain temperatures between 35.6° and 36.0°C during ischemia (Schwartz et al., 1994). Under these conditions, complete degeneration of area CA1 was achieved. However, brain temperature was not measured in the current studies because of the presence of an intrastriatal thermistor or electrode produces protection in the ipsilateral hippocampus (Armstrong et al., 1989 and our own observations). The protection may result from spreading depression elicited by the probe implanted several days before the ischemic episode (Verhaegen et al., 1992). After 3 minutes of occlusion, the halothane concentration was reduced to 0.5%. Five minutes after the onset of ischemia, the clamps were removed and reperfusion in each artery was verified visually. The incision was closed and a topical antibacterial ointment was applied to the wound. Sham surgery involved the same procedures except that the arteries were not clamped. All animals regained consciousness after 15 to 20 minutes and recovered in separate cages.
Drug injections
A sedating dose of diazepam (10 mg/kg intraperitoneally) or vehicle (33% polyethylene glycol in saline) was injected 30 and 90 minutes after the onset of ischemia, except in the therapeutic window study. This dosing regimen was previously shown to produce maximal neuroprotection in gerbils when assessed 7 days after ischemia (Schwartz et al., 1994). Imidazenil (1 to 6 mg/kg intraperitoneally) was injected 30 and 90 minutes after ischemia as well, except as noted otherwise. In the diazepam group, rectal temperature was monitored for 8 hours from the onset of ischemia (hypothermia lasted about 5 hours). In a previous study, we showed that the brain temperature was ~1.5°C below the rectal temperature in the presence of diazepam (Schwartz et al., 1994). We did not maintain normothermia after the diazepam injections, since this eliminates the pharmacodynamic action of diazepam (i.e., the gerbils wake up; Schwartz et al., 1994). Instead, gerbils were made hypothermic to simulate the hypothermia produced by diazepam (see later). In a separate experiment, gerbils were injected with the benzodiazepine antagonist flumazenil (10 mg/kg intraperitoneally) 15 minutes before each diazepam injection.
Hypothermia studies
In a separate group of gerbils, hypothermia was induced after ischemia by spraying the shaved back of the gerbils with ice water, as described previously (Colbourne and Corbett, 1994). The degree and duration of hypothermia were produced to mimic the degree and duration of hypothermia produced by diazepam. Rectal temperature was lowered at a rate of approximately 1.0°C every 10 minutes to ~30.5°C, where it was maintained for 5 hours. After 5 hours of hypothermia, the gerbils were warmed slowly with an incandescent lamp at a rate of 1.0°C every 10 minutes to achieve normothermia. Once gerbils reached 36°C, rectal temperature recording was halted because of excessive movement. Gerbils were killed either 7 or 35 days after ischemia for histologic analysis.
Histology
At day 7 or day 35 after ischemia, the gerbils were sedated deeply by an intraperitoneal injection of pentobarbital (Nembutal, 25 mg) and perfused with phosphate-buffered saline containing heparin (2 U/mL) followed by phosphate-buffered 4% paraformaldehyde solution. After perfusion, the brains were removed, stored in paraformaldehyde solution for 1 to 7 days, and transferred to a 30% sucrose–paraformaldehyde solution for 3 to 7 days. The brains were sliced into 30-μm sections using a freezing microtome, and the sections were mounted on slides and stained with thionin or cresyl violet. Stained slices at the level of the dorsal hippocampus 2.0 mm posterior to bregma were analyzed for damage to the CA1 sector. An experimenter, blind to the treatments, counted the number of viable cell bodies remaining in three portions of area CA1 (medial, intermediate, and lateral) stratum pyramidale (total area counted was 0.96 or 1.44 mm2, depending on the experiment). The number of surviving neurons in these three areas of CA1 were counted in the left and right hemispheres of the brain from at least two dorsal hippocampal slices per animal, and the values were averaged for each animal.
Working memory performance
Preischemia training. Gerbils were trained and tested for working memory using an eight-arm radial maze, as previously reported (Volpe et al., 1989; Levin et al., 1993), with modifications. The maze was constructed with eight arms of 45-cm length and 30-cm height, radiating from a central area diameter of 60 cm. It was located in a testing room with abundant extra-maze visual cues to aid in identification of individual arms of the maze. Gerbils were adapted to the maze before training was initiated; they were placed in a black plastic cylinder in the center of the maze with eight Noyes food pellets (45 mg) for 5 minutes or until all of the pellets were eaten. Adaptation lasted for 4 successive days when each gerbil was consuming all eight pellets in less than 5 minutes. Training consisted of 1 session/day, 5 days/week, between 12 and 4
Postischemia retesting. After ischemia, the gerbils were housed in individual cages for an additional 35-day survival period. For the first 21 days, the gerbils had unrestricted access to both food and water. On the 21st day, the gerbils were returned to the room where the initial training took place and were placed on a restricted diet to return them to approximately 85% of their ad libitum weight. Twenty-eight days after ischemia, testing was resumed using the same radial arm maze procedure used during the preischemia training period. Testing was carried out “blind” to the surgical procedures undergone by each gerbil. Postischemia testing lasted for five sessions (days).
Data analysis
Behavioral and histologic data were analyzed by multiple analysis of variance and post hoc tests (Scheffe's test or Student's t test with Bonferroni correction) to compare individual groups of means and were performed when interactions between variables were significant. Data are reported as the mean ± SD. A probability of P<0.05 was considered to be statistically significant.
RESULTS
Neuroprotection studies with the benzodiazepine full agonist, diazepam
Determination of the therapeutic window. Gerbils were injected with diazepam (10 mg/kg intraperitoneally) either 30 and 90 minutes or 4 and 5 hours after the initiation of forebrain ischemia. Seven days after ischemia, there was significant protection (59.2% ± 30.1%) of hippocampal area CA1 pyramidal cells in the 30- and 90-minute dosing group, but there was no significant protection in the 4- and 5-hour dosing group (Fig. 1). In a separate group, the benzodiazepine antagonist, flumazenil, was injected along with diazepam to demonstrate if the action of diazepam occurred through a benzodiazepine recognition site. Injections of flumazenil alone did not affect the extent of cell death in area CA1 of hippocampus (data not shown). However, flumazenil significantly reduced the neuroprotective efficacy of diazepam; only 16% of gerbils were protected (i.e., more than 75% area CA1 remained viable) in the presence of flumazenil and diazepam compared with 67% in the presence of diazepam alone (P = 0.03).
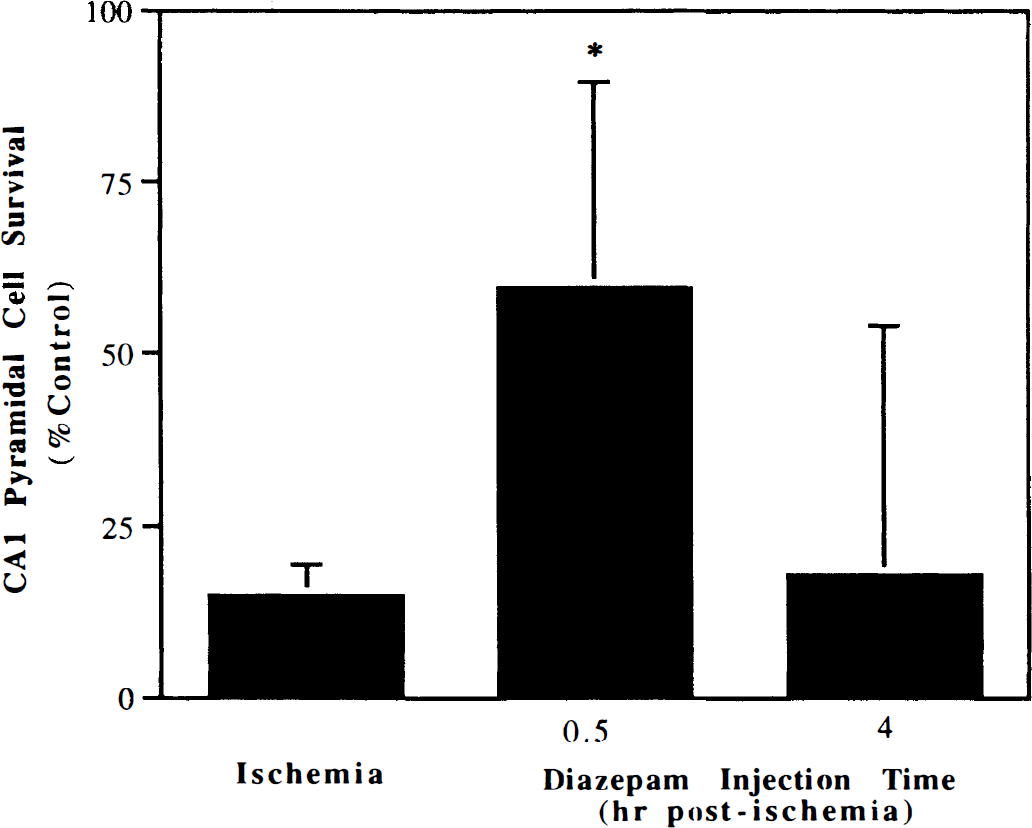
Determination of the therapeutic window for neuroprotection by diazepam. Gerbils were administered two injections of diazepam (10 mg/kg), 1 hour apart at 30 minutes or 4 hours after the onset of reperfusion and killed 7 days later. Values are the mean ± SD (n = 8/group). *P<0.05 compared with ischemia.
Long-term survival studies. Gerbils were injected with diazepam (10 mg/kg) 30 and 90 minutes after 5 minutes of cerebral ischemia. Before the second injection of diazepam, the gerbils became hypothermic as rectal temperature approached 34°C (Fig. 2). After the second injection of diazepam, the rectal temperature decreased to 32.0° ± 2.1°C, similar to our previous studies (Schwartz et al., 1994; 1995a). This degree of hypothermia lasted ~5 hours, and gerbils regained normothermia during the subsequent 2 hours.
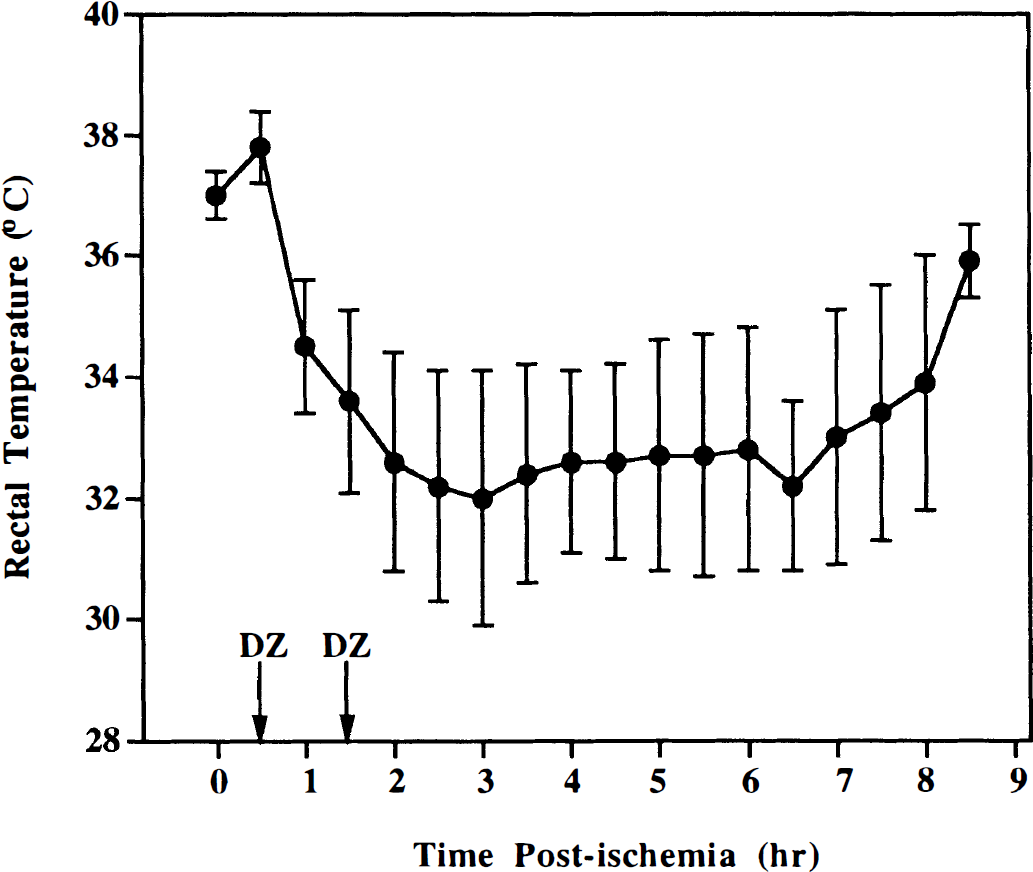
Rectal temperatures recorded during and after ischemia. Gerbils were injected with diazepam (DZ) 30 and 90 minutes after ischemia (arrows). Values are the mean ± SD of 32 gerbils.
By 7 days after cerebral ischemia, there was substantial loss of pyramidal neurons in area CA1 of hippocampus (Fig. 3 and Fig. 4). Approximately 15% of area CA1 pyramidal cells remained by 35 days after ischemia. When administered 30 and 90 minutes after ischemia, diazepam produced significant protection (P = 0.0003) of hippocampal pyramidal cells assessed at 7 days (Fig. 3). Complete protection (more than 80% viable cells) of area CA1 pyramidal cells was achieved in 37.5% of gerbils, partial protection was achieved in 50% of the gerbils, and no protection was observed in one of the eight gerbils. A similar frequency of neuroprotection was maintained 35 days after ischemia, indicating that significant neuroprotection (P<0.0001) persists for at least 5 weeks (Fig. 3). No differences in the degree of protection were observed in the medial, intermediate, or lateral portions of area CA1 (70% ± 31% versus 70% ± 30% versus 74% ± 30% protection, respectively), and the protected pyramidal neurons appeared histologically normal (Fig. 4).
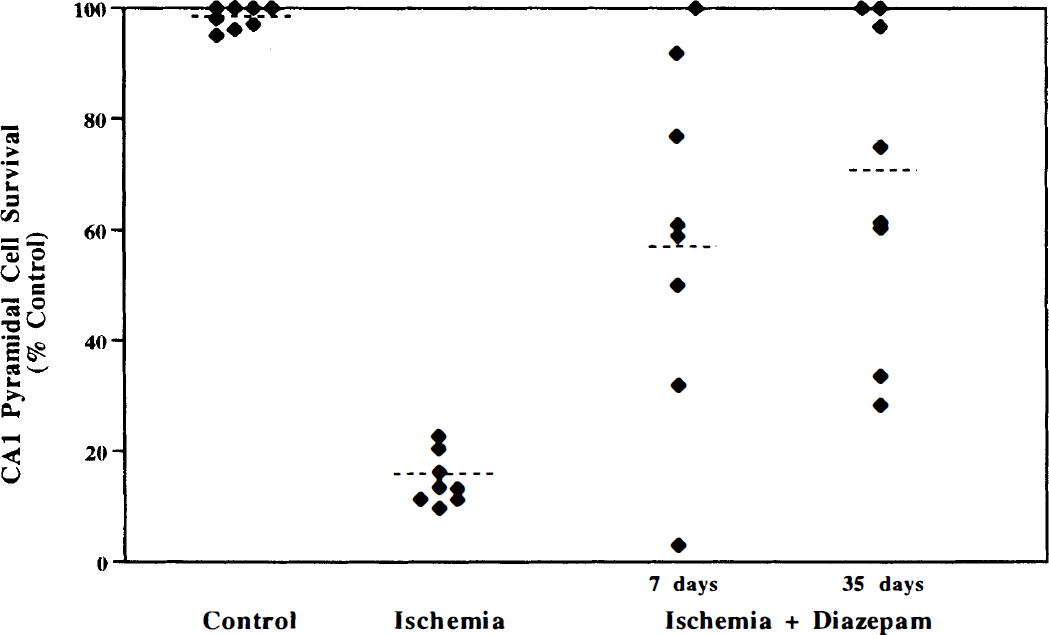
Effect of diazepam on pyramidal cell survival in hippocampal area CA1 7 and 35 days after 5 minutes of cerebral ischemia. Gerbils were injected with diazepam 30 and 90 minutes after ischemia and killed either 7 or 35 days later. Each point represents an animal; the dotted line represents the mean. Significant protection was achieved at both 7 and 35 days after ischemia (P<0.001), ANOVA followed by Scheffe's test.
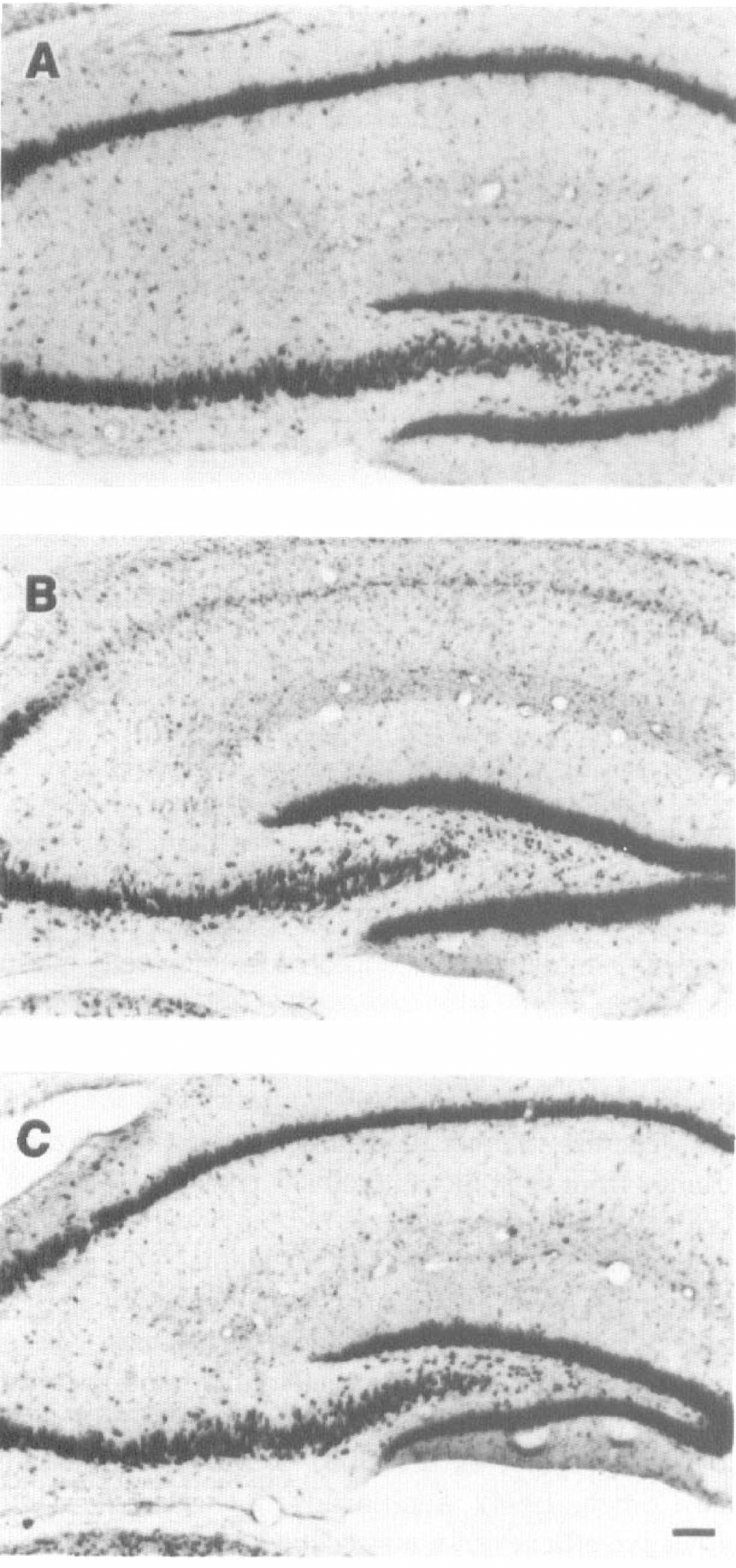
Representative photomicrographs of sections from dorsal hippocampus stained with thionin 35 days after ischemia. Sections are shown from sham-operated gerbils (
Role of hypothermia in short- and long-term neuroprotection
To address whether the neuroprotection by diazepam resulted from its ability to produce hypothermia, we performed several experiments. We previously found that maintaining normothermia in an animal injected with diazepam reduced its pharmacodynamic action, as indicated by a loss of sedation (Schwartz et al., 1994). Therefore, we simulated the degree and duration of diazepam-induced hypothermia by producing hypothermia with a cold-water spray, as described previously (Colbourne and Corbett, 1994). Additionally, we tested the neuroprotective efficacy of a nonsedating benzodiazepine that would be expected to produce little hypothermia (see later).
In the hypothermia studies, the rectal temperature was reduced slowly by a cold-water spray to ~30.5°C and maintained at that level for 5 hours (Fig. 5). The depth of hypothermia was about 2°C deeper than that produced by diazepam, and the return to normothermia took approximately 90 minutes. Although the onset and recovery from hypothermia was slightly earlier than that produced by diazepam, the duration of hypothermia was similar. When gerbils were killed 7 days later, 37.5% were completely protected (P<0.0001) from ischemic damage to the hippocampus (Fig. 6). Twenty-five percent of gerbils had no protection. This degree of protection was similar to that produced by diazepam at 7 days after ischemia (Fig. 3). At 35 days after ischemia, there was no significant neuroprotection by hypothermia (P = 0.125), although 12% of gerbils had partial protection (Fig. 6). This major loss of neuroprotective efficacy of hypothermia 35 days after ischemia was not observed with diazepam.
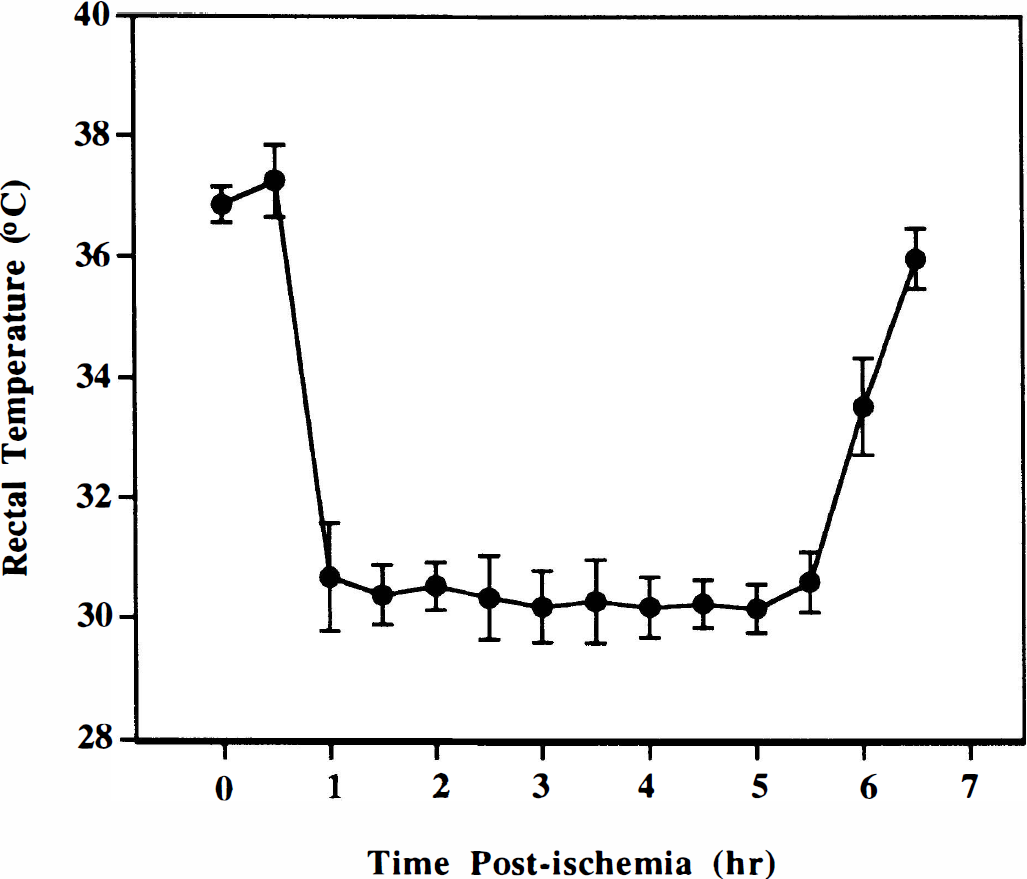
Rectal temperatures recorded during and after ischemia. Gerbils were made hypothermic with a cold-water spray (see Methods). Values are the mean ± SD of eight gerbils.
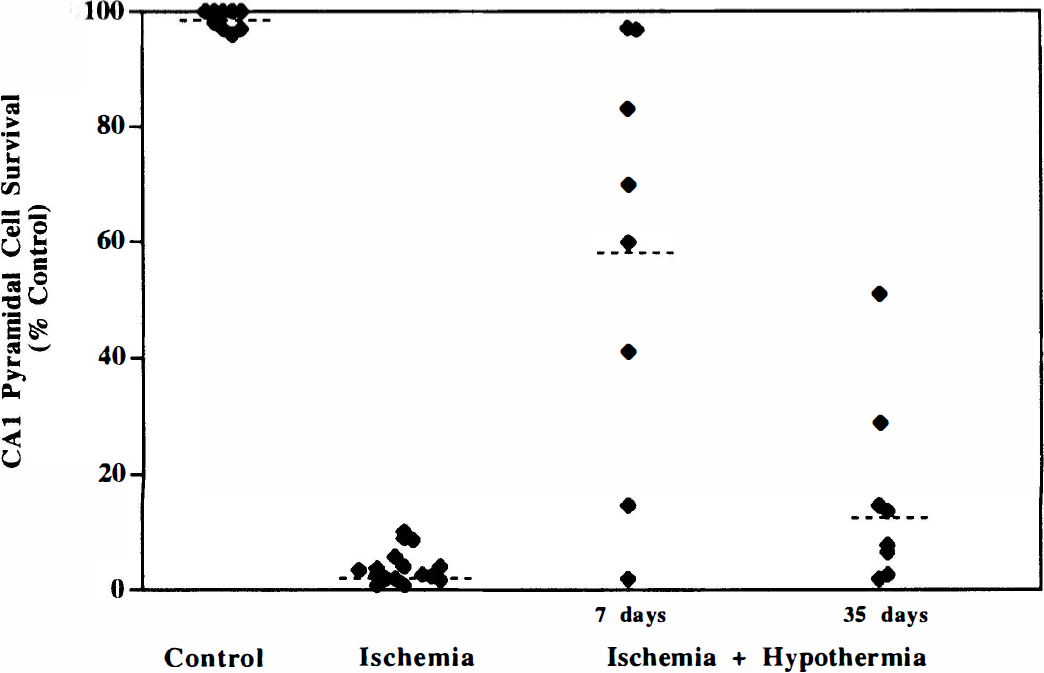
Effect of hypothermia on pyramidal cell survival in hippocampal area CA1 7 and 35 days after 5 minutes of cerebral ischemia. Gerbils were made hypothermic with a cold-water spray (see Methods). Each point represents an animal; the dotted line represents the mean. Significant protection was achieved only at 7 days after ischemia (P<0.0001), ANOVA followed by Scheffe's test.
Neuroprotection studies with the benzodiazepine partial agonist, imidazenil
Dose–response studies
Recently, novel partial allosteric modulators of the γ-aminobutyric acid (GABAA) receptor have been developed with anxiolytic and anticonvulsant activity in the absence of sedation produced by the classical benzodiazepines (Haefely et al., 1992). Imidazenil, a partial benzodiazepine agonist, does not produce ataxia or sedation in rats in doses 30- to 50-fold higher than those that produce anticonflict effects (Giusti et al., 1993). Gerbils were subjected to transient cerebral ischemia and received injections of imidazenil (1 to 6 mg/kg intraperitoneally) 30 and 90 minutes after ischemia. Normothermia was maintained in all subjects during ischemia. No significant effects of 1 mg/kg imidazenil on rectal temperature were observed (Table 1). Rectal temperatures decreased slightly (maximum of 0.9° and 1.8°C, respectively) 30 minutes after injection of 3 and 6 mg/kg imidazenil, respectively. This was followed by a return of temperatures toward preinjection values. The limited decrease in temperature contrasts with experiments in which diazepam (10 mg/kg intraperitoneally) produced marked hypothermia (32°C) after ischemia that lasted for several hours. At doses of 1 to 6 mg/kg, imidazenil did not produce sedation or ataxia. Whereas subjects receiving 3 or 6 mg/kg imidazenil did exhibit decreased spontaneous motor activity during the first 2 hours after ischemia, they were awake and responsive to external stimuli. By 3 hours after ischemia, spontaneous activity had increased considerably.
Effect of imidazenil, administered 30 and 90 minutes after ischemia, on rectal temperature in gerbils
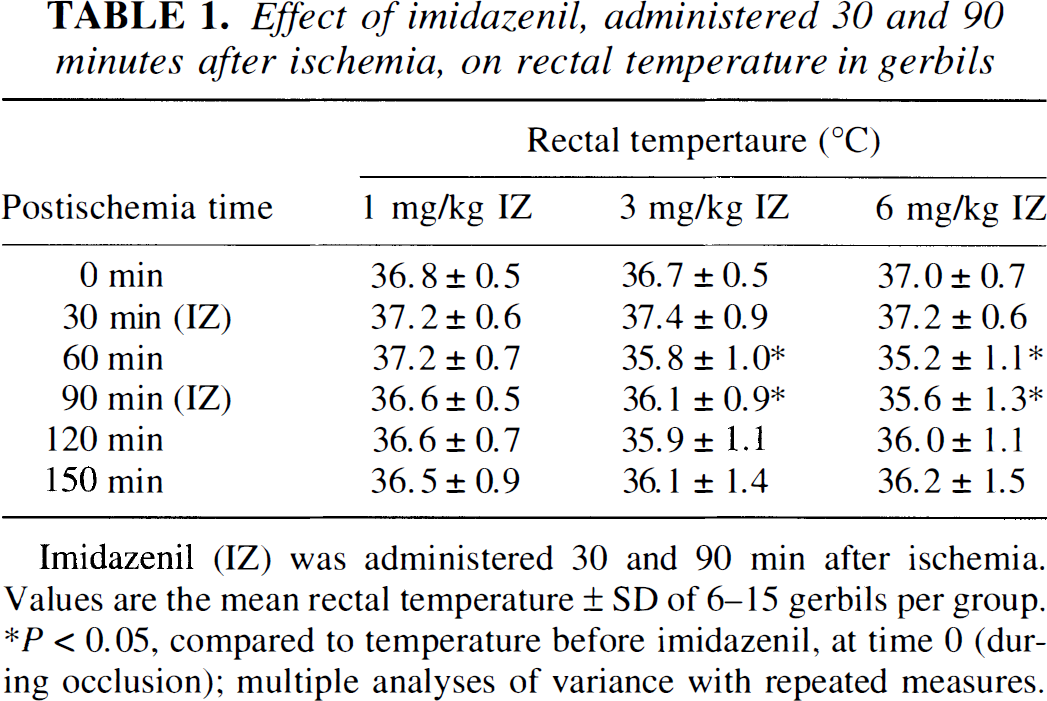
Imidazenil (IZ) was administered 30 and 90 min after ischemia.
Values are the mean rectal temperature ± SD of 6–15 gerbils per group.
P<0.05, compared to temperature before imidazenil, at time 0 (during occlusion); multiple analyses of variance with repeated measures.
Gerbils were killed 7 days after ischemia for histologic examination. The postischemia administration of imidazenil produced a dose-dependent protection of area CA1 pyramidal neurons (Fig. 7). Significant neuroprotection was achieved at 1, 3, and 6 mg/kg imidazenil, but the maximal neuroprotective effect of imidazenil was achieved at a dose of 3 mg/kg (this dose was used in subsequent experiments). In the group given 3 mg/kg, there was an average of 53% of area CA1 pyramidal cells protected (P<0.0001), which was slightly less than that observed with diazepam.
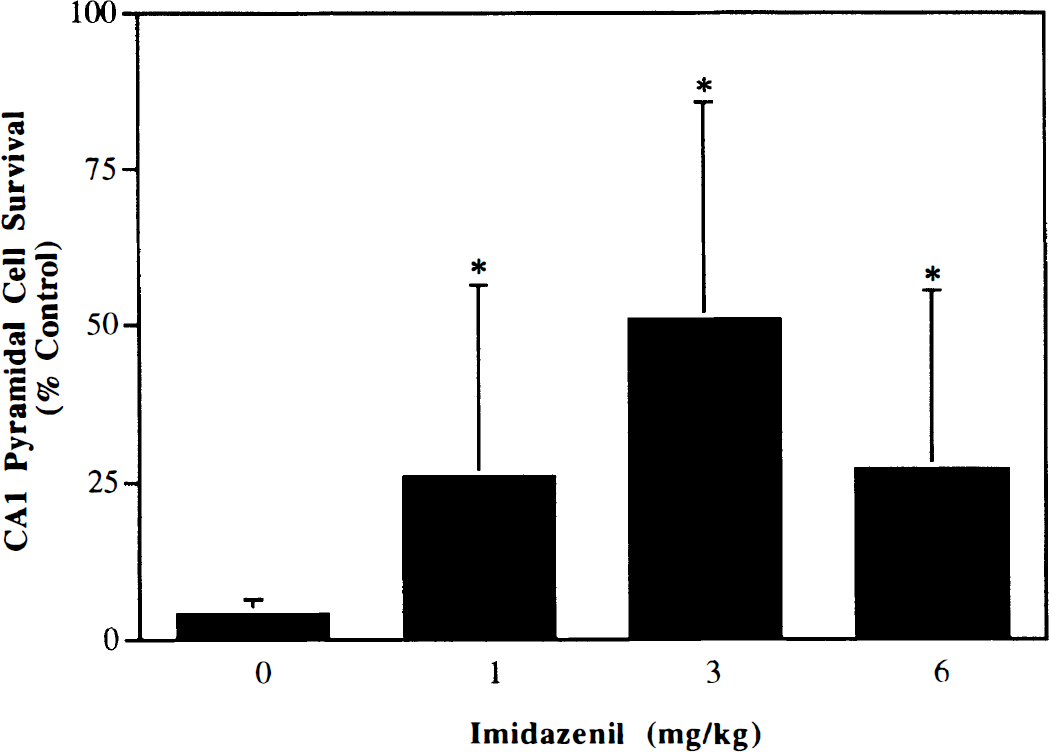
Dose–response for imidazenil neuroprotection in area CA1 hippocampal pyramidal cells 7 days after ischemia. Imidazenil (1, 3, or 6 mg/kg intraperitoneally) was administered 30 and 90 minutes after ischemia. Data are the mean ± SD (n = 8/group). *P<0.05 compared with vehicle, ANOVA followed by Scheffe's test.
Determination of the therapeutic window
When the administration of imidazenil (3 mg/kg) was delayed to 2 and 3 hours or 4 and 5 hours after ischemia, there was no significant protection of area CA1 hippocampal neurons (Fig. 8). This short therapeutic window is similar to that of diazepam, which lost its neuroprotective efficacy when administration was delayed to 4 and 5 hours after ischemia (Fig. 1).
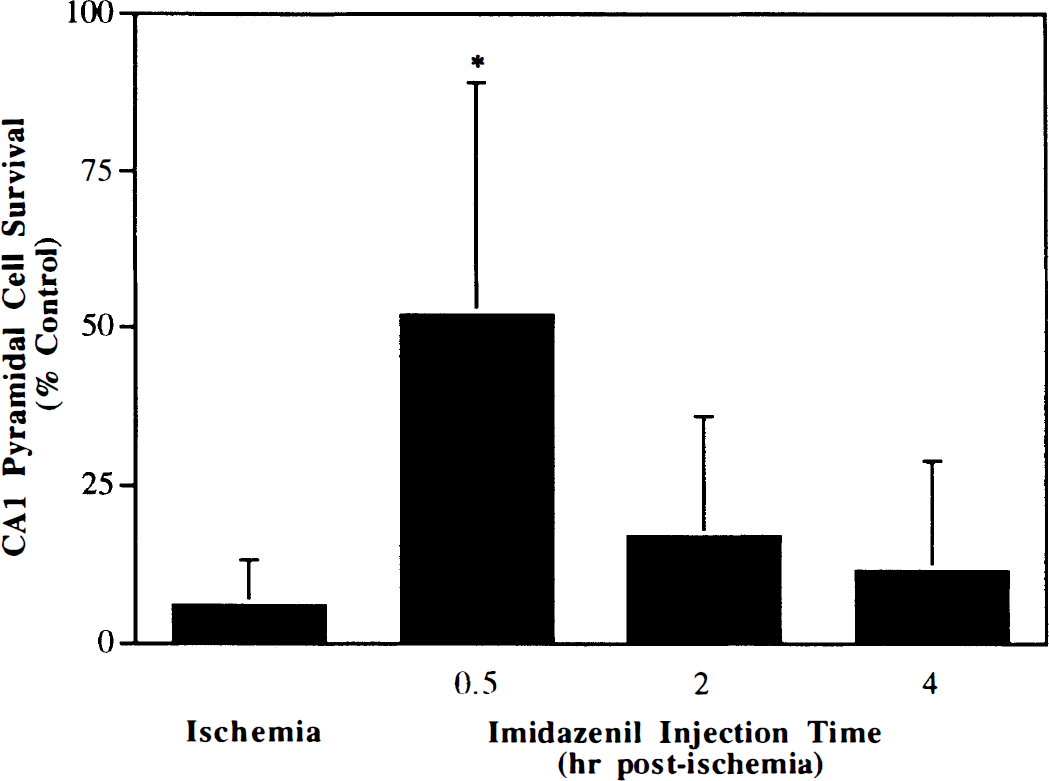
Determination on the therapeutic window for neuroprotection by imidazenil. Gerbils were administered two doses of imidazenil (3 mg/kg), 1 hour apart at either 30 minutes, 2 hours, or 4 hours after ischemia and killed 7 days later. Values are the mean ± SD (n = 8 to 24/group). *P<0.0001, compared with ischemia alone, ANOVA followed by Scheffe's test.
Long-term survival studies
Experiments also were conducted to determine whether imidazenil could produce long-term neuroprotection. Gerbils were killed either 7 or 35 days after ischemia and after having received imidazenil (3 mg/kg). Seven days after ischemia, imidazenil produced significant protection (P<0.0001) of area CA1 pyramidal neurons (Fig. 9). Twenty-five percent of gerbils were completely protected, 62% of gerbils showed partial protection, and one of eight gerbils showed no protection. The frequency of neuroprotection by imidazenil was the same as that by diazepam 7 days after ischemia (compare Fig. 3 and Fig. 9). By 35 days after ischemia, significant protection still was present (P<0.05 compared with ischemia; P = 0.43 compared with 7 days), but it had a bimodal distribution (Fig. 9). Thirty-seven percent of gerbils showed complete protection, and the protected neurons appeared to be normal at 35 days (Fig. 10). However, the remaining 63% of gerbils treated with imidazenil did not show protection in area CA1 of hippocampus. Thus, although the neuroprotective efficacy of imidazenil was similar to that of diazepam 7 days after ischemia, it was less than that produced by diazepam 35 days after ischemia.
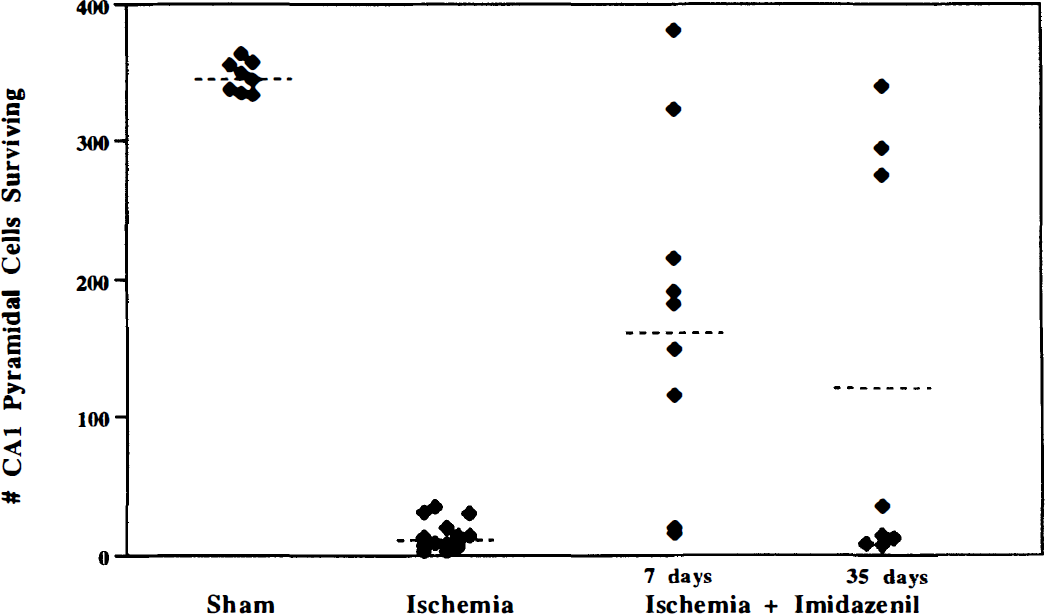
Effect of imidazenil on pyramidal cell survival in area CA1 of the hippocampus 7 and 35 days after 5 minutes of cerebral ischemia. Gerbils were injected with imidazenil (3 mg/kg) 30 and 90 minutes after ischemia and killed either 7 or 35 days later. Each point represents an animal; the dotted line represents the mean. Significant protection was achieved at both 7 and 35 days after ischemia (P<0.0001, ANOVA followed by Scheffe's test).
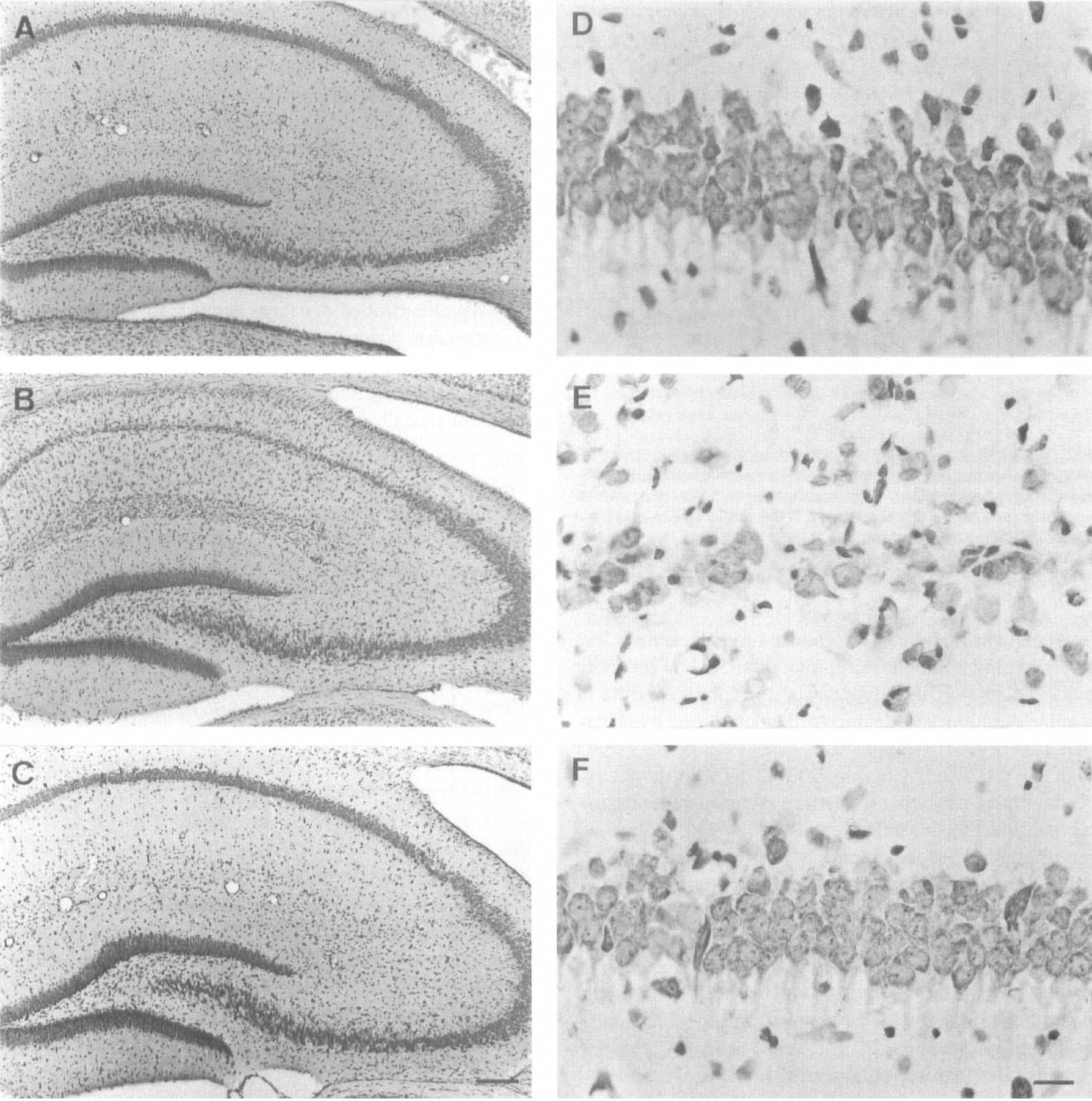
Representative photomicrographs of sections from dorsal hippocampus stained with cresyl violet 35 days after ischemia. Sections are shown from a sham-operated gerbil (
Working memory performance
Gerbils were trained over 20 sessions to perform a working memory task before the induction of cerebral ischemia. Behavior scores from five sequential sessions were grouped together and analyzed by multiple analysis of variance tests. The number of working errors decreased sequentially over the first 15 sessions (Fig. 11). There was no significant difference between the scores from sessions 11 to 15 and sessions 16 to 20, indicating that the gerbils had reached a stable level of performance. Over the 20-session training period, there was a significant decrease in the latency of response (P<0.0005 for sessions 1 to 5 versus sessions 16 to 20), consistent with the development of learning.
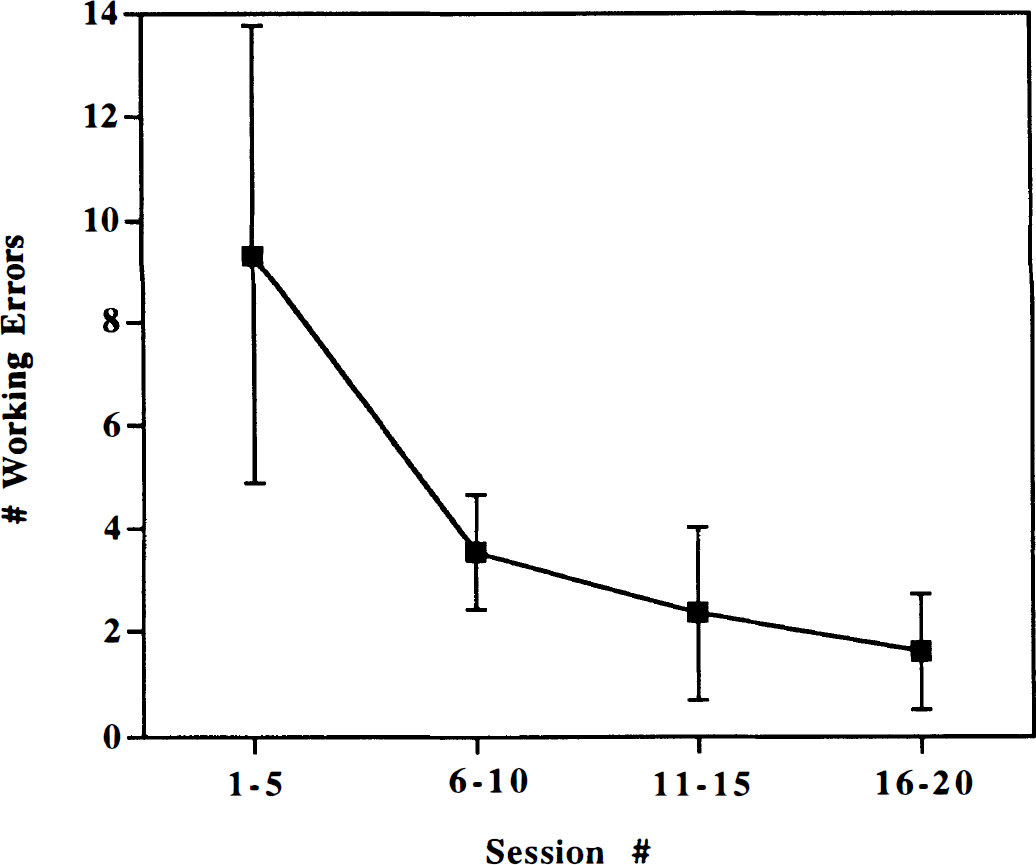
Eight-arm radial maze performance of gerbils during 20 training sessions before the induction of cerebral ischemia. The number of working errors was recorded as a measure of choice accuracy (see Methods). Each point is the mean ± SD value from five consecutive training sessions (over 1 week) (n = 39). See text for statistical analyses.
One month after the ischemia, the gerbils were retested on the eight-arm radial arm maze for five sessions (days). There was no significant interaction between retesting session number and treatment. Therefore, the data from the five retesting sessions were averaged for subsequent group comparisons. There were no significant differences between the sham–vehicle and sham–diazepam groups for the number of working errors, and these groups have been combined as a control group (Fig. 12). Ischemia significantly elevated (246%) the number of working errors (P<0.0001) in the diazepam study, but the 122% increase in working errors in the imidazenil study was not statistically significant, perhaps because of the small sample size. Diazepam, given early in the postischemic period, completely prevented the impairment of working memory assessed 1 month later (Fig. 12). In ischemic gerbils, diazepam reduced the number of working errors (P<0.0002); their performance was not different from controls. No significant difference from control was observed in imidazenil-treated gerbils subjected to ischemia. There were no significant differences in latency among any of the treatment groups.
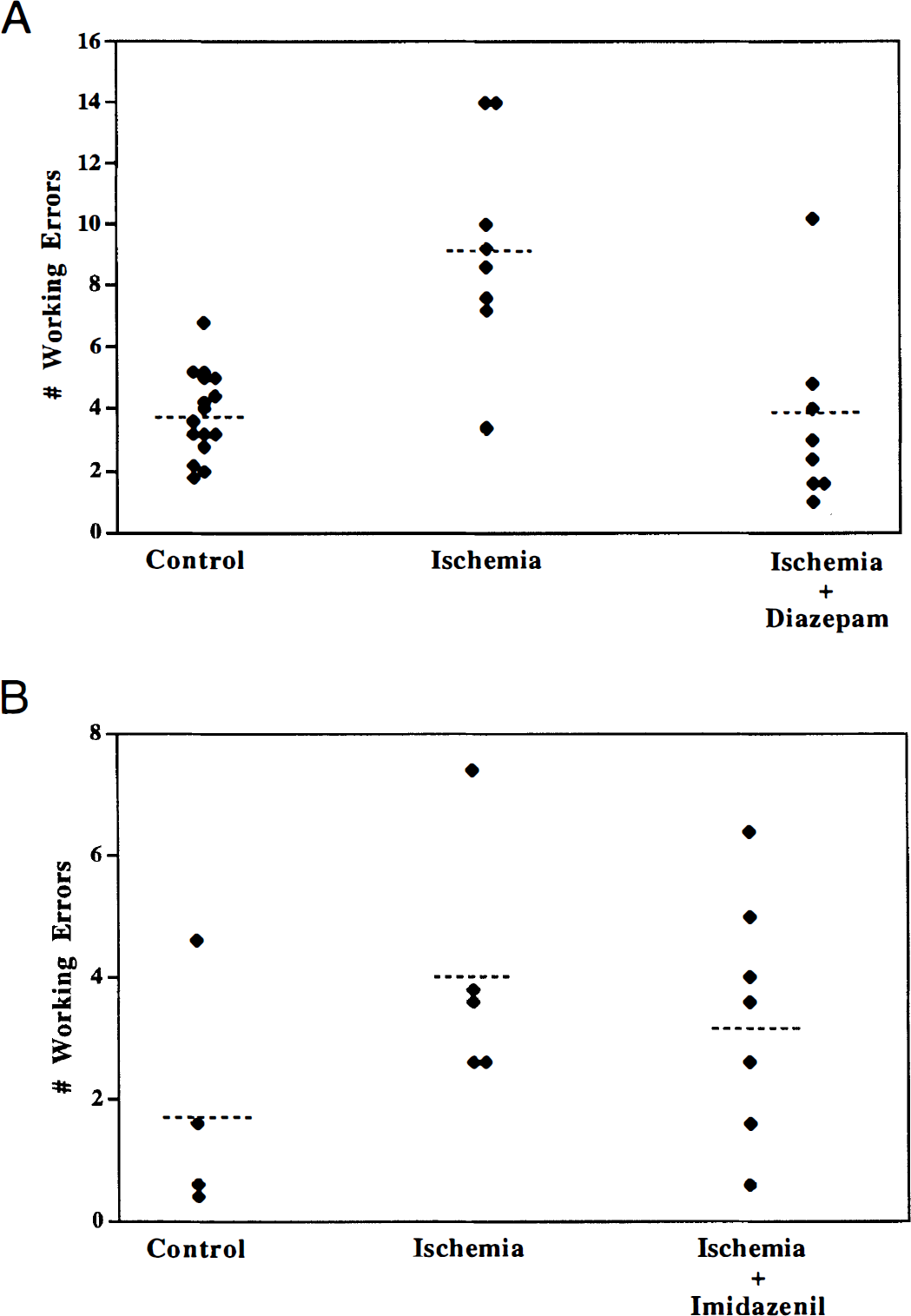
Effect of diazepam and imidazenil on eight-arm radial maze performance of gerbils 1 month after the induction of cerebral ischemia. Each point represents an individual animal's average values from five consecutive testing sessions performed 30 to 34 days after ischemia; the dotted line represents the mean. (
Correlational findings between pyramidal cell viability and behavior
Since behavioral and histologic data were available for most of the gerbils in the behavioral study, we determined the relation between impairment of working memory deficits and the degree of CA1 pyramidal cell survival in the diazepam and imidazenil studies. In the diazepam study, a Spearman rank correlation (r) of the number of working errors (averaged over the 5-day retesting period) versus the number of area CA1 pyramidal cells surviving revealed that the number of working errors increased as the number of surviving CA1 pyramidal cells decreased. Although the correlation was significant, it was weak; r = -0.406, P<0.05. There also was a significant inverse relation in the imidazenil study where r = -0.632, P<0.05.
DISCUSSION
The findings that emerged from this study include the following: (1) the therapeutic window for neuroprotection by diazepam and imidazenil (defined at 7-day survival) is narrow (less than 4 or less than 2 hours after ischemia, respectively); (2) both diazepam or imidazenil administered early during reperfusion protect CA1 hippocampal pyramidal neurons from degeneration for at least 5 weeks after ischemia, but diazepam has better efficacy; (3) hypothermia of similar duration and depth to that produced by diazepam does not produce long-term neuroprotection; and (4) diazepam, a benzodiazepine full agonist, prevents the deficits in working memory assessed 5 weeks after ischemia.
The therapeutic window for both benzodiazepines assessed in the short-term survival groups is limited: delaying administration of diazepam to 4 hours after ischemia or of imidazenil to 2 hours after ischemia is no longer neuroprotective. Interestingly, as long as these drugs were administered early enough, repeated dosing over several hours or days was not required for neuroprotection. Longer therapeutic windows (up to 12 hours after global ischemia) have been obtained with other neuroprotective agents such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor antagonists (Li and Buchan, 1993). The length of time for the therapeutic window for any neuroprotective agent may help to define which cellular processes occurring at a critical time are important to the subsequent development of neuronal death. Thus, depending on the severity of ischemia, neuronal depolarization may persist for several hours after the onset of reperfusion (Hori and Carpenter, 1994). The depolarization triggers the cascade of events leading to cell death by enabling intracellular levels of calcium to rise (Schurr and Rigor, 1992; for review). Increasing neuronal hyperpolarization with a benzodiazepine during this time should be particularly effective at preventing the increase in intracellular calcium. If neuronal hyperpolarization is delayed, the opportunity to reduce or prevent the cascade of events leading to cell death may be missed. Longer therapeutic windows for other agents may signify that neurons can still be rescued after interruption of critical cellular processes occurring “downstream.”
In prior studies, we demonstrated that diazepam protects against ischemia-induced neuronal death for short periods (4 to 7 days) after the ischemic event (Schwartz et al., 1994; 1995). Here we show that such neuronal protection (and the prevention of memory deficits) is maintained for at least 35 days after ischemia. At short survival times, the diazepam-induced hypothermia may play a significant role in the diazepam neuroprotection of the hippocampus. However, we have shown previously that microinjections of diazepam directly into the hippocampus produces neuroprotection in the absence of any hypothermia (Schwartz et al., 1995), indicating that enhancement of GABA neurotransmission directly in the hippocampus can be neuroprotective. In long-term survival animals, the diazepam-induced hypothermia is less likely to play a role in neuroprotection. Several pieces of evidence support this hypothesis. First, hypothermia of similar duration and degree to that produced by diazepam (the degree of hypothermia here was slightly greater than that produced by diazepam) was not neuroprotective 35 days after ischemia. In contrast, significant neuroprotection in diazepam-treated gerbils persisted 35 days after ischemia. Second, neuroprotective efficacy still is achieved with imidazenil, which produces little to no hypothermia for less than 1 hour. Previous studies indicate that even profound hypothermia of less than 3 hours' duration after ischemia is not neuroprotective in gerbils (Welsh and Harris, 1991). However, when hypothermia is prolonged for 24 hours after ischemia, significant neuroprotection still can be observed in gerbils 6 months later (Colbourne and Corbett, 1995).
Compared with diazepam (10 mg/kg) administered with the same dosing regimen, imidazenil (3 mg/kg) has similar neuroprotective efficacy when assessed 7 days after ischemia (60% and 53% protection, respectively). The similar efficacies 7 days after ischemia occur despite the profound hypothermia and sedation produced only by diazepam. Thirty-five days after ischemia, imidazenil's neuroprotective efficacy declined (to 36%) whereas the neuroprotective efficacy of diazepam remained high (69%). Although the average percentage of area CA1 neuroprotection declined with time, the percentage of gerbils (37%) exhibiting complete protection of area CA1 (35 days after ischemia) was the same whether they were treated with diazepam or imidazenil. However, 37% of gerbils receiving diazepam also exhibited partial protection of area CA1, whereas no gerbils receiving imidazenil were partially protected 35 days after ischemia. Thus, the two drugs produce similar short-term efficacy, independent of the presence of hypothermia. Long term, imidazenil still has neuroprotective efficacy, but diazepam may be superior because it also produces partial protection not observed in imidazenil-treated animals.
The long-term neuroprotection by imidazenil (and diazepam) contrasts with another GABA-enhancing drug, tiagabine. Tiagabine, a GABA uptake inhibitor, slowed the development of neuronal death after ischemia; neuroprotection was achieved 4 days after ischemia but was lost by 21 days after ischemia (Inglefield et al., 1995). Thus, increasing GABA neurotransmission with agents that enhance GABAA receptor sensitivity, such as full or partial benzodiazepine agonists, appears to be superior to increasing GABA concentration in the synapse using a GABA uptake inhibitor (tiagabine).
There is considerable evidence that patients who have experienced cerebral ischemia have memory impairments accompanied by degeneration of pyramidal cells in area CA1 of the hippocampus (Cummings et al., 1984; Volpe and Petito, 1985; Zola-Morgan et al., 1986). In rodents, cerebral ischemia produces impairment of working and reference memory, although working memory is more sensitive to the ischemic insult (Davis and Volpe, 1990; Kiyota et al., 1991; Nunn and Hodges, 1994). Several investigators have compared the degree of ischemia-induced damage to area CA1 with the degree of working memory impairment. In most cases, there are weak correlations (but significant) between the degree of area CA1 damage and working memory impairment after transient cerebral ischemia (Olsen et al., 1994b; Whishaw et al., 1994), similar to our findings. However, there also are studies in which no correlations (Nunn et al., 1994) or strong correlations (Kiyota et al., 1992; Volpe et al., 1992) have been observed between the degree of damage to area CA1 after ischemia and the degree of memory impairment. Based on our observations, it appears that at least 75% of cell necrosis in area CA1 must be achieved to obtain a significant deficit in working memory (using our experimental paradigm). Furthermore, choice accuracy in gerbils with only partial neuroprotection was within the range of control gerbils. Such thresholds hamper attempts to find a linear association between the degrees of area CA1 damage and working memory deficits. Thus, we support previous hypotheses that although functional CA1 pyramidal neurons contribute to working memory, there is sufficient redundancy within hippocampal and extrahippocampal circuitry to support such function (Bachevalier and Meunier, 1996). Furthermore, ischemic damage to extrahippocampal areas such as entorhinal and cingulate cortex also may contribute to memory deficits (for review, Bachevalier and Meunier, 1996).
The presence of GABAA receptors on pyramidal neurons provides a therapeutic target for benzodiazepines to enhance synaptic inhibition on these vulnerable cells. We have found that area CA1 interneurons experience ischemia-induced injury (profound dendritic beading), and the dendritic injury is prevented by diazepam long term (Inglefield et al., 1997). Whether the protected pyramidal cells (and interneurons) have normal physiologic function remains to be tested. Based on the current findings, diazepam (given after ischemia) appears to preserve working memory, a major function of the hippocampus. In contrast, the potential clinical usefulness of benzodiazepine partial agonists without the unwanted side effects must be weighed against their reduced long-term neuroprotective efficacy relative to full agonists, such as diazepam.
Footnotes
Acknowledgments
The authors thank Margaret Chen, Francine Fraser, Robin Meray, and Bryan Smith for their excellent technical assistance; and Dr. Alessandro Guidotti for his generosity in supplying imidazenil.