
Abstract
A characteristic event during reperfusion after cerebral ischemia in vivo, and reoxygenation after anoxia in vitro, is hyperoxidation of the electron carriers of the mitochondrial respiratory chain. Current studies have tested the hypothesis that there is a relation among calcium molecules derived from extracellular sources, mitochondrial hyperoxidation, and electrical recovery after anoxia in hippocampal slices. Rat hippocampal slices were superfused with artificial cerebrospinal fluids (ACSF) containing calcium chloride (CaCl2) in concentrations of: 0.5, 1, 2, and 4 mmol/L. Slices were made anoxic and then allowed to recover for 60 minutes. Reduction–oxidation shifts of NADH were measured by rapid-scanning spectrofluorometry. Synaptic activity was indicated by population spike amplitudes in the CA1 pyramidal cell subfield of the hippocampus in response to stimulation of the Schaffer collaterals. Low calcium ACSF concentrations ameliorated NADH hyperoxidation and improved synaptic transmission recovery after anoxia. High calcium ACSF concentrations had opposite effects. These data suggest a link between mitochondrial hyperoxidation and electrical recovery after postanoxia reoxygenation and support the hypothesis that cytosolic calcium overload promotes mitochondrial hyperoxidation and limits electrical recovery.
Keywords
Promising therapies such as administration of thrombolytic agents in patients undergoing a stroke have encouraged efforts to understand the mechanisms of cerebral ischemic damage during reperfusion. Evidence supports that reperfusion itself may promote ischemic injury (Watson and Ginsberg, 1989; Siesjo and Smith, 1991; Choi, 1993; Chan, 1994), and mitochondrial dysfunction may be involved in the injury mechanism (Abe et al., 1995; Ankarcrona et al., 1995; Saris and Eriksson, 1995; Schinder et al., 1996; White and Reynolds, 1996; White and Reynolds, 1997).
A consistent event during reperfusion after ischemia in brain is hyperoxidation of the electron carriers of the mitochondrial respiratory chain (Rosenthal et al., 1976; Duckrow et al., 1981; Welsh et al., 1982; Mayevsky et al., 1985; Paschen et al., 1985; Dora et al., 1986; Tanaka et al., 1986; Welsh et al., 1991; Pérez-Pinzón et al., 1997). These studies suggest that such hyperoxidation signals mitochondrial dysfunction and ensuing pathophysiologic events. For example, NADH hyperoxidation after focal ischemia was directly correlated with histopathologic changes, but inversely related to EEG recovery (Tanaka et al., 1986) and to ATP recovery (Welsh et al., 1991). Also, decreased mitochondrial hyperoxidation was associated with better recovery from ischemia. In rat brain, for example, reperfusion with a slightly “hypoxic” gas mixture lessened postischemic mitochondrial hyperoxidation and promoted greater recovery of electrical activity. Reperfusion with a hyperoxic gas mixture had opposite effects (Feng et al., 1997). In hippocampal slices, NADH hyperoxidation was diminished by antioxidant treatment, which improved recovery of electrical activity after anoxia (Pérez-Pinzón et al., 1997).
Among hypotheses to explain mitochondrial hyperoxidation after anoxia/ischemia is that mitochondria are substrate-limited in the manner of the “state 2” condition of isolated mitochondria (Chance and Williams, 1956; Raffin et al., 1988). This condition could be caused by inhibition of pyruvate dehydrogenase (PDH) (Lundgren et al., 1990; Bogaert et al., 1994; Dimlich and Marangos, 1994). Oxygen free radical production during reperfusion also may be involved in hyperoxidation, since free radical formation is increased with hyperoxia (Feng et al., 1997) and hyperoxidation can be lessened by treatment with antioxidants (Pérez-Pinzón et al., 1997).
Current research sought to determine whether extracellular calcium influenced postanoxic hyperoxidation and electrical recovery in hippocampal slices. This study was suggested by findings that reactive oxygen species may inhibit PDH through protein oxidation, a process that is potentiated by elevated calcium (Bogaert et al., 1994). The current study took advantage of the facts that: 1) mitochondrial hyperoxidation also occurs during reoxygenation after anoxia in hippocampal slices (Rosenthal et al., 1995; Pérez-Pinzón et al., 1997); and 2) hippocampal slices can be manipulated by pharmacologic agents. We report here that lower extracellular calcium blocked NADH hyperoxidation and improved electrical activity recovery after anoxia. In contrast, higher calcium concentrations augmented NADH hyperoxidation and worsened electrical activity recovery.
MATERIALS AND METHODS
Slice preparation
Male Wistar rats (250 to 300 g) were deeply anesthetized with pentobarbital sodium (60 mg/kg), and their systemic core temperature was lowered with ice. The rats then were decapitated and the cranium opened. The brain was superfused with cold artificial cerebrospinal fluid (ACSF). The composition of the ACSF was, in millimolars: 126 NaCl, 3.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 2 MgSO4, 10 glucose, equilibrated with 95% O2 and 5% CO2.
The brain was hemissected, and hippocampi were dissected from the cerebral hemispheres. Slices of 400-μm thickness were sectioned with a motorized Vibroslice microtome from Campden Instruments, Ltd. (Sileby, United Kingdom), oxygenated with 95% O2–5% CO2 in ACSF and stored at 28°C for at least 1 hour before they were transferred to the recording chamber.
Slices were placed in an interface recording chamber to measure extracellular evoked potentials (EP). The upper surfaces were oxygenated with 95% O2–5% CO2 humidified with distilled water; the lower surfaces were bathed at 35°C with ACSF flowing at 1 ml/min. This interface chamber model was chosen over a submerged slice protocol (Pérez-Pinzón et al., 1995) because spreading depression–like depolarization occurred reliably during anoxia and because EP recovery after anoxia of similar duration in this type of chamber already has been described (Pérez-Pinzón et al., 1996).
Evoked and direct current (DC) potentials were measured from the stratum pyramidale of the hippocampal field CA1 with standard micropipettes filled with 150 mmol/L sodium chloride. Orthodromic field potentials were elicited by stimulating the Schaffer collaterals with bipolar, glass-insulated, platinumiridium electrodes separated by approximately 100 μm. Constant-current, square-wave pulses (0.5-millisecond duration) were delivered at 30-second intervals. Field potentials were amplified, low-pass filtered (DC, 10 kHz), converted to digital form, and stored on a microcomputer. Anoxia was produced by switching the gas mixture in the atmosphere above the slice from 95% O2–5% CO2 to 95% N2–5% CO2. Bath P
NADH fluorometry
Redox shifts of intramitochondrial pyridine nucleotide (NAD) were measured in hippocampal slice by rapid-scanning spectrofluorometry, as described previously (Rosenthal and Sick, 1988; Rosenthal et al., 1995). The technique takes advantage of the fact that the reduced pyridine nucleotide (NADH) fluoresces much more intensely than does the oxidized form (NAD+), such that changes in fluorescence emission indicate shifts in the reduction–oxidation ratio of this electron carrier. This technique also takes advantage of previous studies that showed that mitochondrial NADH fluorescence signals were 10 to 20 times that of cytoplasmic NADH. This effect is thought to arise from decreased quenching caused by intramitochondrial binding (Avi-Dor et al., 1962; Estabrook, 1962; Jobsis et al., 1971).
Slices were illuminated with 337-nm excitation light delivered by a pulsed nitrogen laser. Fluorescence emission was measured with a cooled photodiode array detector coupled to a spectrograph. Light was collected from the tissue through the eyepiece of a microscope that imaged the CA1 subfield of the hippocampus (60×). The microscope contained a blocking filter (high-pass 350 nm) to eliminate excitation light. Fluorescence emission spectra were acquired by integrating light emission for 1 second while illuminating slices with light pulses delivered at 10 Hz. Five such spectra were averaged for each measurement period. The short pulse length of the laser (3 nanoseconds) minimized ultraviolet exposure of slices to only 30 nanoseconds per spectrum and 150 nanoseconds per recording. For most slices, spectra were acquired during control conditions, during anoxic depolarization (AD), and at 10-minute intervals during 60 minutes of reoxygenation. Shifts in the ratio of NADH/NAD+ were indicated by shifts in the amplitude of fluorescence intensity at 450 nm. These shifts were expressed as a percentage of the maximal shift toward reduction of NADH, which occurred during the transition from normoxia to anoxia, and these values were used for statistical analysis. Absolute values were transformed to percent reduction values because the geometry and light intensities vary from slice to slice and, thus, the starting NADH fluorescence signals were different in each slice.
Study groups
When anoxia was prolonged beyond approximately 1 minute in hippocampal slices, electrical activity was suppressed, ion homeostasis was lost, and AD occurred (Roberts and Sick, 1988; Pérez-Pinzón et al., 1996). In the current studies, the duration of anoxia was defined as the time from onset of AD to the time of tissue reoxygenation. The time of AD was the same in all study groups (2 minutes). Slices were assigned to one of four groups, which were defined by the concentration of calcium in the ACSF. These groups were slices superfused with 0.5, 1, 2, and 4 mmol/L CaCl2 ACSF. Before anoxia, slices were superfused with normal ACSF (i.e., 2 mmol/L CaCl2) for 10 minutes and then superfused with the new CaCl2 ACSF (e.g., from 2 to 0.5 mmol/L CaCl2 ACSF for group 1) for 30 minutes. Slices were superfused in normal ACSF (i.e., 2 mmol/L CaCl2 ACSF) at the onset of reoxygenation. Events during reoxygenation were recorded for 1 hour.
Statistical analysis
Results provided from experiments of the same group (e.g., 1 mmol/L CaCl2), which were repeated from slices isolated from the same animals, were averaged as n = 1. The significance of differences among groups was established with a two-way analysis of variance (ANOVA) test (time and calcium concentration). Significance in the redox state of NADH within each group at different times (i.e., baseline, anoxia, and reoxygenation) was determined with a Dunnett's post hoc test, where NADH fluorescence signals before anoxia were chosen as controls. Significance was accepted with P < 0.05. All data were expressed as mean ± SD. Significance in the redox state of NADH among different calcium concentrations at specific experimental times (e.g., 60 minutes of reoxygenation) was determined with ANOVA followed by a Scheffe's post hoc test. Significant differences among the four groups on EP activity after 60 minutes after anoxia was determined with ANOVA followed by a Scheffe's post hoc test.
RESULTS
The NADH fluorescence emission spectra, expressed as percent from maximal reduction during normoxic conditions, was not significantly different in the four groups studied. Slices had a NADH percent reduction of 68.0 ± 8.2%, 67.9 ± 7.3%, 62.7 ± 6.1%, and 63.2 ± 2.4% when superfused with 0.5, 1, 2, and 4 mmol/L CaCl2 ACSF, respectively.
Postanoxic hyperoxidation
Examples of NADH fluorescence emission spectra and hippocampal evoked field potentials are shown in Figs. 1 and 2. These figures exhibit data that was derived during normoxia, AD, and after reoxygenation in slices from slices superfused with 0.5, 2, and 4 mmol/L CaCl2 ACSF. Results from slices superfused with 1 mmol/L CaCl2 ACSF showed similarities with those of slices superfused with 0.5 mmol/L CaCl2 ACSF and thus were only shown in Fig. 3.
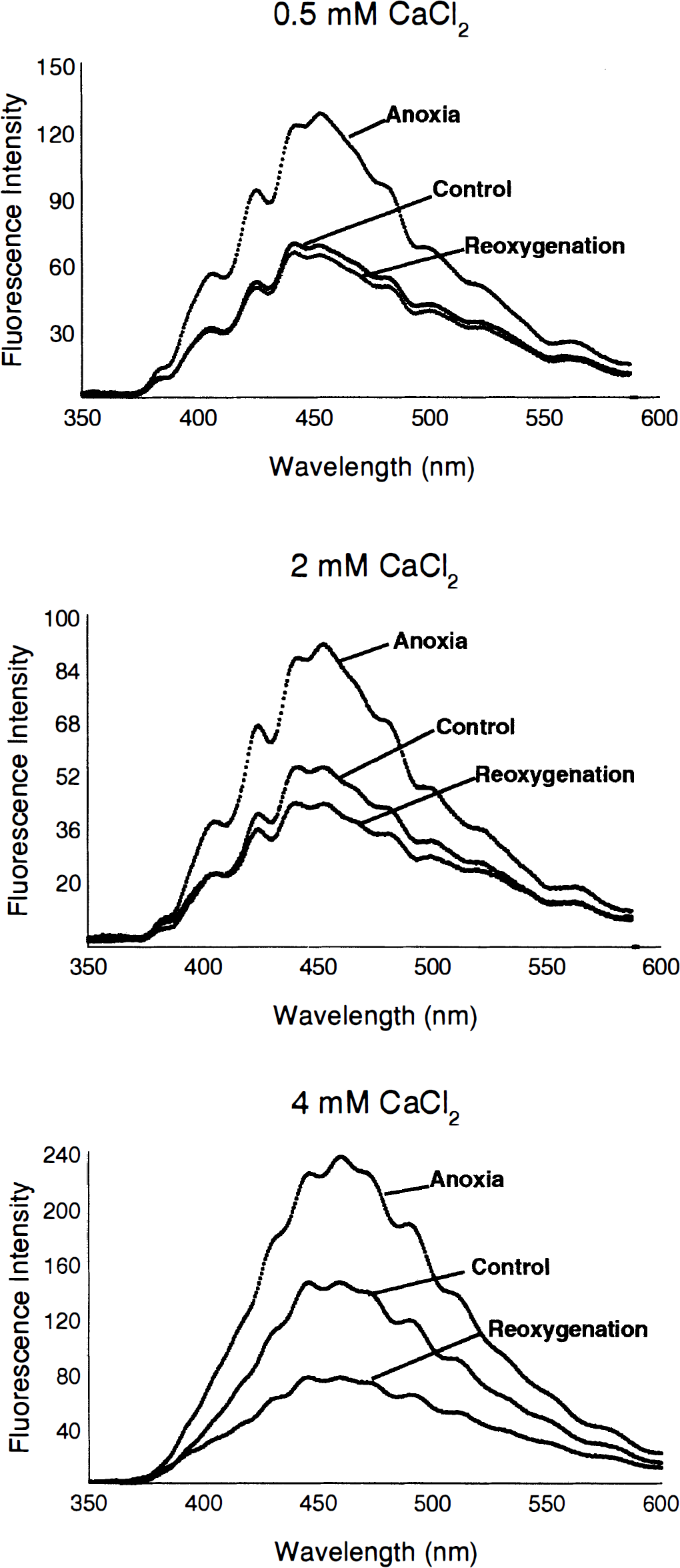
Examples of NADH fluorescence emission spectra recorded from hippocampal slices treated with 0.5,2, and 4 mmol/L CaCl2 ACSF during normoxia, anoxia, and 60 minutes after reoxygenation. Changes in CaCl2 concentration occurred 10 minutes after baseline recording with 2 mmol/L CaCl2 ACSF (e.g., from 2 mmol/L to 0.5 mmol/L CaCl2) and were prolonged for the 30 minutes that included the anoxic insult. Normal ACSF (2 mmol/L CaCl2) was restored at the onset of reoxygenation. Low CaCl2 ACSF lessened the postanoxic hyperoxidation of NADH. Increases in CaCl2 ACSF exacerbated postanoxic hyperoxidation of NADH.
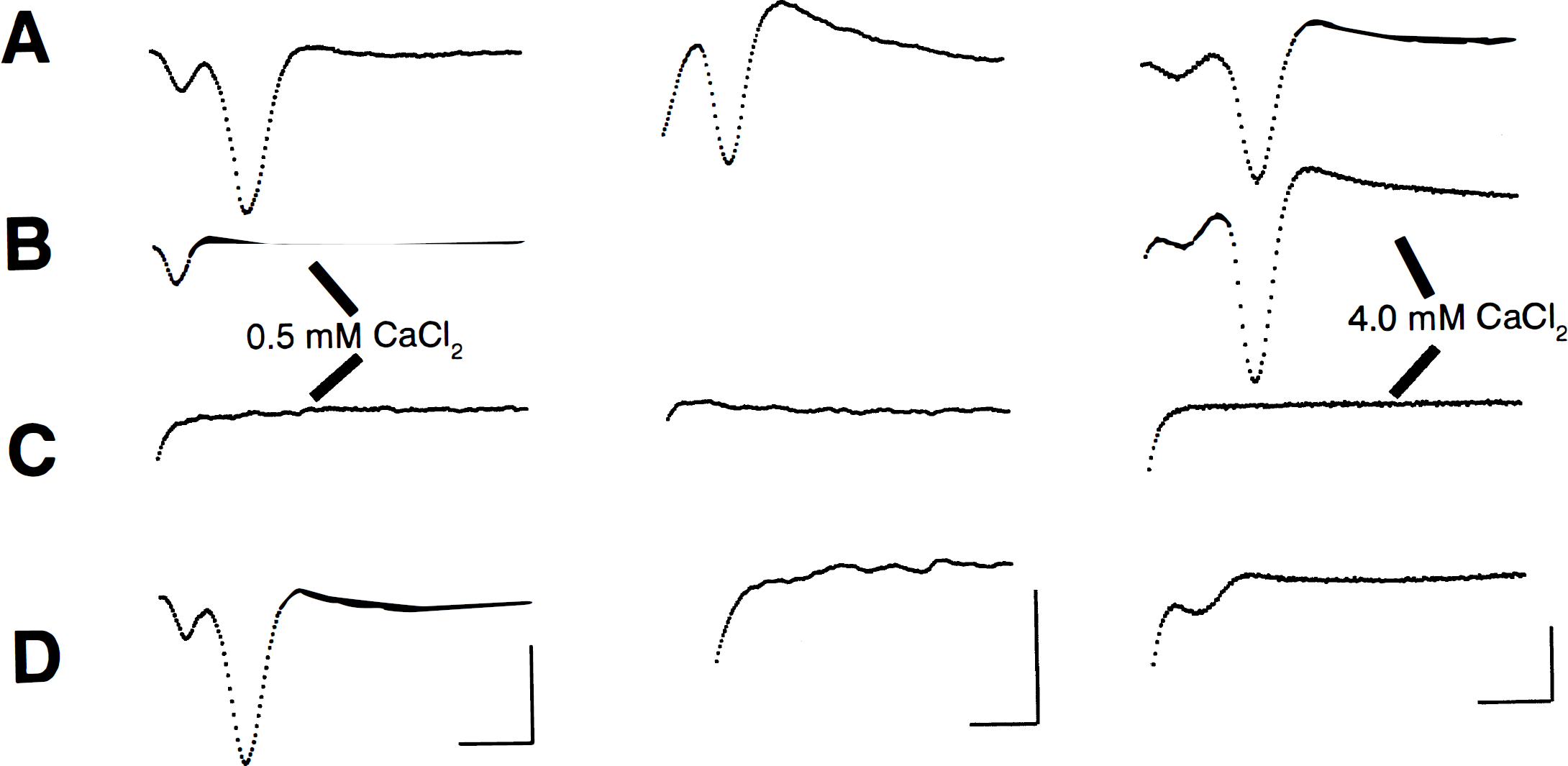
Examples of field potentials recorded from hippocampal slices treated with 0.5, 2, and 4 mmol/L CaCl2 ACSF. All examples of evoked potential records were obtained in ACSF containing 2 mmol/L CaCl2, except those indicated in the figure. Changes in CaCl2 concentration occurred 10 minutes after baseline recording with 2 mmol/L CaCl2 (e.g., from 2 mmol/L to 0.5 mmol/L CaCl2) and were prolonged for the 30 minutes that included the anoxic insult. Normal ACSF (2 mmol/L CaCl2) was restored at the onset of reoxygenation.
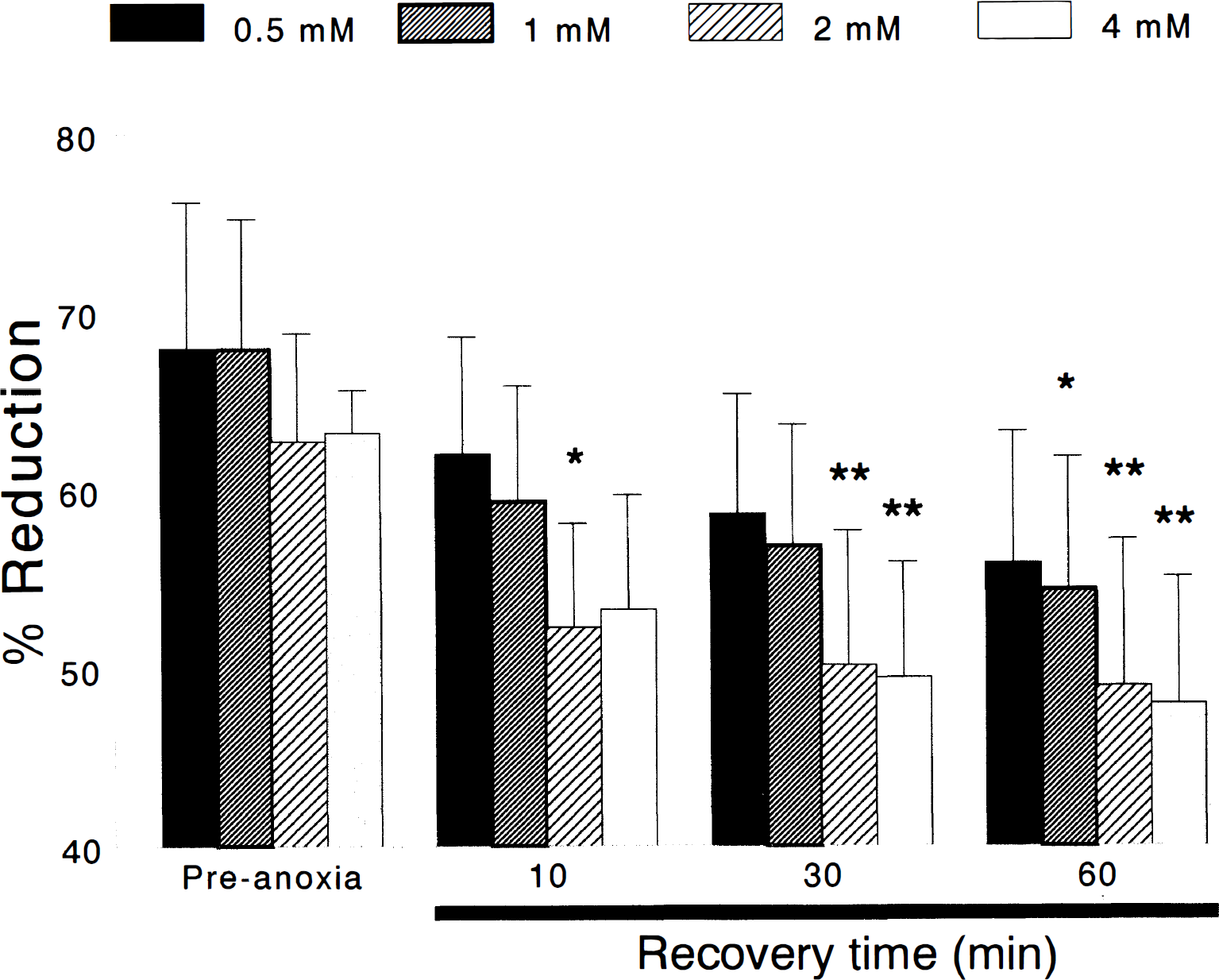
The NADH redox status in hippocampal slices before and after episodes of anoxia sufficient to produce 2 minutes of anoxic depolarization and at 10, 30, and 60 minutes of reoxygenation. These shifts were expressed as a percentage of the maximal shift toward reduction of NADH, which occurred during anoxic depolarization. Slices superfused with 2 and 4 mmol/L CaCl2 showed a long-lasting hyperoxidation (i.e., decrease in reduction) of intramitochondrial NADH/NAD+ for 60 minutes after reoxygenation after anoxia. In slices superfused with 0.5 and 1 mmol/L CaCl2 ACSF, postanoxic hyperoxidation of NADH/NAD+ was lessened (*P < 0.05; **P < 0.01).
In all slices, fluorescence intensity increased (i.e., the NADH/NAD+ ratio increased) during the transition from normoxia to anoxia. The level of reduced NADH reached maximum at approximately the time that the negative shifts of DC potential, which accompanied anoxia, reached their maxima. In all slices, reoxygenation was accompanied by decreases in fluorescence intensity, indicating that oxidation of NADH had occurred (Fig. 1).
Reoxygenation of anoxic slices caused fluorescence intensity to decrease below its preanoxia baseline, indicating hyperoxidation of NADH (c.f. Fig. 1 for spectra). In group 1 slices (0.5 mmol/L CaCl2), NADH was 58.6 ± 6.7% (n = 5) reduced after 30 minutes of reoxygenation. This redox level of NADH was not significantly different from the preanoxia levels, however. After 60 minutes of reoxygenation, NADH was only slightly less reduced than at 30 minutes (55.9 ± 7.4%) but still was not significantly different when compared with preanoxic levels (Figs. 1 and 3).
In group 2 slices (1 mmol/L CaCl2), NADH was 56.8 ± 6.8% (n = 7) reduced after 30 minutes of reoxygenation. This redox level of NADH was not significantly different from the preanoxia levels. After 60 minutes of reoxygenation, NADH was less reduced than at 30 minutes (54.4 ± 7.4%), and reached statistical difference when compared with preanoxic levels (*P < 0.05; Fig. 3).
Exacerbation of NADH hyperoxidation during reoxygenation was observed in groups 3 (2 mmol/L CaCl2) and 4 (4 mmol/L CaCl2). In group 3 slices, NADH was 50.3 ± 7.5% (n = 7) reduced after 30 minutes of reoxygenation. This redox level of NADH was significantly different from the preanoxia levels (**P < 0.01). After 60 minutes of reoxygenation, NADH was less reduced to 49.0 ± 8.2%, which was significantly different when compared with preanoxic levels (**P < 0.01; Figs. 1 and 3).
In group 4 slices, NADH was 49.5 ± 6.4% (n = 6) reduced after 30 minutes of reoxygenation. This redox level of NADH was significantly different from the preanoxia levels (**P < 0.01). After 60 minutes of reoxygenation, NADH was less reduced to 48.0 ± 7.1%, which was significantly different when compared with preanoxic levels (**P < 0.01; Figs. 1 and 3).
As expected, EP amplitude was directly proportional to the ACSF calcium concentration. For example, when ACSF calcium concentration was changed from 2 to 4 mmol/L during normoxia, an increase in the amplitude of EP was observed (Fig. 2). In contrast, decreasing the ACSF calcium concentration from 2 to 0.5 mmol/L during normoxia decreased the amplitude of EP (see Fig. 2).
The effects of anoxia in the different calcium-treated hippocampal slices on EP amplitudes are shown in Fig. 2. In group 1 slices, EP amplitudes recovered to 77.2 ± 48.3% (n = 5) of control after 60 minutes of reoxygenation after anoxia. Recovery of electrical activity was greatly diminished when ACSF calcium was increased, as in groups 2 through 4. In group 2 slices, where ACSF contained 1 mmol/L CaCl2, EP amplitudes recovered to only 45.7 ± 54.0% (n = 7) after 60 minutes of reoxygenation. However, this recovery was not statistically different than that of group 1. Recovery of electrical activity worsened when slices were treated with 2 mmol/L CaCl2 ACSF. After 60 minutes of reoxygenation after anoxia, EP amplitudes were 11.0 ± 23.5% of control (n = 7), which was significantly different from group 1 (*P < 0.05). No recovery of electrical activity in any slice (n = 6) was observed when CaCl2 ACSF was increased to 4 mmol/L (group 4).
DISCUSSION
Cytosolic Ca2+ overload has been proposed as a pathologic mechanism of neuronal death during ischemic insults (Siesjo, 1981; Siesjo, 1986). Such cytosolic Ca2+i overload may result from opening of voltage-dependent calcium channels resulting from decreased ATP and membrane depolarization (Schurr and Rigor, 1992), reversal of the Na+–Ca2+ exchange system (Stys et al., 1991), inhibition of the Ca2+-ATPase (Lipton and Lobner, 1990), and glutamate-induced N-methyl-D-aspartate (NMDA) receptor hyperactivation (Rothman and Olney, 1986; Rothman and Olney, 1987). A link between calcium overload and mitochondrial dysfunction was observed when mitochondrial membrane potential (ΔΨ) exhibited prominent and persistent depolarization in response to NMDA activation, which closely paralleled the incidence of neuronal death (Schinder et al., 1996).
Despite considerable evidence that associates calcium overload with loss of neuronal function and necrosis, little is known of the process by which calcium induces disease. The current study suggests a link between mitochondrial dysfunction and cytosolic calcium overload. We provide direct evidence of a calcium-sensitive change in mitochondrial redox status after reoxygenation of hippocampal slices after brief anoxia. As in earlier studies (Rosenthal et al., 1995; Pérez-Pinzón et al., 1997), the respiratory chain substrate NADH became hyperoxidized when hippocampal slices were reoxygenated after brief anoxia. Results from the current study further corroborate previous studies, which showed that mitochondrial hyperoxidation was associated with poor metabolic and electrical recovery after cerebral ischemia or anoxia (Rosenthal et al., 1976; Duckrow et al., 1981; Mayevsky et al., 1985; Paschen et al., 1985; Dora et al., 1986; Tanaka et al., 1986; Welsh et al., 1991; Pérez-Pinzón et al., 1997). Here we show that elevated extracellular calcium potentiated postanoxic hyperoxidation of NADH whereas lowered extracellular calcium minimized this anoxia-induced change in redox status.
Postanoxic or postischemic oxidation of NADH indicates a state of substrate limitation to mitochondria and may be similar to the state 2 condition described in isolated mitochondria (Chance and Williams, 1956). Substrate limitation may arise from different pathways, which include some key regulatory glycolytic enzymes. One key enzyme thought to play a role is the PDH complex. The role of the PDH complex in hyperoxidation of NADH is supported by previous studies that showed a strong calcium sensitivity of neuronal PDH (Huang et al., 1994). Although increases in cytosolic free calcium during normoxia activates PDH through calcium-induced dephosphorylation of the complex (Huang et al., 1994), increases in cytosolic free calcium during ischemia exerts a different effect on the PDH complex. Bogaert and others (1994) showed that although PDH activity in the frontal cortex was unchanged during cardiac arrest, it was inhibited by 72% after 30 minutes of the insult, and that 65% inhibition persisted at 24 hours after this insult. This study found that inhibition of PDH activity during reperfusion increased dose-dependently to increasing levels of CaCl2 and suggests that a combination of free radical production and cytosolic calcium overload are involved in PDH complex inhibition. These studies also are in agreement with results from the current study and our previous observation that antioxidants protect brain slices against NADH hyperoxidation after anoxia (Pérez-Pinzón et al., 1997). Thus, it is possible that postischemic hyperoxidation, PDH, and free radical production are closely linked to cytosolic calcium overload. Although the precise mechanisms of postischemic inhibition of PDH still are not clearly delineated, evidence from previous and current studies supports that reactive oxygen species induced inhibition of PDH is potentiated by elevated calcium (Bogaert et al., 1994). Thus, it is expected that if inhibition of PDH plays a key role in NADH hyperoxidation, alternative pathways such as pyruvate and lactate may ameliorate this condition.
Another possible mechanism for the reduction in the NADH fluorescence signal during reoxygenation may be degradation of this molecule. Increase free radical production during anoxia may increase pyridine nucleotide catabolism, such as protein ADP ribosylation (Janero et al., 1994; Oppenheimer, 1994). Our previous study in which antioxidants ameliorated NADH hyperoxidation (Pérez-Pinzón et al., 1997) supports the role of this second alternative pathway.
The current study clearly shows that extracellular calcium plays a key role in the hyperoxidation of NADH, but does not rule out the involvement of calcium from cytosolic stores. Previous studies have shown that chelation of cytosolic calcium in reduced buffer calcium solutions protect cortical slices against severe hypoxia (Grohn and Kauppinen, 1996).
Hyperoxidation of NADH may contribute to mitochondrial dysfunction indirectly by increasing the opening probability of the mitochondrial permeability transition (MPT) pore. Support for this hypothesis comes from a study that showed pyridine nucleotide pool (NADH/NAD+) oxidation increased MPT open probability, suggesting that NADH oxidation altered the MPT activation threshold (i.e., the pore opens at more negative mitochondrial membrane potentials; Costantini et al., 1996).
The MPT is characterized by a sudden increase in the permeability of the mitochondrial inner membrane to most molecules, with the consequent collapse of ΔΨ and osmotic swelling of the matrix. If the opening of the pore continues, even matrix proteins are released (Gunter et al., 1994). In mitochondria, pore opening is triggered synergistically by Ca2+, inorganic phosphate, and redox couple oxidation (e.g., NADH/NAD+) (Petronilli et al., 1993; Petronilli et al., 1993; Bernardi and Petronilli, 1996; Chernyak and Bernardi, 1996; Costantini et al., 1996; Nicolli et al., 1996).
These studies support efforts for defining the causes for hyperoxidation, since amelioration of hyperoxidation may inhibit MPT opening and thus the cascade of events that ensued. The decrease of hyperoxidation by low calcium concentration shown in this study points toward the role of hyperoxidation in the pathophysiologic mechanism of ischemia.